

Abstract
Peripheral blood CD4+ CD45RO+ T cells activated in vitro are able to induce expression of tumour necrosis factor-α (TNF-α) in monocytes via a contact-dependent mechanism. Activation is achieved either with interleukin-2 (IL-2)/IL-6/TNF-α over an 8-day period or cross-linking CD3 using anti-CD3 antibody for 48 hr. In this paper, we show that the p38 mitogen-activated protein kinase (MAPK) signalling pathway played different roles in the generation of effector function in these two types of activated T cells. In anti-CD3 activated T cells, p38 MAPK is a negative regulator for anti-CD3 induced cell proliferation and has no significant effect on the acquisition of either the effector function (induction of monocyte-derived TNF-α) or production of T-cell cytokines. In contrast, the p38 MAPK signalling pathway is required for the acquisition of cytokine-induced effector function and promotes cell proliferation and cytokine production.
Keywords: autoimmunity, human, inflammation, rheumatoid arthritis, T cells
Introduction
Contact between activated T cells and monocytes/macrophages induces pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1) and IL-6.1–3 We previously demonstrated that T cells are required to maintain the spontaneous TNF-α production in rheumatoid synovial tissues via a contact-dependent mechanism.2 Evidence suggests that T cells in a chronic inflammatory milieu may favour a T-cell receptor (TCR) -independent mechanism4–6 because of lower expression of CD3ε7 and TCR-ζ,8,9 combined with impaired coupling to downstream signalling pathways.10 Furthermore, we proposed that both the activated phenotype of rheumatoid synovial T cells11 and their effector function (induction of monocyte-derived TNF-α)1,2 are both induced and maintained by cytokines in the local milieu, in a TCR-independent manner. The T lymphocytes stimulated in this manner perpetuate inflammation via the induction of pro-inflammatory cytokines from macrophages and other cell types present in the synovial membrane, creating a vicious cycle of chronicity.
The CD4 memory cells isolated in peripheral blood from healthy individuals can differentiate to resemble rheumatoid T cells phenotypically, when stimulated with TNF-α, IL-2 and IL-6 in vitro for 8 days.11 These cytokine-activated T cells (Tck cells) also acquire the ability to activate monocytes/macrophages that produce cytokines/chemokines in a contact-dependent mechanism (Tck effector function). We have demonstrated that both Tck and rheumatoid T cells use nuclear factor-κB (NF-κB) signalling pathway in the monocytes/macrophages to induce cytokines/chemokines. This induction was negatively regulated by the phosphoinositide 3-kinase pathway.2,12 Although, T cells activated through their TCR (Ttcr cells) were also able to activate monocytes/ to produce TNF-α, the induction was not NF-κB dependent and was positively regulated by the phosphoinositide 3-kinase pathway.2,12 Therefore, Tck cells have been used as a surrogate in vitro model for rheumatoid T cells.
Many signalling cascades can lead to the activation of NF-κB, which includes the p38 mitogen-activated protein kinase (MAPK) pathway. The involvement of p38 in the inflammatory processes of macrophages was first reported by Lee et al. in 1994.13 Since then, the development of specific p38 inhibitors as therapeutic agents has been pursued by many pharmaceutical companies. Despite encouraging pre-clinical and in vitro studies, small molecule p38 inhibitors have never progressed beyond phase II clinical trials (reviewed in refs 14,15). The failure in clinical trials was not only the result of a lack of efficacy [small improvement on American College of Rheumatology 20 (ACR20)16], but also adverse side effects including liver injury and skin disorders both pertained to drug-related effects in different cell types and potential ‘off-target’ effects.
We have previously documented differential signalling pathways induced in monocytes/macrophages following activation by Tck, TCR-activated (Ttcr) or rheumatoid T cells. We have not yet explored the pathways involved in the acquisition of effector function in these distinct subsets. As p38 MAPK activation in response to IL-12/IL-18 stimulation has been reported in both natural killer cells17 and mouse CD4 cells,18 we explored the involvement of p38 MAPK in Tck-activated and Ttcr-activated T cells. We were able to demonstrate a contrasting role for this kinase in the acquisition of effector function.
Materials and methods
Reagents
The following three reagents were generously donated towards our research, human recombinant IL-6 by Novartis (Basel, Switzerland), hrIL-2 by NIH AIDS Research program (Bethesda, MD, USA) and hrTNF-α by Boeringer Ingelheim (Biberach an der Riss, Germany).
Carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes, Breda, the Netherlands) was used to detect cell proliferation, propidium iodide (PI) (Sigma, Dorset, UK) was used for cell cycle analysis. Purified anti-CD8, anti-CD56, anti-CD45RA, anti-CD19 and anti-CD235a (erythrocytes) antibodies used for the negative isolation of CD4+ CD45RO+ (memory cells) were purchased from BD Pharmingen (Oxford, UK). Antibodies for cell phenotype analysis including anti-CD25, anti-CD69, anti-CD62 ligand (CD62L) and anti-CD49d conjugated with FITC, Phycoerythrin (PE), PEcy5, Allophycocyanin (APC), or Pacific-Blue were purchased from BD Pharmingen. Early and late apoptosis was detected by Annexin/PI staining following the manufacturer’s protocol and the apoptosis detection kit was purchased from Alexis Biochemicals, UK. Rabbit polyclonal antibody SAK7 was a kind gift by Professor Saklatvala, Imperial College London. MAPK-activated protein kinase 2 (MAPKAPK2), the substrate of p38, was purchased from Millipore (Watford, UK). SB202190 was purchased from CalBiochem (Nottingham, UK). All reagents and media used were shown to contain < 0·1 units/ml of endotoxin, tested by the Limulus amoebocyte lysate assay (BioWhittaker, Walkersville, MD).
Human CD4+ CD45RO+ cell isolation
Human peripheral blood lymphocytes and monocytes were enriched from single-donor platelet-depleted blood packs purchased from the North London Blood Fusion Service (Colindale, UK) by centrifugal elutriation as described previously.2 Untouched CD4+ CD45RO+ cells were negatively isolated by depleting CD8+, CD19+, CD56+ and CD45RA+ cells using either Miltenyi MACS® isolation protocol as described elsewhere11 or LodeStars beads (Varian Ltd, Oxford, UK). Routinely, CD4+ CD45RO+ cells obtained using these protocols were more than 95% pure. Natural killer and CD8 contaminating cells were negligible (< 0·2%).
Cell stimulation and monocyte co-culture
The Tck cells were generated from purified CD4+ CD45RO+ cells as previously described.2 The Ttcr cells were generated by stimulation with 10 μg/ml plate-bound anti-CD3 antibody (clone UCHT1; BD Pharmingen) for 48 hr. To inhibit p38 MAPK activity, 1 μm SB202190 or an equivalent volume of DMSO (vehicle control) was added to culture 60 min before cytokine stimulation. Supernatants were harvest after day 8 (for Tck) or 48 hr (for Ttcr). Concentrations of interferon-γ (IFN-γ), granulocyte–macrophage colony-stimulating factor (GM-CSF), lymphotoxin-α (LT-α) and IL-10 were measured by sandwich ELISA as described previously.2 The Tck and Ttcr cells were then co-cultured with monocytes at a ratio of 3 : 1 for 18 hr as described previously.2 Levels of TNF-α in the supernatants were determined by ELISA.
Cell phenotype by FACS
Resting CD4+ CD45RO+ cells and Tck and Ttcr cells were stained with different combinations of surface molecule antibodies and analysed using an LSR I flow cytometer (BD Pharmingen) as described elsewhere.11
Cell proliferation by FACS
Freshly isolated CD4+ CD45RO+ cells were labelled with 2·5 μm CFSE as described previously.2 Cells were then analysed by FACScan to ensure uniformed uptake of CFSE before stimulation as described above. On the indicated days, cells were collected and counter-stained with different combinations of antibodies or with 5 μg/ml PI (added 5 min before acquisition) and analysed using LSR I. Routinely, 100 000 events were acquired for analysis using FlowJo.
Cell cycle analysis
Cells were collected on the days indicated and washed twice with PBS before being fixed in ice-cold 70% ethanol. Then, 50 μg/ml PI and 100 μg/ml RNase were added into the cell suspension before acquisition on the FACSCanto. The cell cycle phases were analysed on a gated single-cell population.
p38 kinase assay
Cells were lysed at the times indicated in 800 μl kinase lysis buffer (20 mm HEPES pH 7·4, 50 mm sodium β-glycerol phosphate, 2 mm EGTA, 1% Triton X-100, 10% glycerol, 150 mm NaCl, 10 mm NaF, 1 mm sodium orthovanadate, 2 mm dithiothreitol and 1 mm protease inhibitor cocktail). Protein concentration was determined by bicinchoninic acid assay and the kinase assay was performed as described elsewhere,19 using the in-house SAK7 antibody for p38 immunoprecipitation and 1 μg recombinant MAPKAPK-2 as substrate.
