

Abstract
Integration of solid-state biosensors and lipid bilayer membranes is important for membrane protein research and drug discovery. In these sensors, it is critical that the solid-state sensing material does not have adverse effects on the conformation or functionality of membrane-bound molecules. In this work, pore-spanning lipid membranes are formed over an array of periodic nanopores in free-standing gold films for surface plasmon resonance (SPR) kinetic binding assays. The ability to perform kinetic assays with a transmembrane protein is demonstrated with α-hemolysin (α-HL). The incorporation of α-HL into the membrane followed by specific antibody binding (anti-α-HL) red-shifts the plasmon resonance of the gold nanopore array, which is optically monitored in real time. Subsequent fluorescence imaging reveals that the antibodies primarily bind in nanopore regions, indicating that α-HL incorporation preferentially occurs into areas of pore-spanning lipid membranes.
Introduction
Lipid bilayer membranes are crucial biological structures that form the boundaries of cells and subcellular organelles. These membranes are composed of a variety of lipids, such as phospholipids, sphingolipids, glycolipids or cholesterol, to name a few. Furthermore, these membranes contain numerous peptides and proteins that decorate their surfaces or span across the membrane. The physiological basis for many diseases and conditions can be traced to irregularities in the cell membranes themselves or with membrane-bound proteins. In fact, half of the 100 best selling drugs target membrane-bound proteins.1, 2 Accordingly, the ability to investigate molecular interactions on the surfaces of lipid bilayer membranes is of great importance for basic biology as well as drug discovery.3
In order to interface lipid membranes with biosensors, many groups have employed supported lipid bilayers (SLBs) or black lipid membranes (BLMs).4 In a SLB, the membrane is formed on a solid substrate, whereas in a BLM, the membrane is suspended over a small aperture in a solid substrate. SLBs have been employed in a number of biochemical and biophysical studies on the fundamental properties of lipid membranes and can be formed by different methods.5–9 Because SLBs are formed directly on a solid substrate with a thin layer of water (1–2 nm) between the membrane and the substrate, steric hindrance makes incorporation of integral proteins challenging, and those proteins that are inserted can be denatured.10, 11 In order to overcome this issue, some groups have tethered lipid bilayers to the underlying substrate using linker molecules, or formed a polymer cushion between the membrane and the substrate.12 While these strategies have proven useful for incorporating proteins into lipid bilayers for biosensing,13 the lipid membrane is still only accessible from the top side, limiting their utility for studying transmembrane signaling processes. An alternative approach, suspending membranes over apertures in a substrate in the form of a BLM where both sides of membrane can be accessed, has also been employed for interrogating membranes and membrane-associated proteins. Studies on the electrical properties of lipid membranes and reconstituted ion channels have made extensive use of BLMs, even though the stability of BLMs is notoriously poor. To enhance the long-term stability of BLMs, some groups have employed nanometer-sized apertures for electrical and physical property measurements of membranes and proteins.14–17 This configuration allows for the incorporation of transmembrane proteins into the pore-spanning regions of the membrane; accordingly the ionic transport properties of the membrane proteins can be investigated.18 Similarly, in the present study we demonstrate that lipid bilayer membranes formed by vesicle rupture6 over periodic nanopore arrays in a free-standing metal film can be used for surface plasmon resonance (SPR) sensing.
Surface plasmon resonance biosensing
In conventional SPR instruments such as BIAcore™, analytes binding to capture ligands immobilized on a gold surface changes the local refractive index, which, in turn, induces a shift in the SPR excitation angle or wavelength. Surface plasmons (SPs) are electromagnetic surface waves propagating at the interface of a metallic film and a dielectric medium, and are thereby extremely sensitive to surface properties. While SPR technologies have been successfully used to characterize the interaction between pairs of soluble binding partners,19–22 significant challenges remain to adapt conventional SPR instruments to the needs of membrane proteins.1 In particular, the gold surface prohibits access to the bottom side of the membrane and can partially denature and hinder diffusive mobility of transmembrane proteins.10, 23
Recently, a new class of SPR biosensors based on metallic nanostructures has been demonstrated to overcome the limitations of the currently commercialized technology.24–28
Of particular interest are the SPR sensors based on the extraordinary optical transmission (EOT) effect through periodic nanopore arrays in metallic films.29–31 The periodic array of nanopores can act as a diffraction grating and convert incident light into SP waves at resonance wavelengths, thereby creating a series of intense peaks in the optical transmission spectra. Following an initial demonstration of using the EOT effect for biosensing,32 several groups have shown the potential of this platform for label-free kinetic SPR detection and high-throughput imaging.33–40 While the focus of previous work has mainly addressed the optical design, sensitivity and the development of multiplexing capability, the unique potential of the nanopore geometry, i.e. the ability to naturally integrate SPR biosensing with nano-BLMs and proteins integrated therein, has not been explored. Dahlin et al. demonstrated a glass-supported phospholipid bilayer formed at the bottom of nanopores that are randomly distributed in a 20 nm-thick gold film on a glass substrate for localized surface plasmon resonance (LSPR) biosensing.41, 42 However, the LSPR effect in randomly distributed single nanopores results in a broad extinction spectrum that is weaker than the EOT effect through a periodic nanopore array.31 Also, the LSPR sensors have a limited probing range because the LSPR decay length is about 20–50 nm. In contrast, in the visible range, the sensors based on propagating SP waves such as periodic nanopore arrays can probe about 100–300 nm from the metallic surface.43
In this work, we present periodic nanopore arrays in freestanding Au/Si3N4 films as SPR biosensors to detect the incorporation of a transmembrane protein, α-hemolysin (α-HL), into a pore-spanning membrane and also to detect subsequent antibody binding. α-HL is a water-soluble peptide monomer (33.2 kD) secreted from the pathogenic bacteria Staphylococcus aureus that binds to the plasma membranes of many mammalian cell types.44 Upon binding to the membrane the monomers freely diffuse about the membrane and self-assemble to form a heptameric conductive transmembrane pore.45 The crystal structure of α-HL was determined by Song et al. and shows a membrane-penetrating β-barrel and a mushroom shaped cap domain that protrudes approximately 70 Å above the membrane surface.46 Because α-HL has been so well characterized, it is a commonly used model for studies of transmembrane proteins as well as for nanopore-based biosensing.47, 48
Results and Discussion
EOT-based membrane biosensing
The concept of the EOT-based membrane biosensing platform presented in this work is illustrated in Fig. 1a. A nanopore array is patterned in a free-standing Au/Si3N4 film, followed by conformal encapsulation with a 20-nm-thick silica layer, and a pore-spanning lipid membrane is formed over the silica-coated Au surface of the nanopore array by vesicle rupture. Part of the lipid membrane is thus suspended over the nanopores as a lipid “drum” and is accessible from both sides, more closely mimicking natural cell membranes. The presence of the lipid membrane and the subsequent binding of biomolecules at its surface modulate the local refractive index and the EOT effect through the gold nanopores. This is detected by a shift in the transmission spectrum.
