

Abstract
During early embryogenesis, the phenotype reflecting the embryonic genotype emerges only as maternal proteins are replaced by embryonically encoded forms, a process known as the maternal-to-embryonic transition (MET). Little is understood about MET for most proteins. This study investigates how complete deficiency of the murine dihydrolipoamide dehydrogenase gene (Dld), a gene that encodes an enzyme of mitochondrial energy metabolism, affects the phenotype of the early embryo and how the MET of the DLD protein affects the phenotype. Dld-deficient (−/−) embryos were found to develop similarly to wild-type (+/+) or heterozygous (+/−) embryos throughout the preimplantation period. These three genotypic classes also have comparable rates of glucose uptake (4.9–5.0 pmoles/embryo/h) and lactate production (0.97–1.0 pmoles/embryo/h). Dld-deficient embryos at the end of the preimplantation stage have 44% of DLD enzyme present in oocytes, a proportion similar to that found in +/+ or +/− embryos. This study demonstrates that Dld-deficient preimplantation embryos are phenotypically normal, as the MET for the DLD enzyme is only partially complete by the end of the preimplantation period. These findings have implications for phenotype- or enzyme-based approaches to identify mutations in Dld and other genes that encode proteins with similar MET kinetic profiles.
Keywords: development, dihydrolipoamide dehydrogenase, embryo, metabolism, mutation
Introduction
A unique characteristic of preimplantation embryonic development is that for the early part, and possibly even for the entirety of this period, the phenotype of the embryo is not reflective of its genotype. The early embryo phenotype is largely dictated by the maternal genotype due to the preponderance of maternal proteins inherited from the oocyte. Since the oocyte is diploid and has two active X chromosomes during most of its development, there is bi-allelic representation of most maternal proteins, regardless of the final haploid genotype of the oocyte (Epstein, 1975). It is only after the maternal proteins have been replaced by embryonic forms that the phenotype is truly reflective of the embryonic genotype. This maternal-to-embryonic transition (MET) requires both the degradation of maternal and expression of embryonic proteins.
For most of the thousands of proteins expressed in the oocyte and early embryo, little is known about how long maternal protein persists post conception. There is growing evidence from several non-mammalian species that a number of meiosis-specific proteins are rapidly degraded shortly after fertilization, an essential process that transforms the oocyte into a zygote (DeRenzo and Seydoux, 2004). The longevity of other maternal proteins is less well understood, principally because of difficulties in distinguishing the maternal from embryonic species. Studies of glucose-6-phosphate isomerase (GPI) using parental mice with different isozymes revealed that maternally encoded GPI persists to embryonic day 5.5–6.5, well beyond the 4.5-day preimplantation period (Gilbert and Solter, 1985; West et al., 1986). Evaluations of the activity levels of hypoxanthine-guanine phosphoribosyltransferase, encoded by an X-linked gene, demonstrated sex-related differences in expression by embryonic day 2.5, indicating that maternal enzyme was mostly replaced by this stage (Harper and Monk, 1983). From these studies, it appears that the half-lives of maternal proteins can vary, but studies of more proteins are needed to understand the time course of the MET more globally. Another critical determinant in the emergence of the early embryonic phenotype is the expression of embryonic proteins. Expression of the embryonic genome begins on embryonic day 1 in the mouse and by embryonic day 2 in human embryos (Clegg and Piko, 1982; Flach et al., 1982; Braude et al., 1988). Within a day after onset of transcription, there is an up-regulation of protein synthesis, reflecting production of embryonically encoded proteins (Monesi and Salfi, 1967; Epstein and Smith, 1973). Transcriptional profiles provide some indication of protein expression, but specifics of onset and rate of expression require studies at the protein level. Once again, challenges distinguishing maternal from embryonic forms have limited studies of expression of embryonic proteins.
Understanding the MET kinetics for a given protein is essential for developing a rational approach to identify abnormalities in the associated gene in preimplantation embryos. Many of the commonly accepted and emerging approaches for assessing embryos evaluate phenotypic features. Morphology, metabolism and secreted proteins are all facets of the embryo’s phenotype (Gardner et al., 2000; Ebner et al., 2003; Katz-Jaffe and Gardner, 2007). It is clear that many phenotypic assessments are useful for predicting developmental potential, but it is entirely unclear whether these phenotypic parameters reflect the genotype of the oocyte or embryo. In light of the high incidence of genetic abnormalities in early embryos, particularly aneuploidies, there is a great need for a better understanding of the time course of phenotypic expression of the embryonic genome.
The mutation studied in this report inactivates the dihydrolipoamide dehydrogenase gene (Dld) (Johnson et al., 1997b). The product of this gene, dihydrolipoamide dehydrogenase (DLD) or the E3 component, is a flavoprotein oxidoreductase that transfers reducing equivalents to NAD+ as part of four different multienzyme complexes (Figure 1; Vettakkorumakankav and Patel, 1996). Inactivation of pyruvate dehydrogenase complex prevents conversion of pyruvate to acetyl-CoA, thereby blocking oxidative metabolism of pyruvate, glucose and several amino acids. Deficiency of α-ketoglutarate dehydrogenase interrupts the tricarboxylic acid cycle leading to impairment in oxidative metabolism of fatty acids, several amino acids and a number of other energetic substrates. In summary, DLD deficiency greatly compromises the ability of cells to generate ATP from many of the major energetic substrates. This is supported by the observation that fibroblasts from patients with partial deficiencies in DLD activity, in the range of 10–30% of wild-type activity, produce less ATP from pyruvate and malate than fibroblasts from normal patients (Saada et al., 2000). These DLD-dependent metabolic pathways are also important means for energy production in the embryo (Leese, 1991). The requirement for oxidative metabolism is supported by the observations that murine and bovine embryos cultured in the absence of oxygen or in the presence of high concentrations of inhibitors of mitochondrial metabolism have compromised preimplantation development (Thomson, 1967; Thompson et al., 2000). To compensate for DLD-deficiency, cells up-regulate glycolysis, leading to increased lactate production. Systemic lactic acidosis is a hallmark of DLD deficiency in affected patients (Robinson, 2001).
Figure 1.