Statistical analysis
Statistical analysis was performed using Prism 4.03 software. The Kolmogorov–Smirnov normality test was carried out to determine whether values from a dataset came from a Gaussian distribution. Significance was determined using paired, two-tailed Student’s t-test (when data were normally distributed) and Wilcoxon’s rank test (when data did not come from a normal distribution). P < 0·05 is considered significant.
Results
Differential kinetics of p38 activation in CD4 memory cells activated either by IL-2/IL-6/TNF-α or by TCR ligation
An in vitro kinase assay was used to measure the activity of p38 in CD4 memory cells after stimulation of IL-2/IL-6/TNF-α or CD3 cross-linking. Consistent with published data, the peak activation of p38 in anti-CD3 activated CD4 memory cells occurred within 30 min, as measured by the ability of p38 MAPK to phosphorylate its substrate MAPKAPK-2 (Fig. 1). In comparison, the activity of p38 was bi-phasic in CD4 memory cells stimulated with IL-2/IL-6/TNF-α. A strong peak of p38 activity occurred 5 min after cytokine stimulation, followed by a weaker but distinct second increase in activity at 60 min (Fig. 1).
Figure 1.

Kinetics of p38 activation in cytokine-activated T (Tck) and anti-CD3 activated T (Ttcr) cells. CD4+ CD45RO+ T cells were stimulated with interleukin-2 (IL-2)/IL-6/tumour necrosis factor-α (TNF-α) or plate-bound anti-CD3. Cell lysates were collected at the indicated time. Activity of p38 was determined by phosphorylatation of its substrate mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2) in the kinase assay described (upper panel) and was measured by densitometer analysis and plotted as percentage of maximum response (lower panel). One representative experiment of three independent experiments is shown.
Activation of p38 MAPK is crucial for the acquisition of Tck, but not Ttcr, effector function
To investigate whether early bi-phasic p38 activity played a significant role in the process of Tck effector function acquisition, resting CD4 memory cells were pre-treated with the p38 inhibitor SB202190 for 1 hr before IL-2/IL-6/TNF-α stimulation. Eight days following cytokine stimulation the ability of these cells to activate monocytes, as measured by TNF-α production, was determined. This time-point was chosen based upon our previous observations that cytokine-activated T cells are maximally stimulated at day 8. SB202190 treatment significantly reduced the ability of Tck cells to induce monocyte TNF-α production when compared with Tck cells generated in the presence of DMSO (control) (P < 0·005, Student’s t-test, Fig. 2a). Hence, Tck cells generated in the presence of DMSO (control) activated monocytes to produce an average of 884 ± 1224 pg/ml TNF-α (range 87–3300 pg/ml) whereas, SB202190 pre-treated Tck cells only activated monocytes to produce an average of 248 ± 358 pg/ml TNF-α (range 11–1344 pg/ml). In four out of 13 experiments, inhibition of early p38 activation resulted in 80% reduction of Tck effector function. This suggests that the early bi-phasic activation of p38 in Tck cells is critical for the acquisition of the effector function.
Figure 2.
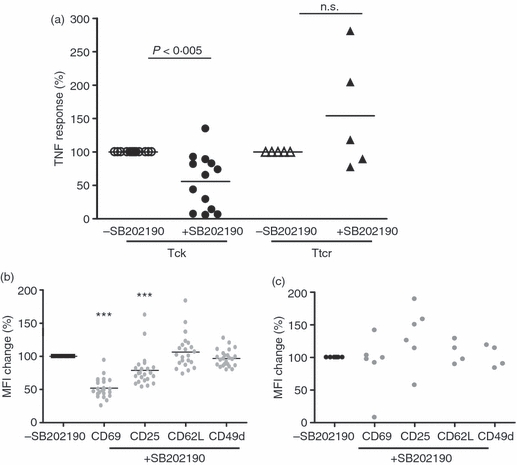
Early p38 activity regulated the effector function of cytokine-activated T (Tck) cells but not anti-CD3 activated T (Ttcr) cells. CD4+ CD45RO+ T cells were stimulated with interleukin-2 (IL-2)/IL-6/tumour necrosis factor-α (TNF-α) for 8 days or with plate-bound anti-CD3 for 48 hr. Each symbol represents an individual donor. All results are presented as a percentage change of those generated without SB202190. (a) The ability of Tck ± SB202190 and Ttcr ± SB20219 to activate monocytes was determined by TNF-α produced after co-culture with autologous monocytes for 18 hr. Tck versus Tck + SB202190: n = 13, P < 0·005; Ttcr versus Ttcr + SB202190: n = 5, not significant. (b) Change of CD69, CD25, CD62L and CD49d expression on Tck ± SB202190. n = 23. p38 inhibition significantly down-regulated mean fluorescence intensity of CD69 (P < 0·0001) and CD25 (P < 0·0005). (c) Expression of CD69 (n = 6), CD25 (n = 6), CD49d (n = 4) and CD62L (n = 4) were not significantly modulated by SB202190 in Ttcr cells.