Figure 1.
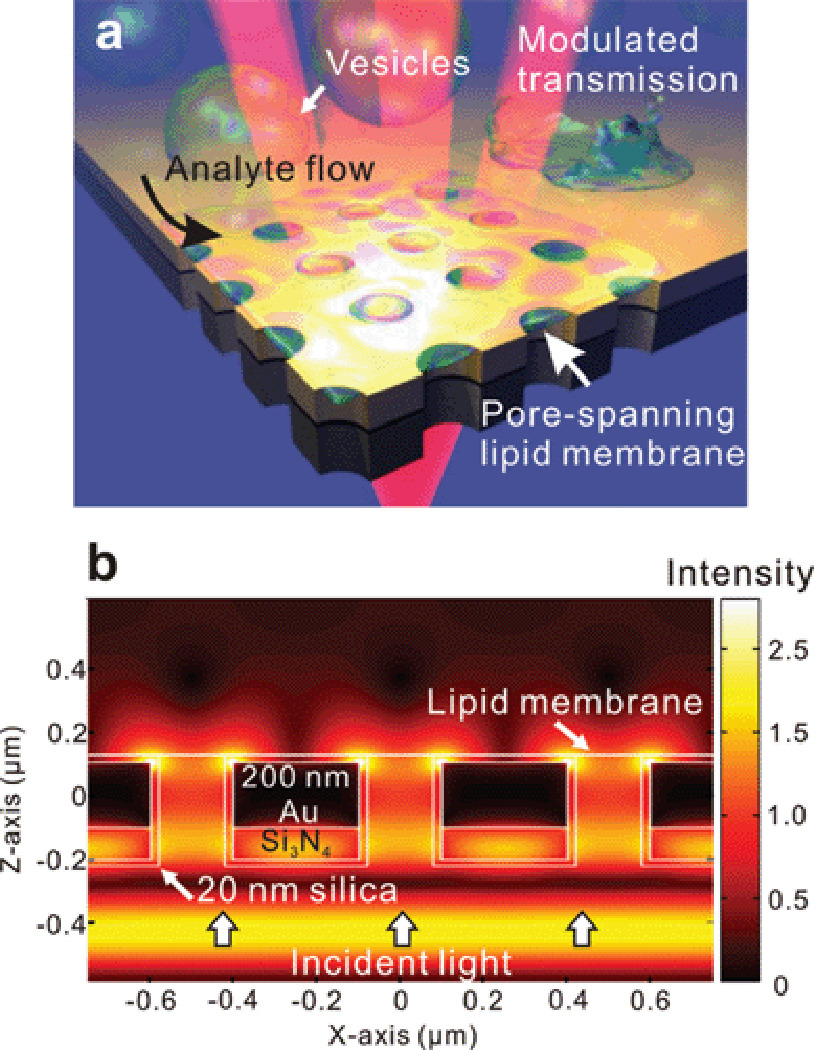
Fig. 1b shows three-dimensional finite-difference time-domain (FDTD) calculations of the field intensity at a transmission resonance (787 nm) in the region of a nanopore array (500 nm pitch). Strong field intensities due to SPs can be observed over the nanopores and near their edges. The intense, highly confined plasmonic fields probe the local refractive index, suggesting high sensitivity in and around the edges of the nanopores49–51 where the pore-spanning lipid membrane forms. The SP fields are, therefore, able to detect the presence of molecules binding to the membrane suspended above the pore.
Fabrication of plasmonic nanopore arrays
Fig. 2a shows a process flow for making our microfluidic SPR detection platform with the nanopore chip. The free-standing Si3N4 film was fabricated by patterning a 100 nm-thick low-stress Si3N4 layer on the backside of a Si wafer and a subsequent anisotropic Si etching process with potassium hydroxide (KOH). Then, a 200 nm thick Au layer was deposited by electron-beam evaporation, along with a 5 nm-thick Cr adhesion layer. The nanopore arrays were milled through the suspended Au and Si3N4 stack with a focused ion beam (FIB). Similar methods were previously used to fabricate free-standing metal films with nanopores to investigate optical transmission, molecular translocation, or flow-through biosensing.49, 52, 53
Figure 2.