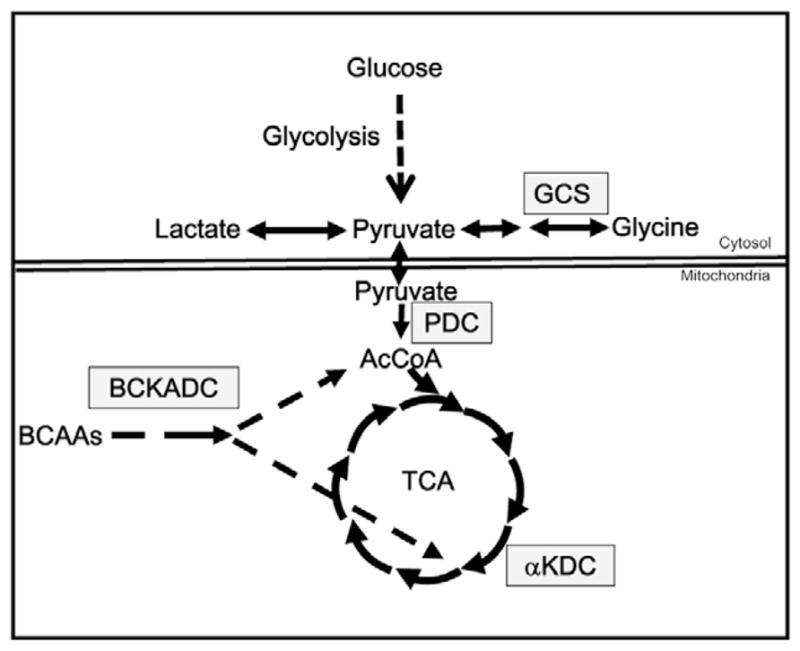
Dihydrolipoamide dehydrogenase (DLD)-dependent metabolic pathways. Several primary pathways of energy metabolism are depicted by arrows. Enzymatic steps that require DLD are shown in boxes. AcCoA = acetyl CoA; αKDC for α-ketoglutarate dehydrogenase; BCAA = branched-chain amino acids; BCKADC = branched-chain ketoacid dehydrogenase; GCS = glycine cleavage system; PDC = pyruvate dehydrogenase complex; TCA = tricarboxylic acid cycle. Reactions above the double lines occur in the cytoplasm whereas those below are mitochondrial.
Complete deficiency of Dld in mice has been shown to result in embryonic lethality in the early post-implantation period (Johnson et al., 1997b). Dld-deficient embryos manifest with developmental delay at embryonic day 7.5 and cease development within the subsequent 1–2 days. The studies in this report evaluate how Dld deficiency affects two of the phenotypic features most likely to be affected, development and metabolism. Studies of embryonic development have assessed whether Dld-deficient embryos have subtle signs of impaired development prior to arresting. Metabolic studies have looked for evidence that Dld-deficient embryos compensate metabolically for this deficiency. Blastocysts have previously been shown to modulate their metabolism in response to different concentrations of energetic substrates (Gardner and Leese, 1988, 1990; Leppens-Luisier and Sakkas, 1997). Studies of DLD enzymatic activity determine how long maternal DLD persists and how much enzyme is produced by the embryo. These studies provide insight into the kinetics of the MET for this mitochondrial enzyme and determine how this transition impacts the early embryonic phenotype.
Materials and methods
Animals
The animals used in these studies carry a targeted null mutation in the dihydrolipoamide dehydrogenase gene (Dldtm1ptl) in a mixed genetic background composed of approximately of 25% 129/Ola, the genetic background of the embryonic stem cell line used for gene targeting, and 75% C57BL/6J (Johnson et al., 1997b). Animals were housed and bred in compliance with the Institutional Animal Care and Use Committee guidelines. The colony was maintained by breeding heterozygous animals and genotyping progeny as previously described (Johnson et al., 1997b).
Embryo collection, culture, biopsy and evaluation
To produce embryos for these studies, Dld heterozygous (+/−) females aged 6–8 weeks were superovulated with 2.5 IU of pregnant mare’s serum gonadotrophin followed 48 h later with 2.5 IU human chorionic gonadotrophin (HCG) (Sigma, St Louis, MO, USA). Immediately following the second injection, females were individually mated with Dld +/− males. Embryos were collected from the oviducts of females at 21 h after mating. For the first 24 h of culture, groups of 10 embryos were cultured together in 20 μl drops of G1 medium (Lane et al., 2003; Gardner and Lane, 2007) supplemented with 5 mg/ml human serum albumin (HSA, VitroLife, Kungsbacka, Sweden) under standard paraffin oil (Ovoil, VitroLife). Cultures were maintained at 37°C with 5% O2 and 6% CO2. After 24 h of culture, embryos at the two-cell stage were individually transferred to 0.5 μl drops of culture medium containing 5 mg/ml HSA. Embryos were transferred to fresh media each day taking care to minimize the carryover volume. Spent media was stored at −80°C until metabolic analyses were performed. For the second day of culture, embryos were maintained in modified G1 medium containing 0.15 mmol/l glucose, 0.3 mmol/l pyruvate and no lactate for purposes of metabolic analysis. For days 3 and 4 of culture, G2 base medium containing 0.5 mmol/l glucose, 0.3 mmol/l pyruvate and no lactate was used. On the morning of day 5, embryos were individually transferred to 40 nl drops of modified G2 medium for a 4-h period of metabolic analysis. Embryos were evaluated for morphology at 24, 48, 72, 96, 104 and 120 h post HCG (Gardner et al., 2005). For differential cell counts, individual blastocysts at 124 h post HCG were stained, counted in 5 μl of phosphate-buffered saline (PBS) on a siliconized slide and then recovered for genotypic analysis (Hardy et al., 1989). For enzymatic analyses, hatching blastocysts with similar sizes of trophectodermal evaginations were chosen for biopsy. The exposed trophectoderm was drawn into a polar body biopsy tool (Humagen, Charlottesville, VA USA) and cell attachments were laser ablated (Research Instruments, Cornwall UK). Biopsied fragments were immediately processed for genotypic analysis.