In contrast, the ability of Ttcr cells to activate monocytes to produce TNF-α was not significantly modulated by pre-treatment with the p38 inhibitor (Fig. 2a). The Ttcr cells generated in the presence of DMSO, activated monocytes to produce 533 ± 461 pg/ml TNF-α (range 86–1260 pg/ml) compared with 650 ± 507 pg/ml TNF-α (range 242–1384 pg/ml) induced by SB202190 pre-treated Ttcr cells.
Expression of CD69 and CD25 on CD4+ CD45RO+ Tck cells is modulated by p38 inhibition
We previously determined that Tck activation causes the up-regulation of several T-cell activation markers including CD25, CD69, CD62L and CD29/CD49d (VLA-4). CD69 and CD49d has previously been implicated in the activation of monocytes by T cells by us11 and others.3 Pre-treatment with SB202190 did not alter the expression levels of CD62L or CD49d on Tck cells (Fig. 2d) or Ttcr cells (Fig. 2e). CD69 was up-regulated on both Tck and Ttcr cells and its expression was significantly attenuated on Tck cells pre-treated with SB202190 (Fig. 2d) but not on the Ttcr cells (Fig. 2e). This suggests that the up-regulation of CD69 was p38 dependent when CD4 memory cells were activated through cytokine receptors but not following TCR ligation.
The p38 inhibition was also found to significantly down-regulate CD25 in Tck cells (Fig. 2d) but not Ttcr cells (Fig. 2e). T regulatory cells (Treg cells) are characterized by their high expression of CD2520 Pre-incubation with SB202190 did not alter the percentage of Treg cells following cytokine-activation (data not shown). This suggested that p38-dependent acquisition of Tck effector function was not carried out through manipulation of Treg cells in culture.
Cytokine secretion by Tck cells is decreased by p38 inhibition
Given the role of p38 in the stabilization of pro-inflammatory cytokines including TNF-α and IFN-γ (reviewed in ref. 21), we investigated the effect of p38 inhibition on the cytokines produced by Ttcr and Tck cells. In agreement with published papers, IFN-γ, LT-α and GM-CSF production by Tck cells after 8 days of culture were reduced significantly by p38 inhibition. Pre-treating CD4 memory cells with SB202190 before IL-2/IL-6/TNF-α stimulation reduced the concentration of IFN-γ from 10658 ± 28115 to 1443 ± 2897 pg/ml (P < 0·006, Student’s t-test); LT-α from 1241 ± 2181 to 851 ± 1776 pg/ml (P < 0·005, Wilcoxon rank test); GM-CSF from 3579 ± 2299 to 586 ± 1394 pg/ml (P < 0·0001, Student’s t-test); and IL-10 from 11 ± 16 to 2 ± 3 pg/ml.
In contrast, p38 MAPK activation showed no significant modulation of the cytokine production of Ttcr cells (Wilcoxon paired rank test). Production of IFN-γ was 8306 ± 5708 pg/ml by Ttcr cells compared with 37751 ± 33533 pg/ml by Ttcr generated in the presence of SB202190; LT-α production was 8192 ± 17949 pg/ml compared with 15048 ± 32677 pg/ml; GM-CSF was 9247 ± 8965 pg/ml compared with 12439 ± 12497 pg/ml and IL-10 was 948·6 ± 1250 pg/ml compared with 1086 ± 930·4 pg/ml.