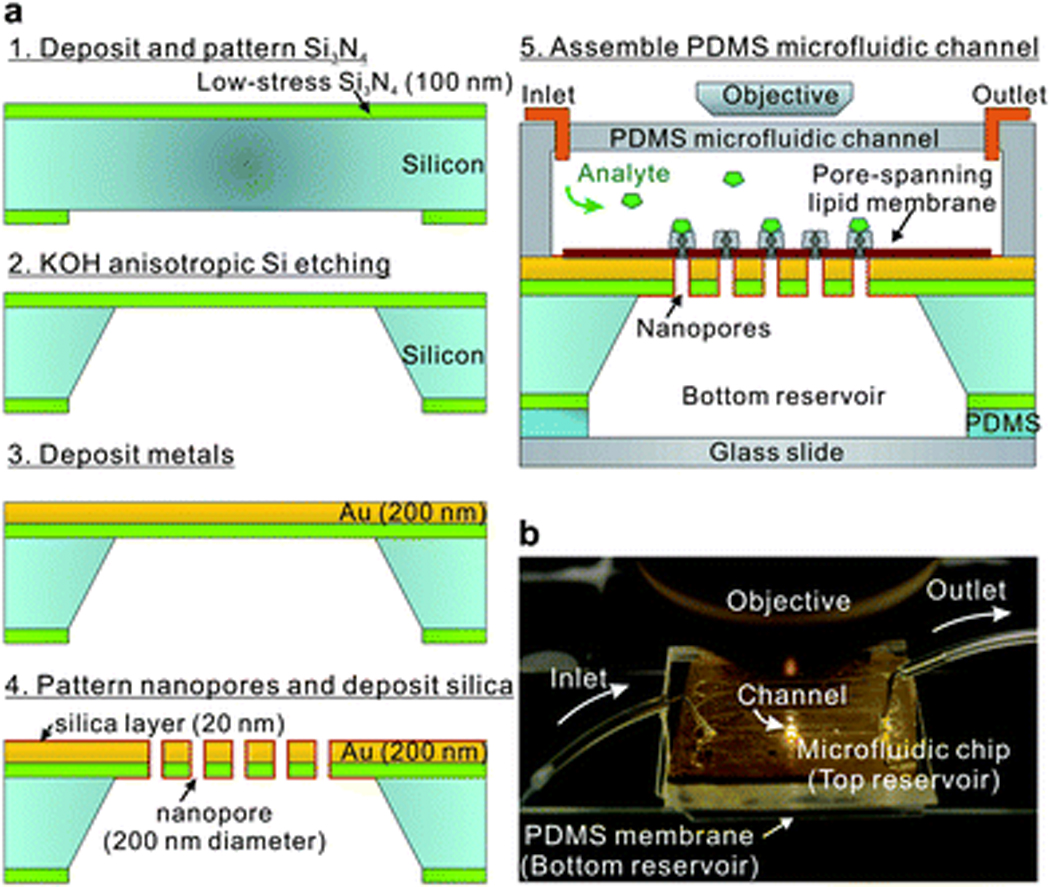
A polydimethylsiloxane (PDMS, Sylgard) microfluidic channel and a thin PDMS membrane sealed the top and bottom sides of nanopore chip, allowing both sides of the chip, the nanopores, and the lipid membrane to be immersed in aqueous solution. After forming a pore-spanning lipid bilayer via vesicle rupture, analytes were injected through the top-side microfluidic channel for real-time binding kinetic measurements as shown in Fig. 2b. For EOT spectral measurements, a tungsten-halogen lamp illuminated the nanopore arrays from the top-side through a 50× microscope objective. The transmitted light is then collected with a fiber-optic spectrometer.36
Fig. 3a shows a scanning electron microscope (SEM) image of the backside of the free standing Au/Si3N4 film. The bottom reservoir is created by anisotropic wet etching of Si with KOH. Fig. 3b shows a cross-sectional SEM image of the nanopore array milled through the Au/Si3N4 suspended layers. The array consists of 16 × 16 nanopores with the pore diameter of 200 nm and periodicity of 500 nm.
Figure 3.

The Au sensing surface was then coated with a 20 nm-thick silica layer as shown in Fig. 3c using atomic layer deposition (ALD),54 which deposits a conformal layer covering nanosized structures with precise thickness control. The silica layer uniformly covered the top surface and the sidewall inside each nanopore, which promoted vesicle rupture on its hydrophilic surface, allowing the formation of the suspended lipid bilayer over the nanopore array. In general, propagating SP-based periodic nanopore arrays show sensing ranges of about 100–300 nm in the visible wavelengths, several times longer than LSPR based sensors with randomly distributed nanopores.55 Therefore, the presence of the 20 nm thick silica layer does not severely degrade the EOT detection sensitivity.56
Pore-spanning lipid membrane formation
For pore-spanning membrane formation, a phosphate buffered saline (PBS) solution was injected through the microfluidic channel, flowing for at least 1 hour to ensure that both sides of nanopores were filled with buffer. This guarantees that the membrane would be contacted by buffer on both sides, closely mimicking a natural cell membrane environment and increasing the stability of the pore-spanning lipid membrane. Then the channel was rinsed and filled with NaTrisCa buffer before injecting lipid vesicles. A solution containing vesicles was injected with a slow flow rate of 10 µL/hr for at least 1 hour to allow vesicle rupture and formation of the pore-spanning lipid membrane. (Supplementary Fig. S1) The channel was then rinsed with NaTrisCa buffer followed by PBS.
The vesicle rupture pathway, while somewhat dependent on the lipid composition and the nature of the substrate, proceeds through the following general steps. First, a small number of vesicles adsorb to the surface. Then when a critical number of vesicles populate the surface, they begin to rupture, forming a SLB. Consequently, SLB grows by rupture of additional vesicles at the edge of the supported bilayer, driven by hydrophobic interactions between the SLB edge and adjacent vesicles.57 A variety of substrates, such as glass and SiO2 are compatible with vesicle rupture.57, 58 Noble metal surfaces can also be used, provided they have chemical functionalization that promotes vesicle/surface interactions and subsequent rupture.59 The vesicles used in this study averaged 361 nm in diameter, determined by dynamic light scattering. The rupture of a vesicle this size leads to a planar membrane disc that is 722 nm in diameter, which is more than sufficient to cover an individual nanopore.
After lipid membrane formation, fluorescence recovery after photobleaching (FRAP) was used to confirm the formation of a continuous lipid membrane on the substrate. FRAP is capable of quantifying the two dimensional lateral diffusion of lipid components on the membrane and is widely used to characterize supported or suspended lipid membranes.
For our experiments, a drop of vesicle solution was placed on the nanopore array surface and the substrate was placed in a humidified box for at least 1 hour to allow vesicle rupture and membrane formation. After exposure to the fluorescently-labeled (Rho-PE) vesicle solution and surface washing, a homogenous fluorescent layer was readily apparent indicating that vesicle rupture occurred and a membrane layer had been formed. The surfaces of the sample were kept immersed in aqueous solution throughout the course of the experiment. In the FRAP experiments, a small circular area of membrane (~21.5 µm diameter) was photobleached with an intense laser beam at a wavelength of 405 nm. Diffusion of unbleached Rho-PE back into the previously bleached area indicates a continuous membrane layer. Fig. 4a depicts a FRAP experiment over an array of 200 nm holes with 500 nm periodicity. The nanopores are not visible in the fluorescence images, but their presence was confirmed by observing the sample in transmission mode. Four fluorescence images of the sample show the array before photobleaching, right after photobleaching, at 40% recovery and at maximum recovery. Immediately after bleaching a dark spot is evident; the fluorescence of which begins to recover immediately and nears full recovery (~ 90%) after approximately 50 seconds, indicating that a continuous membrane is present. Fig. 4b presents the recovery curves for two different experimental conditions: an egg PC membrane over a nanopore array and an egg PC membrane supported on a flat silica surface. Qualitatively, the recovery curves from the two conditions are quite similar, indicating that the presence of the nanopore array not significantly change the continuity or fluidity of the membrane. Quantitatively, from the recovery curve we are able to calculate the diffusion coefficient D for lipids in the membrane, using the equation D = R2/4τD, where R is the radius of the photobleached spot, and τD is the characteristic diffusion time.60, 61 We determine τD from fits of the recovery curves as described by Soumpasis.60 For membranes over nanopore arrays and supported on flat silica, the diffusion coefficients were calculated to be 1.99 ± 0.14 µm2/s and 1.94 ± 0.14 µm2/s, respectively which are in the range of previous FRAP results on silica surfaces.42, 62, 63 The similarity between D values suggests that a pore-spanning lipid membrane is formed over the nanopore array, and that the pores do not limit lipid diffusion.64
Figure 4.