Genotypic analysis
Individual stained blastocysts or trophectodermal biopsies were rinsed in PBS, transferred to polymerase chain reaction (PCR) microtubes containing 2.5 μl of 200 mmol/l KOH, 50 mmol/l dithiothreitol (DTT) lysis buffer and stored at −80°C. When the PCR was to be performed, samples were thawed and denatured at 65°C for 10 min. After cooling to 4°C, samples were neutralized with 2.5 μl of 900 mmol/l Tris–HCl, pH 8.3, 300 mmol/l KCl and 200 mmol/l HCl. To this 5 μl sample, 45 μl of a PCR cocktail was added that included primers Dld Ext Com S1 5′-GTGTTACACTGCCTGGTTTTATGAGATT-3′, Dld Ext Targ AS1 5′-GTAGGCCCAGATGTACAGGAATAGC-3′ and Dld Ext WT AS1 5′-GAGCCAAAAATATCATGCACATAACATT-3′. Samples were subjected to 20 cycles of amplification with an annealing temperature of 63°C. One microlitre of this PCR reaction was then added to a second 20 μl reaction with the internal primers Dld Int Com S1 5′-CTTCTGGTGGTAAAGCTGAAGTTAT-3′, Dld Int Targ AS1 5′-CCCATTCAATGTCTCGGTTA-3′ and Dld Int WT AS1 5′-TCTGTTATTGACTGGAATTCTACCT-3′. Following 35 cycles with an annealing temperature of 63°C, samples were subjected to agarose gel electrophoresis. Amplification of the mutant allele produced a 168-base pair fragment whereas the wild-type allele product was 140 base pairs in length.
Metabolic analysis
Spent media from day 4 (96–120 h post HCG) and day 5 (120–124 h) of culture were analysed for changes in glucose and lactate consumption using ultramicrofluorometric analytic methods described previously (Gardner and Leese, 1990; Gardner, 2007). For each assay, 21.8 nl of a cocktail containing enzyme, buffer and reagents was placed on a siliconized slide under mineral oil using a constriction pipette. The cocktail contains all the necessary components to catalyse a NAD(P)H-linked reaction once the substrate is added. To the cocktail drop, 2.2 nl of spent medium was added and the resulting change in fluorescence from consumption or production of NAD(P)H was determined after 3-min incubation. Fluorescence readings were performed using a Nikon TE3000 microscope equipped with a photomultiplier tube, Solamere EFX2 microfluorimeter and MacLab data-recording hardware and software. The concentrations of substrate in spent media and control drops (not incubated with an embryo) were determined by comparing the changes in fluorescence to standard curves. If it is assumed that all consumed glucose enters the glycolytic pathway and that all lactate released into the medium is derived from glucose, then it is possible to present the data as a proportion of consumed glucose that is converted to lactate. This proportion is computed by dividing the lactate production rate by double the glucose consumption rate since 2 moles of lactate are generated from 1 mole of glucose.
Enzymatic analysis
Unfertilized ovulated oocytes and biopsied blastocysts were collected from culture media, washed in PBS three times and placed in Eppendorf tubes containing 0.5 μl of enzyme extraction buffer made up of 20 mmol/l potassium phosphate, pH 7.4; 1% Triton X-100; 2 mmol/l EDTA and 1× concentration of protease inhibitor cocktail (Sigma). The samples were flash frozen in liquid nitrogen and stored at −80°C. Upon thawing, samples were aspirated from storage tubes and placed under mineral oil on a siliconized glass slide using a finely pulled Pasteur pipette and stored at 4°C. A microfluorometric assay was developed by miniaturizing the standard enzymatic assay (Patel et al., 2000). In preparation for the assay, 21.8 nl of enzyme cocktail containing 100 mmol/l potassium phosphate, pH 8.0, 1.5 mmol/l EDTA, 1 mmol/l DTT, 0.5 mg/ml Triton X-100, 3 mmol/l NAD and 3 mmol/l dihydrolipoamide (all reagents from Sigma, except dihydrolipoamide, which was generously provided by Dr Douglas Kerr at Case Western Reserve University, USA) was placed on a siliconized glass slide with an oil overlay and prewarmed to 37°C. To assess enzymatic activity of the sample in the extraction buffer, 2.2 nl of cell lysate was added to a drop of enzyme cocktail and then immediately placed on an inverted epifluorescence microscope with a heated stage. Changes in fluorescence were recorded every 5 s for a 2-min period using a Cascade CCD camera controlled by Metamorph software. Enzymatic activities were assessed in duplicate. Only samples in which both recordings yielded curves with correlation coefficients of 0.95 or greater were included in the data (roughly 95% of all data). To estimate the portion of enzymatic activity removed by biopsy, the number of cells in biopsied specimens was determined by phase contrast microscopy and the amount of enzymatic activity in biopsied and unbiopsied wild-type blastocysts were compared. Enzymatic activities of biopsied specimens were adjusted to account for the biopsy-associated sample loss. Enzymatic activity was normalized to total protein using previously published values of 29.9 ng for an unfertilized oocyte and 25.2 ng for a late blastocyst (Sellens et al., 1981). By comparing the activity levels in oocytes from heterozygous females to those in Dld-deficient blastocysts, it was possible to derive a theoretical half-life using the standard formula for first-order decay, half-life = (time × log2)/(log(initial activity/final activity)). The differences in mean DLD activities of Dld-deficient blastocysts compared with those of heterozyogous and wild-type blastocysts reflect embryonic expression from one and two alleles, respectively.
Statistics
Proportions of embryos in specific morphological categories were compared by the Fisher’s exact test. Cell count and metabolic data were compared using a one-way analysis of variance (ANOVA) with Bonferroni correction.
Results
Dld-deficient embryos develop normally
Serial evaluation of embryonic morphology revealed that the majority of embryos for all three genotypic classes reached the 2-cell stage by 24 h (data not shown), 8-cell or compacting stage by day 3, early or full blastocyst stage by the morning of day 4, expanded or full blastocyst by the afternoon of day 4 (104 h) and expanded or hatching blastocyst stage by day 5 (120 h; Figure 2a). At all of these observational time points, homozygous mutant embryos had rates of development that did not differ significantly from +/+ or +/− embryos. The ratio of genotypes of blastocysts, 27:47:24, was similar to the expected Mendelian ratio of 1:2:1, confirming that Dld-deficient embryos did not arrest in development before the blastocyst stage. Differential cell counts at the conclusion of the culture period (124 h) revealed no statistically significant differences in trophectodermal and inner cell mass cell numbers between the three genotypic classes (Figure 2b).
Figure 2.

Developmental assessment of Dld-deficient embryos. (a) Assessment of morphological progression. The results from four morphological assessments are shown in the graph. The genotypes of the embryos are presented below. The numbers in each genotypic category were: +/+, 27; +/−, 47; −/−, 24. The times of evaluation and a designation of the morphological categories are shown under the graph. 8c-8-cell; com = compacting; EB = early blastocyst (less than two-thirds of the volume of the embryo); B = blastocyst (greater than or equal to two-thirds of the volume of the embryo); ExB = expanded blastocyst; H = hatching blastocyst. (b) Differential cell counts according to genotypes and cell type. TE = trophectoderm; ICM = inner cell mass.