SB202190 inhibits proliferation of CD4+ CD45RO+ Tck cells but promotes proliferation of Ttcr cells
The proliferation of CD4 memory cells upon IL-2/IL-6/TNF-α stimulation was p38 dependent (Fig. 3a,b). Tck cells started to divide 3 days after cytokine stimulation and at least six cell divisions were detected by CFSE dilution by day 8 and pre-treatment with SB202190 resulted in a 50% reduction in the proportion of Tck cells that underwent proliferation. Annexin V analysis showed that there was no pronounced difference in the percentage of apoptotic cells between SB202190-treated and DMSO-treated Tck cells (Fig. 3c). The actual cell numbers at day 8 were comparable between Tck cells generated in the presence or absence of SB202190 (Fig. 3e), this implies that Tck cells generated in the presence of SB202190 were non-proliferative but viable. This was reflected by the fact that fewer cells entered into S and G2 phases of the cell cycle when Tck cells were pre-treated with SB202190 (Fig. 3d) (at day 6, a reduction from 5% to 3% was seen following SB202190 treatment in Tck cells) suggesting that more cells were arrested in G0/G1 phase. The reduction of Tck cells entering into the cell cycle following SB202190 treatment was reproduced in at least four individual donors.
Figure 3.
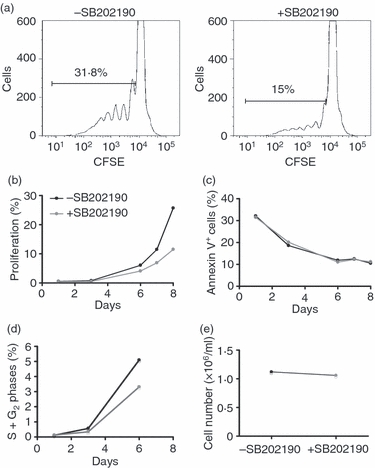
p38 activation promoted cytokine-activated T (Tck) cell proliferation. CFSE-labelled Tck cells were differentiated in the presence or absence of SB202190. (a) Proliferation of CFSE-labelled Tck ± SB202190 after 8 days. A representative of seven experiments is shown. (b) and (c) Proliferation and apoptosis of Tck ± SB202190 over 8 days. (d) The proportion of Tck ± SB202190 in S and G2 phases of cell cycle at indicated days (a representative of four experiments). (e) Cell number in Tck ± SB202190 cultures at day 8.
In contrast, inhibition of p38 MAPK activity resulted in an increased proliferation of CD4 memory cells stimulated with anti-CD3 antibody (Fig. 4a,b). The Ttcr cells started to divide 3 days after anti-CD3 stimulation and 35% of cells had undergone cell division measured by CFSE dilution at day 8. CD4 memory cells pre-treated with SB202190 were hyperproliferative with nearly twice as many Ttcr cells (> 70%) undergoing cell division. This increase in cell proliferation was reflected in more Ttcr cells passing through G0/G1 to S phase transitional check points when p38 activity was inhibited (at day 5 SB202190 pre-treatment increased the proportion of Ttcr cells in S/G2 phases from 10% to 30%) (Fig. 4d). Analysis of Annexin V showed that SB202190 suppressed apoptosis of Ttcr cells (Fig. 4c). This resulted in more than twice the number of cells (Fig. 4e). This finding was reproduced in four individual donors.
Figure 4.
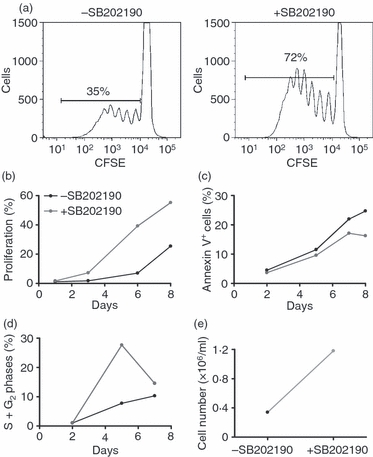
p38 activation suppressed anti-CD3 activated T (Ttcr) cell proliferation. CFSE-labelled Ttcr cells were differentiated in the presence or absence of SB202190. (a) Proliferation of CFSE-labelled Ttcr ± SB202190 after 8-day culture. A representative of four experiments is shown. (b, c) Proliferation and apoptosis of Ttcr ± SB202190 over 8 days. (d) Proportion of Ttcr cells entered into S and G2 phases of cell cycle at indicated days. (e) The day 8 cell number in Ttcr ± SB202190 cultures.
Discussion
p38 MAPK as a therapeutic target for inflammatory diseases was first proposed when Lee et al. discovered its role in the regulation of TNF-α production.13 However, clinical trials of its inhibitors have been largely disappointing because of adverse side effects. In this study, we have demonstrated distinct roles of p38 MAPK in the acquisition of effector function and cell cycle regulation of T cells activated by cytokines or TCR ligation. We were also able to demonstrate that p38 positively regulates the effector function of Tck cells (an in vitro model for rheumatoid T cells) but played a minimal role in TCR-activated T cells (Ttcr cells).