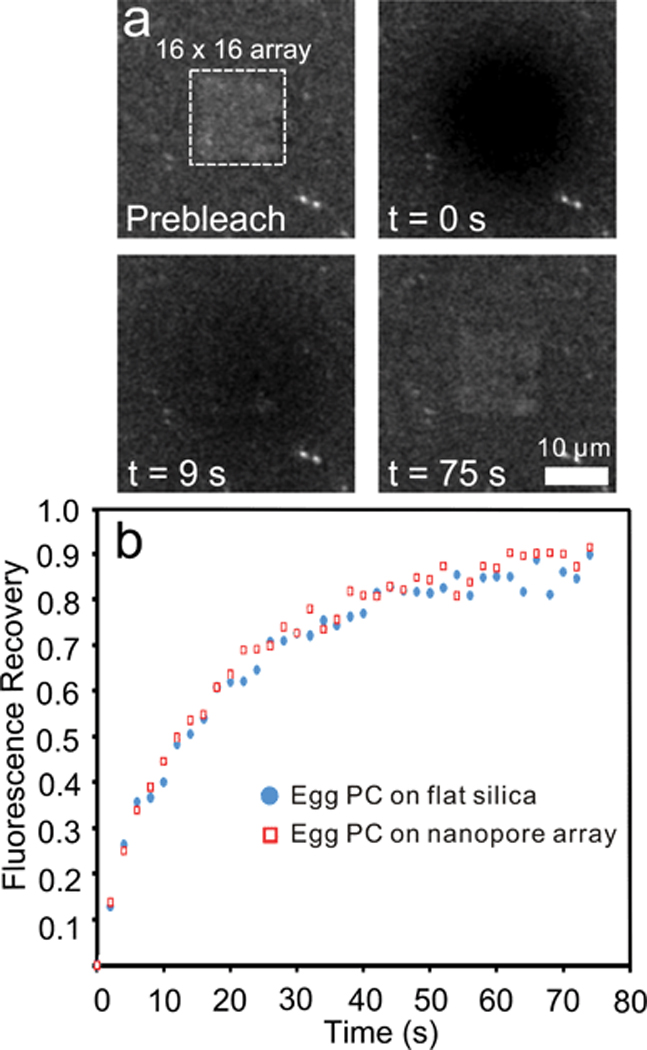
Recently Jönsson and coworkers performed FRAP analysis on membranes spanning random arrays of 80 nm-diameter wells in a silica substrate.64 Their theoretical analysis of diffusion coefficients in a well-spanning membrane suggests that the apparent diffusion coefficient should be 1.1-times the diffusion coefficient for a membrane totally supported by underlying layer of silica. In reality, they measured an apparent diffusion coefficient that was nearly identical (0.99-times) to the diffusion coefficient of a supported membrane on silica. They suggest the slight discrepancy between theory and experiment may be due to small amount of the membrane conforming to the nanowell wall. The experimental parameters presented here are quite similar to those presented by Jönsson et al. In our work, 12% of the surface area of a unit cell of the nanopore array is occupied by a nanopore. The diffusion coefficient calculated for a membrane over a nanopore array is experimentally equivalent or even a bit larger than the diffusion coefficient of a fully supported membrane, which agrees well with the theoretical and experimental results in Jönsson et al., indicating that a pore-spanning membrane is formed. Without the formation of pore-spanning lipid membrane, the effective diffusion coeffient would be smaller than the value for a SLB on flat silica.64
Additional confirmation of pore-spanning membrane formation was provided by imaging the array from the side opposite that which the lipid membrane is formed. (Supplementary Fig. S2) Fluorescence in the shape of a nanopore array is visible from this viewpoint. Because the chip is opaque, fluorescence excitation and emission can only pass through the nanopores. Fluorescence visible from this viewpoint can only come from fluorophores over the array in the form of a pore-spanning membrane. Taken in combination with FRAP data, this provides strong evidence that the nanopores are covered with a lipid membrane and that the membrane is continuous.
Real-time kinetic binding assays
In periodic nanopore-based plasmonic biosensors, SP-mediated EOT is measured through the nanopores as shown in Fig. 5a. The SP waves sense local changes in refractive index, and the resonant transmission wavelengths red-shift with increasing amounts of adsorbate.
Figure 5.