Dld-deficient embryos have normal glucose and lactate metabolism
To determine if Dld-deficient embryos were metabolically normal, analysis of glucose uptake and lactate production was performed on spent media from day 4 (96–120 h post-HCG) and for several hours during day 5 (120–124 h) of culture. Glucose consumption was similar for all genotypic classes at both time points (Figure 3a). Rates of glucose uptake on day 5 were found to be generally lower than those on day 4. In evaluating the production of lactate at these same time points, it was found that there were no differences between genotypes or time of development (Figure 3b). As an approximation of glycolytic activity, the proportion of consumed glucose excreted as lactate was also determined (Figure 3c). The proportions were relatively low, ranging between 9.2% and 13.1%, and none of the groups differed significantly. As developmental staging and cell count data were available for these embryos, it was also possible to normalize metabolic activities to cell number or to compare only between similarly staged embryos (data not shown). Even with these corrections, metabolic activities were not significantly different between the three genotypic classes.
Figure 3.

Metabolic assessment of Dld-deficient embryos. (a) Glucose consumption. (b) Lactate production. (c) Proportion of glucose converted to lactate. Data with different letters are statistically different (P < 0.05).
Dld-deficient embryos have substantial residual maternal DLD enzyme
To determine if the apparently normal development and metabolism were due to the presence of maternal enzyme, a microfluorometric assay for DLD enzymatic activity was developed. Repeating this assay five times on a blastocyst lysate produced data with a coefficient of variation of 6%, suggesting that the assay results were highly reproducible. Wild-type oocytes were found to have activity levels comparable with several cultured cell types such as fibroblasts and lymphocytes (Table 1). These activity levels were substantially lower than those found in embryonic stem cells or most tissues. Oocytes from females that carry the Dld mutation have roughly half the activity of oocytes from wild-type females (Table 1, Figure 4a). To estimate the proportion of enzymatic activity removed from the blastocyst, wild-type embryos with the same genetic background as the experimental embryos were biopsied. Attempts to normalize DLD activity to isocitrate dehydrogenase (IDH) activity were unsuccessful due to the extremely low IDH activity found in oocytes and embryos as has been previously reported for bovine oocytes (Cetica et al., 2003). In 10 blastocysts biopsied, activity was found to be on average 15% lower than in unbiopsied blastocysts (27.7 ± 12.5 versus 23.5 ± 16.3 pmoles/embryo/h). These results were consistent with counts of cells removed during initial biopsy testing (8.1 ± 4.4), which corresponds to 13.6–15.4% of total cell numbers (Figure 2b). Consequently, recorded enzymatic activities were corrected by a factor of 1.18 to account for the cells removed by biopsy. Total DLD activities in all three genotypic classes of blastocysts were found to be significantly lower than the activity levels in oocytes (Figure 4a). Between these three genotypic classes of blastocysts, there were no statistically significant differences in total DLD activities, although there was a positive trend between the activity levels and the number of functional Dld alleles. The change in activity between oocytes from heterozygous females and homozygous mutant blastocysts (44.3 to 19.7 pmoles/embryo/h) represents the elimination of maternal enzyme over the course of the preimplantation period. Assuming that the enzyme was degraded according to first-order kinetics, the half-life of maternal DLD was estimated to be 106.1 h (4.4 days).
Table 1.
Dihydrolipoamide dehydrogenase enzymatic activity of various cell types and tissues.
| Tissue/cell type | Mean total protein (mIU/mg) |
|---|---|
| +/+ Oocytea | 42.7 ± 15.9 |
| +/− Oocytea | 26.4 ± 9.1 |
| +/+ Day-5 blastocysta | 17.7 ± 9.9 |
| −/− Day-5 blastocysta | 12.4 ± 4.7 |
| Mouse ESC (E14.1 line)b | 210 ± 22 |
| +/− Mouse ESCb | 90 ± 35 |
| Rat heart tissuec | 723 |
| Rat skeletal muscle tissuec | 161 |
| Rat brain tissuec | 287 |
| Human cultured fibroblastsd | 62 ± 13 |
| Human lymphocytesd | 50 ± 17 |
ESC = embryonic stem cell.
Data from this study;
data from MT Johnson and MS Patel, unpublished results;
Figure 4.

Dihydrolipoamide dehydrogenase (DLD) enzymatic activity in oocytes and embryos. (a) Activity level in oocytes from wild-type and heterozygous females and from blastocysts of three different genotypes. The additional horizontal line in the box-whisker plots represents the means for the group, and groups that are statistically different (P < 0.05) are indicated with different letters. (b) Model for maternal-to-embryonic transition for DLD enzyme. The half-life of maternal DLD was estimated based on the loss of activity from the oocyte to the blastocyst in Dld-deficient embryos. Embryonic production of DLD enzyme was estimated by determining differences in enzyme activity in −/− and +/− or +/+ embryos, which reflects expression from one or two alleles, respectively.
Discussion
The first part of this study evaluated the development of Dld-deficient embryos. Previous studies had only shown that Dld-deficient blastocysts were present in the expected Mendelian proportion (Johnson et al., 1997b). Serial morphological assessment of mouse or human embryos is a better gauge of embryonic development than blastocyst formation (Ebner et al., 2003; Neuber et al., 2006). A good indicator of preimplantation development is the proportion of blastocysts on the afternoon of day 4 (104 h, i.e., the time that the blastocyst stage is reached in vivo) (Gardner et al., 2005). A further indicator of embryonic development is the differential cell count of the blastocyst, particularly the number of inner cell mass (ICM) cells which has been linked to subsequent fetal development (Fleming, 1987; Lane and Gardner, 1997). Even with this level of scrutiny, no differences in developmental parameters were found between Dld-deficient embryos and wild-type or heterozygous embryos. Larger numbers of embryos would need to be analysed to determine if the Dld-deficient embryos differ significantly in any of these parameters. Most importantly, even if significant differences were found, they would be expected to be small and clinically unimportant. For example, using the expanded/hatching stage as a selection criterion at 104 h would only reduce the proportion of Dld-deficient blastocysts from 25% in a non-selected group to 16% in this selected group using the proportions of morphologies in Figure 2a. The absence of a distinct morphological phenotype in Dld-deficient embryos was not unexpected. It is well recognized that embryos carrying a wide variety of genetic abnormalities, including gross changes in chromosomal complement, can develop normally throughout the preimplantation period (Sandalinas et al., 2001; Rubio et al., 2007). Furthermore, the development of Dld-deficient embryos into the post-implantation period is entirely consistent with normal preimplantation development (Johnson et al., 1997b).