In this paper, we showed that the up-regulation of CD69 was p38-dependent in Tck cells but not in TCR-stimulated T cells. We reported previously that blockade of CD69 interaction with its unknown ligand(s) significantly inhibited the Tck contact-dependent activation of monocytes and spontaneous TNF-α production in rheumatoid synovial membrane ex vivo cultures.11 It was also reported by others that, induction of IL-1 in monocytes/THP-1 by anti-CD3-activated T-cell lines were also partially CD69 dependent.22,23 In addition, activation of macrophages by IL-15-stimulated T cells and rheumatoid T cells were also CD69 dependent.24 Taking this into account, p38-dependent acquisition of Tck effector function may be partly to the result of regulation of CD69 by p38 MAPK.
Tck cells require cell division to acquire effector function (Nicola MG Smith, paper in preparation) and we have shown that p38 activation promoted the transition from G0/1 phase to S phase, leading to cell division. The observation that p38 MAPK is a negative regulator of cell proliferation and suppressed G0/1 to S phase transition in Ttcr cells was not expected. These findings contradict published data by Geginat et al.25 showing that p38 did not have an inhibitory effect on cell proliferation in human primary CD4+ cells activated by anti-CD3 ligation. The expression of p27kip1 (a cell cycle inhibitor) was decreased in both Tck and Ttcr cells; however, p38-mediated cell cycle control in Tck cells was not p27kip1 dependent (data not shown). Exreacellular-regulated kinase 1 (Erk1) and Erk2 have been shown to be the main regulators of the mammalian cell cycle (reviewed in refs 26,27) and the involvement of p38 MAPK signalling is not clearly defined as yet. In fibroblast cell lines, p38 activation led to cell cycle arrest by blocking G1 to S phase transition28 by inhibition of activator protein 1 and ETS1 dependent synthesis of cyclin D.29 A similar function of p38 has not been reported in CD4+ T cells. Indirect evidence suggests that activity of p38 MAPK is elevated in anergic mouse T cells30,31 and that p38 negatively regulated the Erk-dependent IL-2 transcription in human T cells by ‘weak’ anti-CD3/anti-CD28 stimulation (at sub-optimal concentrations).32
Our data clearly demonstrate that p38 MAPK acted as a suppressor of cell division in Ttcr cells, supporting the T-cell anergy model whereby activation of p38 MAPK is important to maintain the non-proliferative status of T cells when stimulated through TCR cross-linking without co-stimulation. Notably, with regard to p38 MAPK as a therapeutic target for inflammatory diseases, these data indicate that p38 MAPK inhibition may lead to adverse effects such as the reversal of T-cell clonal anergy.
The p38-dependent up-regulation of CD69, cell cycle arrest and reduction of effector function appears to be exclusively related to cytokine-activated Tck cells. As rheumatoid T cells are functionally mimicked by Tck cells,2,11 these data suggest that p38 inhibition in rheumatoid T cells may be of therapeutic value. We previously showed that delivery of p38 dominant-negative virus to rheumatoid synovial cultures significantly inhibited spontaneous TNF-α production.33 It is intriguing to speculate that this inhibition was both through p38 inhibition in macrophages (blocking TNF-α production) and through blockade of Tck-like activity in rheumatoid arthritis synovial membrane.
In conclusion, this study demonstrates for the first time that p38 MAPK differentially regulates effector function of TCR-dependent and TCR-independent activation of T cells. Distinct temporal and spatial p38 phosphorylation patterns in Tck and Ttcr cells may reflect the two activation pathways (MAPK kinase-dependent and -independent pathways) reported by Salvador et al.34 and Yang et al.18 These studies describe that, in addition to the Gadd45β/Gadd45γ/MEKK4 signal axis described in cytokine-activated T cells,18,35 an alternative TCR/Lck/ZAP70/pTyr232p38 signal pathway34 was found in T cells activated by anti-CD3/CD28. Both pathways ultimately lead to Thr180/Tyr182 phosphorylation of p38 but, with different phenotypic outcomes. This suggests that the identification of the downstream events of p38 MAPK activation may lead to more specific therapeutic targets for the treatment of chronic inflammatory diseases.
Acknowledgments
This work was supported by a core grant to Kennedy Institute of Rheumatology division by ARC. We wish to offer our thanks to Professor J Saklatvala for providing the anti-p38 antibody SAK7 and to Dr A Clark, Dr A. Andersson and Dr B. Gregory for their critical review of the manuscript. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: recombinant human IL-2 from Fisher BioServices.