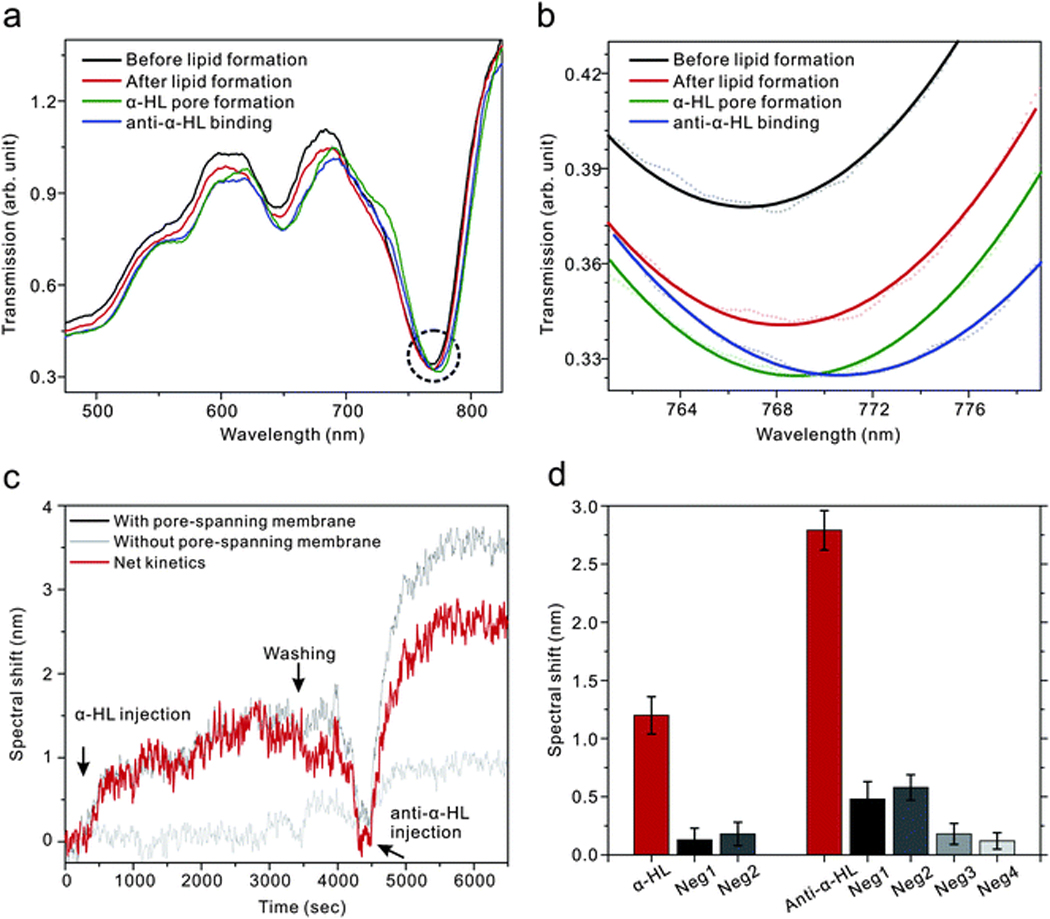
The spectral shifts from the nanopore arrays were observed at the minima (Fig. 5b) in the transmission spectra marked by the dashed circle in Fig. 5a. After forming pore-spanning lipid membrane, the resonance wavelength red-shifted. The position shifted further after transmembrane proteins (α-HL) were incorporated into the lipid membrane and subsequent binding of biotinylated anti-α-HL to the α-HL. The real-time kinetics were measured by monitoring the spectral shift of the transmission as a function of time while molecules bind to the surface.
Fig. 5c shows real-time kinetic measurements of α-HL binding on the membrane surface and subsequent binding of anti-α-HL with α-HL. For a positive control, 1 µM of α-HL was injected and incubated for an hour, sufficient to form transmembrane pores.65 Then 300 nM of anti-α-HL was injected to bind with α-HL after PBS washing. A first negative control was performed with the same solutions on a lipid-free surface where the nanopore surface was coated by a monolayer of 3-Aminopropyl-triethoxysilane (APTES). By subtracting the spectral response of the negative control from that of positive control, it is possible to eliminate experimental artifacts such as bulk refractive index changes and non-specific binding.40
To confirm the incorporation of α-HL with the lipid membrane and specific binding between α-HL and anti-α-HL, the spectral shifts obtained from the positive control were compared with, in all, four negative controls: (1) a lipid-free environment with APTES from Fig. 5c; (2) a lipid-free environment where the pore-spanning membrane is replaced by a layer of BSA; (3) the same pore-spanning lipid environment as the positive control, but with the absence of α-HL; (4) the same pore-spanning lipid environment as the positive control, but α-HL is replaced by BSA. As shown in Fig. 5d, spectral shifts from those three negative controls were less than 20% of the shift in the positive control for both α-HL and anti-α-HL binding, indicating that α-HL can be incorporated only with the lipid membrane environment and that specific binding of anti-α-HL can occur only with the presence of α-HL on the membrane surface.
One of distinguishing advantages of SPR biosensing compared to other sensing techniques is that it enables the measurement of the binding affinity of protein-protein interactions from their real-time label-free association and dissociation binding kinetic curves.19
Fig. 6a shows real-time kinetic measurements with different concentrations (50, 100, and 200 nM) of anti-α-HL. After 5 min of baseline with a PBS solution, an anti-α-HL solution was injected to the channel with a constant flow rate of 30 µL/min. The association of binding was measured for 15 min and then the channel was washed by PBS solution with the same flow rate of 30 µL/min for dissociation. A nonlinear least squares analysis method66 was used to determine the dissociation constant, KD, from the kinetic measurements, giving a value of (1.9 ± 1.0) × 10−8 M. This value is fairly close to the previously reported value with monoclonal anti-α-HL binding.67 We also determined the limit of detection to be 26 nM from the signal at 3 times the standard deviation of baseline noise.
Figure 6.
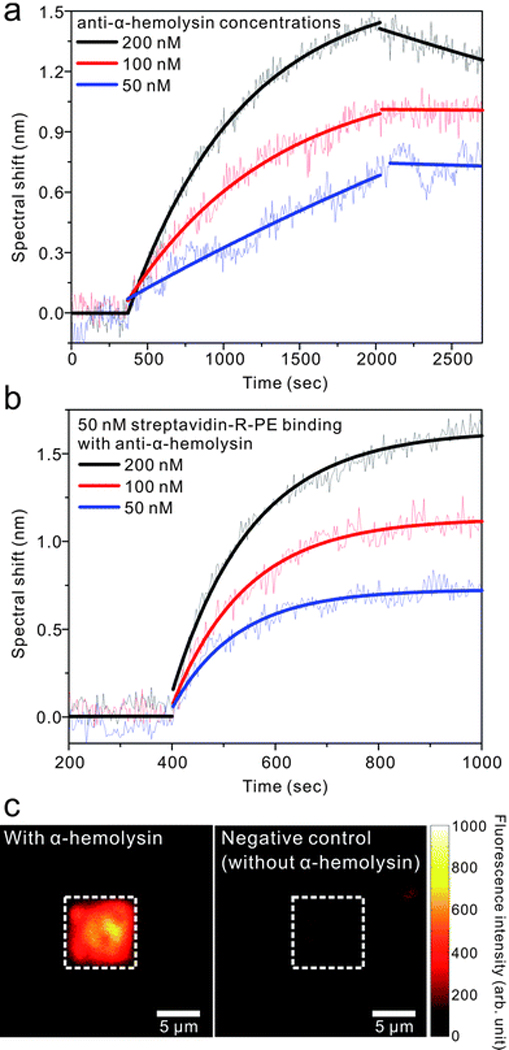
For further confirmation of the specific binding of anti-α-HL with α-HL on the pore-spanning lipid membrane, 50 nM of streptavidin-R-phycoerythrin (SAPE) was injected to bind with the biotinylated anti-α-HL used in Fig. 6a.