To determine whether Dld-deficient embryos differ in their metabolism from heterozygous and wild-type embryos, glucose consumption and lactate production were measured at the late preimplantation/peri-implantation period. This period was chosen because it would be the least likely to be influenced by maternal enzyme. From the little that is known about maternally encoded proteins, it appears that there is a steady decline in these proteins, predominantly due to elimination of maternal transcripts (Hamatani et al., 2004). By determining glucose consumption and lactate production rates, glycolytic activity could be approximated. Glycolytic activity was evaluated since it would be expected that Dld-deficient embryos would up-regulate this pathway in the face of the crippled oxidative metabolic pathways. When evaluating glycolytic activity, it is important to take into account the concentration of glucose in the culture medium. Gardner and Leese (1988) have shown there to be a positive correlation between glucose concentration and glucose uptake or lactate production. With a concentration of 0.5 mmol/l glucose in this study, the rates of glucose uptake (3.7–5.0 pmoles/embryo/h) and lactate production (0.69–1.0 pmoles/embryo/h) are similar to previously reported values of 3.5–4.5 and 1.6–2.2 pmoles/embryo/h, respectively (Gardner and Leese, 1988). It is likely that differences in glucose concentration also account for the differences in glucose consumption observed for the two time points (Figure 3a). Since the 24-h metabolic assessment used 500 nl of medium and the 4-h evaluation used only 40 nl, the effective glucose concentration at the end of the 4-h period would be roughly half of that for the 24-h period, 0.125 versus 0.25 mmol/l, respectively. The lower concentrations of glucose used in these metabolic assessments may also explain the relatively low proportions of glucose that are converted to lactate. More pyruvate may be directed towards oxidative metabolism to generate ATP when there is reduced availability of glucose. The findings of similar glucose consumption and lactate production rates strongly suggest that Dld-deficient embryos have no metabolic abnormalities. As mentioned previously, increased lactate production due to increased glycolysis is a cardinal biochemical marker of Dld-deficiency. The lack of a biochemical phenotype is also consistent with the normal embryonic development.
Of all the potential explanations for the lack of developmental and biochemical phenotypes in Dld-deficient embryos, the persistence of maternally encoded DLD enzyme seemed most plausible. To assess the amount of maternal DLD in oocytes and embryos, a microfluorometric DLD enzyme assay was developed. The activity measured in this assay is likely to reflect maximal enzymatic activity due to the high concentrations of enzyme and NAD+ used and the 25,000-fold dilution of the cell extract. To date, enzymatic activities of less than 20 enzymes have been measured in single human or mouse embryos and none have been performed in combination with genotypic analysis (Chi et al., 1988; Martin et al., 1993). Of the enzymes previously evaluated, only three are strictly mitochondrial: citrate synthase, α-hydroxyacyl-CoA dehydrogenase and α-ketoglutarate dehydrogenase. This highly sensitive DLD assay is likely to have applications in evaluating Dld-deficiency and other disorders that affect mitochondrial metabolism. The levels of DLD activity in oocytes are similar to those found in cultured fibroblasts and lymphocytes, standard specimens for evaluating enzymatic deficiency (Table 1). In contrast, DLD activity levels in oocytes and embryos were much lower than in embryonic stem cells or various tissues. These findings were somewhat surprising based on the observation that oocytes have roughly twice the mitochondria/cell volume of other cell types (Piko and Matsumoto, 1976). The reported enzyme activities could be falsely low if either the values used as estimates of protein content for oocytes and embryos (Sellens et al., 1981) are too high for these specimens or the procedure used to solubilize the enzyme was not effective, although this technique is routinely used for a variety of tissues and cell types. The finding that oocytes from heterozygous females have roughly half the activity compared with oocytes from wild-type females is consistent with previous observations that Dld heterozygotes have roughly 50% activity in various somatic tissues (Johnson et al., 1997b). It has also been shown that oocytes from female mice with two X chromosomes have twice the activity of the X-linked enzymes hypoxanthine-guanine phosphoribosyltransferase, gluocose-6-phosphate dehydrogenase and phosphoglycerate kinase as compared with oocytes from females with only one X chromosome (Epstein, 1975).
Enzymatic analysis of embryos at the end of the preimplantation period revealed two important observations: (i) −/− blastocysts have similar levels of enzymatic activity to the other two genotypic classes; and (ii) −/− blastocysts still have 44% of the DLD activity present in oocytes (Figure 4a). The similar values of DLD enzymatic activity among all three genotypic classes provides a likely explanation for why there are no phenotypic differences between these three groups. The DLD activity in −/− blastocysts is almost certainly from maternal DLD protein since this mutation completely ablates enzymatic activity and there are numerous gene (Feigenbaum and Robinson, 1993; Johnson et al., 1997a) and protein (Williams, 1991; Hinman et al., 1989) studies in a variety of mammalian species indicating that no other protein can replace the function of DLD. Previous studies of Dld heterozygous mice and clinical data also strongly support that 44% residual activity is sufficient for normal cellular function and it has been shown that embryonic stem cells with 50% DLD activity can be maintained in culture and can contribute to all tissues of the mouse following blastocyst injection (Johnson et al., 1997b). Based on the 13 confirmed clinical cases of Dld deficiency, it appears that residual activity must be less than 30% of control activity to cause symptoms (Cameron et al., 2006).
The general decline in DLD enzymatic activity throughout the preimplantation period is not without precedent. Previous studies of more than 20 enzymes have shown that enzymatic activity can rise, fall or remain unchanged throughout the preimplantation period (Biggers and Stern, 1973). A number of metabolic enzymes, including lactate dehydrogenase, glucose-6-phosphate dehydrogenase, glutamate dehydrogenase and phosphofructokinase decrease in activity during this period, some falling by more than one order of magnitude. The difference in activity between oocytes from heterozygous females and Dld-deficient blastocysts reflects the decline in maternal DLD. Over the course of the first 5 days of development, there is a loss of 56% of maternal DLD. If one assumes first-order kinetics, the half-life of maternal DLD would be 106.1 h and would decline exponentially as shown in Figure 4b. Although this curve is useful for presenting the diminution of maternal enzyme, the decline in DLD activity is unlikely to adhere strictly to first-order kinetics. Studies of maternal glucose-6-phosphate isomerase and hypoxanthine-guanine phosphoribosyl transferase have shown that these enzymes increase in activity, presumably due to a burst of translation of maternal message, before declining precipitously during the preimplantation period (Harper and Monk, 1983; West et al., 1986). The estimated half-life for DLD in the embryo is somewhat longer than the 43 h recorded in pigeon breast muscle and cultured 3T3-L1 pre-adipocytes (Furuta, 1979; Carothers et al., 1988). Using this model to extrapolate to embryonic day 7.5, when Dld-deficient embryos arrest, it would be estimated that the embryos would have approximately 30% of the amount of enzyme originally present in the oocyte (Johnson et al., 1997b). As mentioned before, a threshold of 30% residual activity is associated with development of symptoms in humans (Cameron et al., 2006). As far as is known, this is the first report evaluating the longevity of the maternal form of a mitochondrial enzyme. It is expected that the maternal forms of many other mitochondrial proteins will last beyond the preimplantation period due to similarly long half lives, which average 3.8 days (Grisolia et al., 1985). In support, several other mutations affecting nuclear-encoded mitochondrial proteins were found not to affect preimplantation viability (Li et al., 2000; Huang et al., 2004; Piruat et al., 2004).