Glossary
Abbreviations:
- FWB
FACS wash buffer
- M ratio
T cell to monocyte ratio
- Tck
cytokine-activated T cells
- Ttcr
anti-CD3 activated T cells
Disclosures
We declare no financial and commercial conflict of interest.
References
- 1.Sebbag M, Parry SL, Brennan FM, Feldmann M. Cytokine stimulation of T lymphocytes regulates their capacity to induce monocyte production of tumor necrosis factor-alpha, but not interleukin-10: possible relevance to pathophysiology of rheumatoid arthritis. Eur J Immunol. 1997;27:624–32. doi: 10.1002/eji.1830270308. [DOI] [PubMed] [Google Scholar]
- 2.Brennan FM, Hayes AL, Ciesielski CJ, Green P, Foxwell BM, Feldmann M. Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum. 2002;46:31–41. doi: 10.1002/1529-0131(200201)46:1<31::AID-ART10029>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 3.Burger D. Cell contact-mediated signaling of monocytes by stimulated T cells: a major pathway for cytokine induction. Eur Cytokine Netw. 2000;11:346–53. [PubMed] [Google Scholar]
- 4.Cope AP. Studies of T-cell activation in chronic inflammation. Arthritis Res. 2002;4(Suppl. 3):S197–211. doi: 10.1186/ar557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isomaki P, Clark JM, Panesar M, Cope AP. Pathways of T cell activation and terminal differentiation in chronic inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:287–93. doi: 10.2174/1568010054022042. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Gorman CL, Vermi AC, et al. TCRzetadim lymphocytes define populations of circulating effector cells that migrate to inflamed tissues. Blood. 2007;109:4328–35. doi: 10.1182/blood-2006-12-064170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg L, Ronnelid J, Klareskog L, Bucht A. Down-regulation of the T cell receptor CD3 zeta chain in rheumatoid arthritis (RA) and its influence on T cell responsiveness. Clin Exp Immunol. 2000;120:174–82. doi: 10.1046/j.1365-2249.2000.01180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang B, Myers LK, Rosloniec EF, Whittington KB, Stuart JM, Kang AH. Characterization of signal transduction through the TCR-zeta chain following T cell stimulation with analogue peptides of type II collagen 260–267. J Immunol. 1998;160:3135–42. [PubMed] [Google Scholar]
- 9.Isomaki P, Panesar M, Annenkov A, Clark JM, Foxwell BM, Chernajovsky Y, Cope AP. Prolonged exposure of T cells to TNF down-regulates TCR zeta and expression of the TCR/CD3 complex at the cell surface. J Immunol. 2001;166:5495–507. doi: 10.4049/jimmunol.166.9.5495. [DOI] [PubMed] [Google Scholar]
- 10.Gringhuis SI, Leow A, Papendrecht-Van Der Voort EA, Remans PH, Breedveld FC, Verweij CL. Displacement of linker for activation of T cells from the plasma membrane due to redox balance alterations results in hyporesponsiveness of synovial fluid T lymphocytes in rheumatoid arthritis. J Immunol. 2000;164:2170–9. doi: 10.4049/jimmunol.164.4.2170. [DOI] [PubMed] [Google Scholar]
- 11.Brennan FM, Smith NM, Owen S, et al. Resting CD4+ effector memory T cells are precursors of bystander-activated effectors: a surrogate model of rheumatoid arthritis synovial T-cell function. Arthritis Res Ther. 2008;10:R36. doi: 10.1186/ar2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beech JT, Andreakos E, Ciesielski CJ, Green P, Foxwell BM, Brennan FM. T-cell contact-dependent regulation of CC and CXC chemokine production in monocytes through differential involvement of NFkappaB: implications for rheumatoid arthritis. Arthritis Res Ther. 2006;8:R168. doi: 10.1186/ar2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–46. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 14.Gaestel M, Kotlyarov A, Kracht M. Targeting innate immunity protein kinase signalling in inflammation. Nat Rev Drug Discov. 2009;8:480–99. doi: 10.1038/nrd2829. [DOI] [PubMed] [Google Scholar]
- 15.Xu JJ, Hendriks BS, Zhao J, de Graaf D. Multiple effects of acetaminophen and p38 inhibitors: towards pathway toxicology. FEBS Lett. 2008;582:1276–82. doi: 10.1016/j.febslet.2008.01.063. [DOI] [PubMed] [Google Scholar]