Fig. 6b shows the binding kinetics of 50 nM of SAPE to the membrane-bound anti-α-HL antibodies with the three different concentrations from Fig. 6a. Each curve shows different maximum signal value, Rmax, which corresponds to the maximum capacity of immoblized ligand that binds with analytes in the injected solution. It indicates that the amount of anti-α-HL bound on the surface is different, since Rmax values have a linear relation to the concentration of anti-α-HL injected in Fig. 6a. (Supplementary Fig. S3)
After consequent binding of 100 nM of anti-α-HL and 50 nM of SAPE on the lipid membrane surface, the fluorescent images were taken as shown in Fig. 6c. Positive and negative controls are with and without α-HL on the membrane surface, respectively. The images are shown with the same intensity scale. While the negative control shows partial dim spots due to non-specific binding, the positive control shows strong contrast of fluorescent intensity between inside and outside of array. This means that more α-HL was incorporated on the nanopore array where pore-spanning membranes were formed over the nanopores and more anti-α-HL and SAPE bind to those areas.68 These α-HL and anti-α-HL kinetic measurements and subsequent binding with SAPE demonstrate successful formation of pore-spanning lipid membrane incorporated with transmembrane protein on a free-standing nanopore arrays that are able to detect antibody binding events in real-time using an SPR sensing technique.
Conclusions
We have integrated kinetic SPR sensors based on periodic nanopores in a gold film with a pore-spanning lipid membrane that is in contact with buffer on both sides. The part of the lipid membrane spanning the nanopore is freely suspended and is thus more similar to natural cellular membranes than a supported membrane. Lipid membranes were formed using vesicle rupture facilitated by conformal encapsulation of the gold sensing surface with a 20-nm-thick ALD-grown silica layer. FRAP measurements demonstrated that the lipid membrane formed over the nanopore arrays is homogeneous and continuous. The detection of anti-α-HL binding on α-HL incorporated lipid membranes demonstrates the functionality of our integrated platform for lipid membrane SPR sensing. Because our method can exploit the SPR sensing capability of gold films while avoiding their undesirable side effects for cell membranes, i.e. denaturing membrane proteins or hindering the formation of stable lipid bilayers, it provides a natural platform for real-time label-free SPR imaging40 with cell membranes. Furthermore, our technique for making pore-spanning lipids does not require organic solvents, thus it is compatible with cell plasma membranes and membrane proteins integrated therein, demonstrating that this platform could potentially be employed for characterizing ligand binding to transmembrane proteins such as G protein-coupled receptors (GPCRs) and ion channels. In addition, SPR could be combined with electrical measurements, such as impedance spectroscopy, to monitor and quantify insertion of ion channels. Recent advances in high-throughput nanofabrication techniques for patterning large-area periodic nanopores37, 69, 70 can also extend the capability of the present platform toward microarray-based membrane biosensing and SPR imaging.40
Methods
Sample Fabrication
The 100 nm-thick low-stress Si3N4 layer was initially deposited on a (100) Si wafer using low pressure chemical vapor deposition (LPCVD). After patterning Si3N4 layer on the backside of Si wafer using reactive ion etching (RIE), the Si substrate was subsequently etched from the backside with anisotropic KOH etching, stopping at the Si3N4 layer, and creating a suspended Si3N4 film with dimensions ranging from 50 × 50 µm2 to 300 × 300 µm2. Then a 5 nm chromium adhesion layer and a 200 nm-thick gold film were deposited onto the nitride layer by an e-beam evaporator (CHA, SEC 600). Nanopores through the Au and Si3N4 layers with a diameter of 200 nm were made into regular arrays using focused ion beam (FIB, FEI Quanta 200 3D) milling. The FIB conditions were an acceleration voltage of 30 kV with a current of 500 pA. The surface was coated by a 20 nm-thick silica layer using atomic layer deposition (ALD, Cambridge Nano Tech, Inc., Savannah) at 100 °C. The detailed fabrication process is described in the supplementary information.
FDTD Calculations
Commercial FDTD software (FullWAVE, RSoft Design Group) was used. For computational efficiency, periodic boundary conditions were used, simulating an infinite nanopore array. The refractive index of the 100 nm-thick nitride membrane was set to 2.05, and the refractive indices of the 200 nm-thick gold and 5 nm-thick chromium layers were simulated as a Drude/Lorentz model fit to experimental optical constants.71, 72 A non-uniform mesh grid was also used, with a 10 nm grid step in the surrounding liquid buffer areas (refractive index = 1.33), tapering to 1 nm at the edges and corners of the structures, and incorporating at least 10 grid point within the 5 nm-thick lipid (refractive index 1.46)13 and chromium layers. The 20 nm-thick conformal ALD layer had a refractive index of 1.45. The entire x-y-z simulation domain was 500-by-500-by-2000 nm, periodic in x and y. A pulsed-wave simulation was performed first, finding the location of the EOT resonance peak, followed by a continuous-wave simulation to extract the steady-state field distributions in and around the nanopores.
Vesicle Preparation
The vesicles employed for biosensing experiments were composed of L-α-phosphatidylcholine (egg PC) and from Avanti Polar Lipids (Alabaster, AL USA).
For FRAP, 1% (w/w) 1, 2 dimyristoyl-sn-glycero-3-phosphatidylethanolamine-N-(lissamine Rhodamine B sulfonyl) ammonium salt (Rho-PE) was included with the Egg PC. To form the vesicles, chloroform solutions with the proper amounts of egg PC and Rho-PE were placed in a glass vial and the chloroform was evaporated under vacuum for 3 hours. The dry lipid film was then rehydrated with NaTrisEDTA (10 mM NaCl, 10 mM Tris, 10 mM EDTA, pH = 8) and refrigerated overnight. The rehydrated lipid suspension was vortex mixed then sonicated for 10 minutes in a room temperature water bath. The resulting vesicles had a diameter of 361 ± 215 nm, as determined by dynamic light scattering. The vesicles were then transferred to NaTrisCa buffer (10 mM NaCl, 10 mM Tris, 10 mM CaCl2, pH = 8) at a total lipid concentration of 200 µg/mL. NaTrisCa buffer has been employed by other groups to promote vesicle rupture on silica surfaces.42
FRAP Measurements
For FRAP measurements, vesicles containing 1% Rho-PE were allowed to rupture on a silica-coated chip with milled nanopore arrays. FRAP was carried out on an Olympus FV1000 upright confocal microscope controlled by Olympus Fluoview software. A small spot of membrane was photobleached with an intense laser beam at 405 nm, and the fluorescence recovery was monitored as a function of time by rastering a laser spot at 534 nm across the sample. During fluorescence recovery image frames were recorded at 0.5 or 0.33 Hz..