The finding that wild-type (+/+) Dld embryos did not have significantly higher levels of enzyme than homozygous mutant (−/−) embryos indicates that there is no or little expression from the embryonic Dld alleles during preimplantation development. As seen in Figure 4a, there is a positive relationship between the number of functional Dld alleles and the mean activity level. This observation warrants further study to determine if these differences are significant in larger data sets. Given the fact that the Dld gene is expressed at major zygotic genome activation (Hamatani et al., 2004), it would be expected that variations in gene copy number would cause differences in enzymatic activity levels. Of note, the difference in activity between +/+ and −/− embryos (i.e., difference of two functional alleles) is almost exactly twice that of the difference between +/− and −/− embryos (difference in one functional allele) as would be expected. To illustrate how differences in the number of functional Dld alleles can affect total enzymatic activity, the differences in mean activities of the three genotypic classes of blastocysts were incorporated into the hypothetical model presented in Figure 4b. Assuming that expression of DLD protein is uniform and that protein is produced beginning at the second day of development (roughly 24 h after zygotic genome activation), it is possible to generate a hypothetical curve for embryonic DLD expression (Figure 4b). By adding the maternal and embryonic curves, a model for changes in total DLD activity can also be generated. This figure serves to provide a graphical representation of the kinetics of the DLD MET and highlights the small contribution of embryonically encoded enzyme throughout the preimplantation period.
The findings in this report have important implications for preimplantation genetic screening and diagnostics. The case of Dld deficiency serves as an example of how a protracted MET can completely obscure the embryonic genotype throughout the preimplantation period. In this scenario, any phenotypic evaluation, whether it be morphological, metabolic or enzymatic, would not distinguish Dld-deficient from wild-type embryos. Given the relatively similar development and metabolism of mouse and human embryos, it is reasonable to propose that human preimplantation embryos deficient in DLD would also have normal phenotypes, although studies are needed to test this hypothesis. The only feasible approach to identify Dld-deficient embryos would be through biopsy and direct detection of the mutation. This would also be the case for other proteins with similar MET profiles. The small contribution of embryonically encoded enzyme to total enzymatic activity also suggests that increases in gene copy number would be unlikely to cause phenotypic abnormalities in the early embryo. Increases in gene copy number could occur in the context of trisomies, a genetic disorder commonly seen in embryos. Using the model described in Figure 4b, triplication of the Dld gene would only increase total DLD activity at the blastocyst stage by 5.2% assuming that this condition arose in an oocyte that originally had two normal copies of this gene.
Based on the observation that the majority of mutations affecting critical cellular genes in mice lead to post-implantation lethality, it is likely that many genetic deficiencies will have very subtle or no phenotypic manifestations during the preimplantation period (Copp, 1995). The delay in the onset of phenotypic manifestations for many of these mutations is likely to be caused by abundance of maternal protein. The mammalian oocyte may be endowed with large amounts of critical cellular proteins to allow the embryo to proceed through early development without having to rely on embryonic expression. This strategy would afford the embryo with many days and possibly even more than 1 week to express its own cellular machinery. Such an arrangement might seem reproductively disadvantageous since conceptuses carrying many doomed mutations would implant, but the relatively low incidence of mutations would probably have little effect on fecundity, particularly in poly-ovulatory species. This study provides important insight into the MET for a vital cellular enzyme. As more is learned about the METs of other proteins, more informed strategies to identify mutations in preimplantation embryos will follow.
Acknowledgments
The authors acknowledge the excellent technical support from Amanda Chapman, Courtney Sheehan, Ann Janesch. MTJ was supported by NIH grant K08 HD047431. Animals were kindly provided by Dr Mulchand Patel.
Biography
 Dr Johnson received his MD and PhD degrees from Case Western Reserve University, USA. As part of his Doctoral thesis project, he generated the null mutation used in these studies. Dr Johnson became interested in reproductive genetics during residency training in pediatrics and medical genetics. Following clinical training, he joined the Department of Genetics at Case Western and spent several years working with Dr David Gardner as a visiting scientist at the Fertility Laboratories of Colorado. He recently took a faculty position in the Department of Obstetrics and Gynecology at the University of Michigan where he continues to study how genetic abnormalities affect the early mammalian embryo.
Dr Johnson received his MD and PhD degrees from Case Western Reserve University, USA. As part of his Doctoral thesis project, he generated the null mutation used in these studies. Dr Johnson became interested in reproductive genetics during residency training in pediatrics and medical genetics. Following clinical training, he joined the Department of Genetics at Case Western and spent several years working with Dr David Gardner as a visiting scientist at the Fertility Laboratories of Colorado. He recently took a faculty position in the Department of Obstetrics and Gynecology at the University of Michigan where he continues to study how genetic abnormalities affect the early mammalian embryo.
Footnotes
Declaration: Pa Vang is an employee of VitroLife, Inc., a supplier of embryology culture media and supplies. The other authors report no financial or commercial conflicts of interest.