- 16.Ding C. Drug evaluation: VX-702, a MAP kinase inhibitor for rheumatoid arthritis and acute coronary syndrome. Curr Opin Investig Drugs. 2006;7:1020–5. [PubMed] [Google Scholar]
- 17.Mavropoulos A, Sully G, Cope AP, Clark AR. Stabilization of IFN-gamma mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood. 2005;105:282–8. doi: 10.1182/blood-2004-07-2782. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM. IL-18-stimulated GADD45 beta required in cytokine-induced, but not TCR-induced, IFN-gamma production. Nat Immunol. 2001;2:157–64. doi: 10.1038/84264. [DOI] [PubMed] [Google Scholar]
- 19.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–9. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–11. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark AR, Dean JL, Saklatvala J. Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 2003;546:37–44. doi: 10.1016/s0014-5793(03)00439-3. [DOI] [PubMed] [Google Scholar]
- 22.Manie S, Kubar J, Limouse M, et al. CD3-stimulated Jurkat T cells mediate IL-1 beta production in monocytic THP-1 cells. Role of LFA-1 molecule and participation of CD69 T cell antigen. Eur Cytokine Netw. 1993;4:7–13. [PubMed] [Google Scholar]
- 23.Isler P, Vey E, Zhang JH, Dayer JM. Cell surface glycoproteins expressed on activated human T cells induce production of interleukin-1 beta by monocytic cells: a possible role of CD69. Eur Cytokine Netw. 1993;4:15–23. [PubMed] [Google Scholar]
- 24.McInnes IB, Leung BP, Sturrock RD, Field M, Liew FY. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat Med. 1997;3:189–95. doi: 10.1038/nm0297-189. [DOI] [PubMed] [Google Scholar]
- 25.Geginat J, Sallusto F, Lanzavecchia A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4+ T cells. J Exp Med. 2001;194:1711–9. doi: 10.1084/jem.194.12.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773:1299–310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson MG, Millar JB. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J. 2000;14:2147–57. doi: 10.1096/fj.00-0102rev. [DOI] [PubMed] [Google Scholar]
- 28.Molnar A, Theodoras AM, Zon LI, Kyriakis JM. Cdc42Hs, but not Rac1, inhibits serum-stimulated cell cycle progression at G1/S through a mechanism requiring p38/RK. J Biol Chem. 1997;272:13229–35. doi: 10.1074/jbc.272.20.13229. [DOI] [PubMed] [Google Scholar]
- 29.Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–16. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 30.Ohkusu-Tsukada K, Tominaga N, Udono H, Yui K. Regulation of the maintenance of peripheral T-cell anergy by TAB 1-mediated p38 alpha activation. Mol Cell Biol. 2004;24:6957–66. doi: 10.1128/MCB.24.16.6957-6966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohkusu-Tsukada K, Tominaga N, Udono H, Yui K. Regulation of the maintenance of peripheral T-cell anergy by TAB 1-1 mediated p38alpha activation. Mol Cell Biol. 2005;25:8763. doi: 10.1128/MCB.25.19.8763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kogkopoulou O, Tzakos E, Mavrothalassitis G, Baldari CT, Paliogianni F, Young HA, Thyphronitis G. Conditional up-regulation of IL-2 production by p38 MAPK inactivation is mediated by increased Erk1/2 activity. J Leukoc Biol. 2006;79:1052–60. doi: 10.1189/jlb.0705418. [DOI] [PubMed] [Google Scholar]
- 33.Campbell J, Ciesielski CJ, Hunt AE, et al. A novel mechanism for TNF-alpha regulation by p38 MAPK: involvement of NF-kappa B with implications for therapy in rheumatoid arthritis. J Immunol. 2004;173:6928–37. doi: 10.4049/jimmunol.173.11.6928. [DOI] [PubMed] [Google Scholar]
- 34.Salvador JM, Mittelstadt PR, Guszczynski T, Copeland TD, Yamaguchi H, Appella E, Fornace AJ, Jr, Ashwell JD. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol. 2005;6:390–5. doi: 10.1038/ni1177. [DOI] [PubMed] [Google Scholar]
- 35.Lu B, Ferrandino AF, Flavell RA. Gadd45beta is important for perpetuating cognate and inflammatory signals in T cells. Nat Immunol. 2004;5:38–44. doi: 10.1038/ni1020. [DOI] [PubMed] [Google Scholar]