Analysis of FRAP image sequences was done with ImageJ software version 1.42q. In each frame of the image sequence the fluorescence intensity of the bleached spot was averaged and fractional fluorescence recovery was calculated as described by Axelrod et al.73 and the recovery curve was fit as described by Soumpasis60 using Matlab. From the fit of the recovery curve, the characteristic diffusion time was determined. The diffusion coefficient (D) was calculated from the average of three experiments with the equation D = R2/4τD, where R is the radius of the bleached spot and τD is the characteristic diffusion time.
Experimental Setup
For spectral measurements, a tungsten-halogen lamp illuminated the nanopore arrays from the top-side through a 50× microscope objective (NA = 0.55). The transmitted light is then collected using an optical fiber (200 µm diameter core), and transmission spectrum was analyzed with an Ocean Optics Fiber Optic Spectrometer. The integration time is 1 sec. The measured transmission spectra were normalized by a reference spectrum of the tungsten-halogen lamp. For kinetic experiments, the spectral shifts of minimum transmission around 770 nm were monitored. During kinetic measurements, the spectra were recorded every 5 sec, except streptavidin binding which was recorded every 2 sec because of the fast binding between biotin and streptavidin. For binding kinetics of anti-α-HL and streptavidin shown in Fig. 6a and 6b, the solutions were injected with a constant flow rate of 30 µL/min. For dissociation of binding, PBS was injected with the same flow rate of 30 µL/min. The flow rate is controlled by a syringe pump (Harvard Apparatus PHD-2000). The molecules used in this study (α-HL, anti-α-HL, and strepavidin-R-phycoerythrin) were obtained from Sigma-Aldrich, Co., Abcam, Inc., and Invitrogen, respectively.
Supplementary Material
Acknowledgements
This work was supported by grants from the NIH (R01 GM092993-01) and the NSF IDBR Program (DBI-0964216). H.I. and N.C.L. acknowledge 3M Fellowship and UMN Doctoral Dissertation Fellowship, respectively. S.H.O. acknowledges the Minnesota Partnership Award for Biotechnology and the 3M Faculty Award. Device fabrication was performed at the U. of Minnesota Nanofabrication Center (NFC), which receives support from NSF through the National Nanotechnology Infrastructure Network.
Footnotes
Electronic Supplementary Information (ESI) available: Details of fabrication process for making a free-standing nanopore array chip integrated with microfluidic channels and supplementary figures. See DOI: 10.1039/b000000x/
Notes and references
- 1.Cooper MA. J. Mol. Recognit. 2004;17:286–315. doi: 10.1002/jmr.675. [DOI] [PubMed] [Google Scholar]
- 2.Drews J. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 3.Maynard JA, Lindquist NC, Sutherland JN, Lesuffleur A, Warrington AE, Rodriguez M, Oh SH. Biotechnol. J. 2009;4:1542–1558. doi: 10.1002/biot.200900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sackmann E. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 5.Castellana ET, Cremer PS. Surf. Sci. Rep. 2006;61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brian AA, McConnell HM. Proc. Nat. Acad. Sci. U.S.A. 1984;81:6159–6163. doi: 10.1073/pnas.81.19.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamm LK, McConnell HM. Biophys. J. 1985;47:105–113. doi: 10.1016/S0006-3495(85)83882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cremer PS, Boxer SG. J. Phys. Chem. B. 1999;103:2554–2559. [Google Scholar]
- 9.Keller CA, Glasmastar K, Zhdanov VP, Kasemo B. Phys. Rev. Lett. 2000;84:5443–5446. doi: 10.1103/PhysRevLett.84.5443. [DOI] [PubMed] [Google Scholar]
- 10.Salafsky J, Groves JT, Boxer SG. Biochemistry. 1996;35:14773–14781. doi: 10.1021/bi961432i. [DOI] [PubMed] [Google Scholar]
- 11.Glazier SA, Vanderah DJ, Plant AL, Bayley H, Valincius G, Kasianowicz JJ. Langmuir. 2000;16:10428–10435. [Google Scholar]
- 12.Sinner EK, Knoll W. Curr. Opin. in Chem. Biol. 2001;5:705–711. doi: 10.1016/s1367-5931(01)00269-1. [DOI] [PubMed] [Google Scholar]
- 13.Vockenroth IK, Atanasova PP, Jenkins ATA, Köper I. Langmuir. 2008;24:496–502. doi: 10.1021/la7030279. [DOI] [PubMed] [Google Scholar]
- 14.Han X, Studer A, Sehr H, Geissbühler I, Di Berardino M, Winkler FK, Tiefenauer LX. Adv. Mater. 2007;19:4466–4470. [Google Scholar]
- 15.Hennesthal C, Steinem C. J. Am. Chem. Soc. 2000;122:8085–8086. [Google Scholar]
- 16.White R, Zhang B, Daniel S, Tang J, Ervin E, Cremer P, White H. Langmuir. 2006;22:10777–10783. doi: 10.1021/la061457a. [DOI] [PubMed] [Google Scholar]
- 17.Reimhult E, Kumar K. Trends Biotechnol. 2008;26:82–89. doi: 10.1016/j.tibtech.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 18.White R, Ervin E, Yang T, Chen X, Daniel S, Cremer P, White H. J. Am. Chem. Soc. 2007;129:11766–11775. doi: 10.1021/ja073174q. [DOI] [PubMed] [Google Scholar]