References
- Biggers JD, Stern S. Metabolism of the preimplantation mammalian embryo. Advances in Reproductive Physiology. 1973;6:1–59. [PubMed] [Google Scholar]
- Braude P, Bolton V, Moore S. Human gene expression first occurs between the four- and eight-cell stages of preimplantation development. Nature. 1988;332:459–461. doi: 10.1038/332459a0. [DOI] [PubMed] [Google Scholar]
- Cameron JM, Levandovskiy V, Mackay N, et al. Novel mutations in dihydrolipoamide dehydrogenase deficiency in two cousins with borderline-normal PDH complex activity. American Journal of Medical Genetics A. 2006;140:1542–1552. doi: 10.1002/ajmg.a.31313. [DOI] [PubMed] [Google Scholar]
- Carothers DJ, Pons G, Patel MS. Induction of dihydrolipoamide dehydrogenase in 3T3-L1 cells during differentiation. Biochemical Journal. 1988;249:897–902. doi: 10.1042/bj2490897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetica P, Pintos L, Dalvit G, et al. Involvement of enzymes of amino acid metabolism and tricarboxylic acid cycle in bovine oocyte maturation in vitro. Reproduction. 2003;126:753–763. [PubMed] [Google Scholar]
- Chi MM, Manchester JK, Yang VC, et al. Contrast in levels of metabolic enzymes in human and mouse ova. Biology of Reproduction. 1988;39:295–307. doi: 10.1095/biolreprod39.2.295. [DOI] [PubMed] [Google Scholar]
- Clegg KB, Piko L. RNA synthesis and cytoplasmic polyadenylation in the one-cell mouse embryo. Nature. 1982;295:343–344. doi: 10.1038/295342a0. [DOI] [PubMed] [Google Scholar]
- Copp AJ. Death before birth: clues from gene knockouts and mutations. Trends in Genetics. 1995;11:87–93. doi: 10.1016/S0168-9525(00)89008-3. [DOI] [PubMed] [Google Scholar]
- DeRenzo C, Seydoux G. A clean start: degradation of maternal proteins at the oocyte-to-embryo transition. Trends in Cell Biology. 2004;14:420–426. doi: 10.1016/j.tcb.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Ebner T, Moser M, Sommergruber M, et al. Selection based on morphological assessment of oocytes and embryos at different stages of preimplantation development: a review. Human Reproduction Update. 2003;9:251–262. doi: 10.1093/humupd/dmg021. [DOI] [PubMed] [Google Scholar]
- Epstein CJ. Gene expression and macromolecular synthesis during preimplantation embryonic development. Biology of Reproduction. 1975;12:82–105. doi: 10.1095/biolreprod12.1.82. [DOI] [PubMed] [Google Scholar]
- Epstein CJ, Smith SA. Amino acid uptake and protein synthesis in preimplantation mouse embryos. Developmental Biology. 1973;33:171–184. doi: 10.1016/0012-1606(73)90172-3. [DOI] [PubMed] [Google Scholar]
- Feigenbaum AS, Robinson BH. The structure of the human dihydrolipoamide dehydrogenase gene (DLD) and its upstream elements. Genomics. 1993;17:376–381. doi: 10.1006/geno.1993.1335. [DOI] [PubMed] [Google Scholar]
- Flach G, Johnson MH, Braude PR, et al. The transition from maternal to embryonic control in the 2-cell mouse embryo. EMBO Journal. 1982;1:681–686. doi: 10.1002/j.1460-2075.1982.tb01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming TP. A quantitative analysis of cell allocation to trophectoderm and inner cell mass in the mouse blastocyst. Developmental Biology. 1987;119:520–531. doi: 10.1016/0012-1606(87)90055-8. [DOI] [PubMed] [Google Scholar]
- Furuta S. Turnover of pigeon breast muscle pyruvate dehydrogenase complex. Journal of Biochemistry. 1979;86:183–190. [PubMed] [Google Scholar]
- Gardner DK. Noninvasive metabolic assessment of single cells. Methods in Molecular Medicine. 2007;132:1–9. doi: 10.1007/978-1-59745-298-4_1. [DOI] [PubMed] [Google Scholar]
- Gardner DK, Lane M. Embryo culture systems. In: Gardner DK, editor. In Vitro Fertilization: A Practical Approach. InformaHealthcare; New York: 2007. pp. 221–282. [Google Scholar]
- Gardner DK, Leese HJ. Concentrations of nutrients in mouse oviduct fluid and their effects on embryo development and metabolism in vitro. Journal of Reproduction and Fertility. 1990;88:361–368. doi: 10.1530/jrf.0.0880361. [DOI] [PubMed] [Google Scholar]
- Gardner DK, Leese HJ. The role of glucose and pyruvate transport in regulating nutrient utilization by preimplantation mouse embryos. Development. 1988;104:423–429. doi: 10.1242/dev.104.3.423. [DOI] [PubMed] [Google Scholar]
- Gardner DK, Reed L, Linck D, et al. Quality control in human in-vitro fertilization. Seminars in Reproductive Medicine. 2005;23:319–324. doi: 10.1055/s-2005-923389. [DOI] [PubMed] [Google Scholar]
- Gardner DK, Pool TB, Lane M. Embryo nutrition and energy metabolism and its relationship to embryo growth, differentiation, and viability. Seminars in Reproductive Medicine. 2000;18:205–218. doi: 10.1055/s-2000-12559. [DOI] [PubMed] [Google Scholar]
- Gilbert SF, Solter D. Onset of paternal and maternal Gpi-1 expression in preimplantation mouse embryos. Developmental Biology. 1985;109:515–517. doi: 10.1016/0012-1606(85)90477-4. [DOI] [PubMed] [Google Scholar]
- Grisolia S, Hernandez-Yago J, Knecht E. Regulation of mitochondrial protein concentration: a plausible model which may permit assessing protein turnover. Current Topics in Cellular Regulation. 1985;27:387–396. doi: 10.1016/b978-0-12-152827-0.50040-2. [DOI] [PubMed] [Google Scholar]
- Hamatani T, Carter MG, Sharov AA, et al. Dynamics of global gene expression changes during mouse preimplantation development. Developmental Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- Hardy K, Handyside AH, Winston RM. The human blastocyst: cell number, death and allocation during late preimplantation development in vitro. Development. 1989;107:597–604. doi: 10.1242/dev.107.3.597. [DOI] [PubMed] [Google Scholar]
- Harper MI, Monk M. Evidence for translation of HPRT enzyme on maternal mRNA in early mouse embryos. Journal of Embryology and Experimental Morphology. 1983;74:15–28. [PubMed] [Google Scholar]
- Hinman LM, Sheu KF, Baker AC, et al. Deficiency of pyruvate dehydrogenase complex (PDHC) in Leigh’s disease fibroblasts: an abnormality in lipoamide dehydrogenase affecting PDHC activation. Neurology. 1989;39:70–75. doi: 10.1212/wnl.39.1.70. [DOI] [PubMed] [Google Scholar]
- Hong YS, Kerr DS, Craigen WJ, et al. Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Human Molecular Genetics. 1996;5:1925–1930. doi: 10.1093/hmg/5.12.1925. [DOI] [PubMed] [Google Scholar]
- Huang G, Lu H, Hao A, et al. GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I. Molecular and Cellular Biology. 2004;24:8447–8456. doi: 10.1128/MCB.24.19.8447-8456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Yang HS, Johanning GL, et al. Characterization of the mouse dihydrolipoamide dehydrogenase (Dld) gene: genomic structure, promoter sequence, and chromosomal localization. Genomics. 1997a;41:320–326. doi: 10.1006/geno.1997.4670. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Yang HS, Magnuson T, et al. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proceedings of the National Academy of Sciences of the United States of America. 1997b;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz-Jaffe MG, Gardner DK. Embryology in the era of proteomics. Theriogenology. 2007;68 (Suppl 1):S125–S130. doi: 10.1016/j.theriogenology.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Lane M, Gardner DK. Differential regulation of mouse embryo development and viability by amino acids. Journal of Reproduction and Fertility. 1997;109:153–164. doi: 10.1530/jrf.0.1090153. [DOI] [PubMed] [Google Scholar]
- Lane M, Gardner DK, Hasler MJ, et al. Use of G1.2/G2.2 media for commercial bovine embryo culture: equivalent development and pregnancy rates compared to co-culture. Theriogenology. 2003;60:407–419. doi: 10.1016/s0093-691x(03)00030-x. [DOI] [PubMed] [Google Scholar]
- Leese HJ. Metabolism of the preimplantation mammalian embryo. Oxford Reviews of Reproductive Biology. 1991;13:35–72. [PubMed] [Google Scholar]
- Leppens-Luisier G, Sakkas D. Development, glycolytic activity, and viability of preimplantation mouse embryos subjected to different periods of glucose starvation. Biology of Reproduction. 1997;56:589–596. doi: 10.1095/biolreprod56.3.589. [DOI] [PubMed] [Google Scholar]
- Li K, Yucheng L, Shelton J, et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- Martin KL, Hardy K, Winston RM, et al. Activity of enzymes of energy metabolism in single human preimplantation embryos. Journal of Reproduction and Fertility. 1993;99:259–266. doi: 10.1530/jrf.0.0990259. [DOI] [PubMed] [Google Scholar]
- Monesi V, Salfi V. Macromolecular synthesis during early embryonic development in the mouse embryo. Experimental Cellular Research. 1967;46:632–635. doi: 10.1016/0014-4827(67)90397-7. [DOI] [PubMed] [Google Scholar]
- Neuber E, Mahutte NG, Arici A, et al. Sequential embryo assessment outperforms investigator-driven morphological assessment at selecting a good quality blastocyst. Fertility and Sterility. 2006;85:794–796. doi: 10.1016/j.fertnstert.2005.08.064. [DOI] [PubMed] [Google Scholar]
- Patel MS, Hong YS, Kerr DS. Genetic defects in E3 component of alpha-keto acid dehydrogenase complexes. Methods in Enzymology. 2000;324:453–464. doi: 10.1016/s0076-6879(00)24253-5. [DOI] [PubMed] [Google Scholar]
- Patel MS, Vettakkorumakankav NN, Liu TC. Dihydrolipoamide dehydrogenase: activity assays. Methods in Enzymology. 1995;252:186–195. doi: 10.1016/0076-6879(95)52022-8. [DOI] [PubMed] [Google Scholar]
- Piko L, Matsumoto L. Number of mitochondria and some properties of mitochondrial DNA in the mouse egg. Developmental Biology. 1976;49:1–10. doi: 10.1016/0012-1606(76)90253-0. [DOI] [PubMed] [Google Scholar]
- Piruat JI, Pintado CO, Ortega-Saenz P, et al. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Molecular and Cellular Biology. 2004;24:10933–10940. doi: 10.1128/MCB.24.24.10933-10940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BH. Lactic acidemia: disorders of pyruvate carboxylase and pyruvate dehydrogenase. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. II. McGraw-Hill; New York: 2001. pp. 2275–2295. [Google Scholar]
- Rubio C, Rodrigo L, Mercader A, et al. Impact of chromosomal abnormalities on preimplantation embryo development. Prenatal Diagnosis. 2007;27:748–756. doi: 10.1002/pd.1773. [DOI] [PubMed] [Google Scholar]
- Saada A, Aptowitzer I, Link G, et al. ATP synthesis in lipoamide dehydrogenase deficiency. Biochemical and Biophysical Research Communications. 2000;269:382–386. doi: 10.1006/bbrc.2000.2310. [DOI] [PubMed] [Google Scholar]
- Sandalinas M, Sadowy S, Alikani M, et al. Developmental ability of chromosomally abnormal human embryos to develop to the blastocyst stage. Human Reproduction. 2001;16:1954–1958. doi: 10.1093/humrep/16.9.1954. [DOI] [PubMed] [Google Scholar]
- Sellens MH, Stein S, Sherman MI. Protein and free amino acid content in preimplantation mouse embryos and in blastocysts under various culture conditions. Journal of Reproduction and Fertility. 1981;61:307–315. doi: 10.1530/jrf.0.0610307. [DOI] [PubMed] [Google Scholar]
- Thompson JG, McNaughton C, Gasparrini B, et al. Effect of inhibitors and uncouplers of oxidative phosphorylation during compaction and blastulation of bovine embryos cultured in vitro. Journal of Reproduction and Fertility. 2000;118:47–55. [PubMed] [Google Scholar]
- Thomson JL. Effect of inhibitors of carbohydrate metabolism on the development of preimplantation mouse embryos. Experimental Cell Research. 1967;46:252–262. doi: 10.1016/0014-4827(67)90063-8. [DOI] [PubMed] [Google Scholar]
- Vettakkorumakankav NN, Patel MS. Dihydrolipoamide dehydrogenase: structural and mechanistic aspects. Indian Journal of Biochemistry and Biophysics. 1996;33:168–176. [PubMed] [Google Scholar]
- West JD, Leask R, Green JF. Quantification of the transition from oocyte-encoded to embryo-encoded glucose phosphate isomerase in mouse embryos. Journal of Embryology and Experimental Morphology. 1986;97:225–227. [PubMed] [Google Scholar]
- Williams CH. Chemistry and Biochemistry of Flavoenzymes. CRC Press; Boca Raton: 1991. pp. 121–211. [Google Scholar]