- 19.Liedberg B, Nylander C, Lundstrom I. Sens. Actuators. 1983;4:299–304. [Google Scholar]
- 20.Smith EA, Corn RM. Appl. Spectrosc. 2003;57:320A–332A. doi: 10.1366/000370203322554446. [DOI] [PubMed] [Google Scholar]
- 21.Shumaker-Parry JS, Aebersold R, Campbell CT. Anal. Chem. 2004;76:2071–2082. doi: 10.1021/ac035159j. [DOI] [PubMed] [Google Scholar]
- 22.Ouellet E, Lausted C, Lin T, Yang CWT, Hood L, Lagally ET. Lab Chip. 2010;10:581–588. doi: 10.1039/b920589f. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka M, Rossetti FF, Kaufmann S. Biointerphases. 2008;3:FA12–FA16. doi: 10.1116/1.2905233. [DOI] [PubMed] [Google Scholar]
- 24.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nat. Mater. 2008;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 25.Galush WJ, Shelby SA, Mulvihill MJ, Tao A, Yang PD, Groves JT. Nano Lett. 2009;9:2077–2082. doi: 10.1021/nl900513k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao H, Yang J-C, Lin JY, Stuparu AD, Lee MH, Mrksich M, Odom TW. Nano Lett. 2010;10:2549–2554. doi: 10.1021/nl101165r. [DOI] [PubMed] [Google Scholar]
- 27.Odom TW. MRS Bullein. 2010;35:66–73. [Google Scholar]
- 28.Gordon R, Brolo AG, Sinton D, Kavanagh KL. Laser Photonics Rev. 2010;4:311–335. [Google Scholar]
- 29.Ebbesen TW, Lezec HJ, Ghaemi HF, Thio T, Wolff PA. Nature. 1998;391:667–669. [Google Scholar]
- 30.Gao HW, Henzie J, Odom TW. Nano Lett. 2006;6:2104–2108. doi: 10.1021/nl061670r. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Vidal FJ, Martin-Moreno L, Ebbesen TW, Kuipers L. Rev. Mod. Phys. 2010;82:729–787. [Google Scholar]
- 32.Brolo AG, Gordon R, Leathem B, Kavanagh KL. Langmuir. 2004;20:4813–4815. doi: 10.1021/la0493621. [DOI] [PubMed] [Google Scholar]
- 33.Stark PRH, Halleck AE, Larson DN. Methods. 2005;37:37–47. doi: 10.1016/j.ymeth.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 34.Tetz KA, Pang L, Fainman Y. Opt. Lett. 2006;31:1528–1530. doi: 10.1364/ol.31.001528. [DOI] [PubMed] [Google Scholar]
- 35.Stewart ME, Mack NH, Malyarchuk V, Soares J, Lee TW, Gray SK, Nuzzo RG, Rogers JA. Proc. Nat. Acad. Sci. U.S.A. 2006;103:17143–17148. doi: 10.1073/pnas.0606216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lesuffleur A, Im H, Lindquist NC, Oh SH. Appl. Phys. Lett. 2007;90:243110. [Google Scholar]
- 37.Henzie J, Lee MH, Odom TW. Nat. Nanotechnol. 2007;2:549–554. doi: 10.1038/nnano.2007.252. [DOI] [PubMed] [Google Scholar]
- 38.Yang JC, Ji J, Hogle JM, Larson DN. Nano Lett. 2008;8:2718–2724. doi: 10.1021/nl801043t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindquist NC, Lesuffleur A, Im H, Oh S. Lab Chip. 2009;9:382–387. doi: 10.1039/b816735d. [DOI] [PubMed] [Google Scholar]
- 40.Im H, Lesuffleur A, Lindquist NC, Oh SH. Anal. Chem. 2009;81:2854–2859. doi: 10.1021/ac802276x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dahlin A, Zäch M, Rindzevicius T, Käll M, Sutherland DS, Höök F. J. Am. Chem. Soc. 2005;127:5043–5048. doi: 10.1021/ja043672o. [DOI] [PubMed] [Google Scholar]
- 42.Jonsson MP, Jonsson P, Dahlin AB, Höök F. Nano Lett. 2007;7:3462–3468. doi: 10.1021/nl072006t. [DOI] [PubMed] [Google Scholar]
- 43.Yang JC, Gao HW, Suh JY, Zhou W, Lee MH, Odom TW. Nano Lett. 2010;10:3173–3178. doi: 10.1021/nl102078j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhakdi S, Tranum-Jensen J. Microbiol. Mol. Biol. Rev. 1991;55:733–751. doi: 10.1128/mr.55.4.733-751.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gouaux JE, Braha O, Hobaugh MR, Song L, Cheley S, Shustak C, Bayley H. Proc. Nat. Acad. Sci. U.S.A. 1994;91:12828–12831. doi: 10.1073/pnas.91.26.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 47.Bayley H, Cremer PS. Nature. 2001;413:226–230. doi: 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- 48.Gu LQ, Shim JW. Analyst. 2010;135:441–451. doi: 10.1039/b907735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eftekhari F, Escobedo C, Ferreira J, Duan X, Girotto EM, Brolo AG, Gordon R, Sinton D. Anal. Chem. 2009;81:4308–4311. doi: 10.1021/ac900221y. [DOI] [PubMed] [Google Scholar]
- 50.Ferreira J, Santos MJL, Rahman MM, Brolo AG, Gordon R, Sinton D, Girotto EM. J. Am. Chem. Soc. 2009;131:436–437. doi: 10.1021/ja807704v. [DOI] [PubMed] [Google Scholar]
- 51.Jonsson MP, Dahlin AB, Feuz L, Petronis S, Höök F. Anal. Chem. 2010;82:2087–2094. doi: 10.1021/ac902925e. [DOI] [PubMed] [Google Scholar]
- 52.Mansuripur M, Zakharian AR, Lesuffleur A, Oh SH, Jones RJ, Lindquist NC, Im H, Kobyakov A, Moloney JV. Opt. Exp. 2009;17:14001–14014. doi: 10.1364/oe.17.014001. [DOI] [PubMed] [Google Scholar]
- 53.Chansin GAT, Mulero R, Hong J, Kim MJ, deMello AJ, Edel JB. Nano Lett. 2007;7:2901–2906. doi: 10.1021/nl071855d. [DOI] [PubMed] [Google Scholar]
- 54.Hausmann D, Becker J, Wang SL, Gordon RG. Science. 2002;298:402–406. doi: 10.1126/science.1073552. [DOI] [PubMed] [Google Scholar]
- 55.Haes AJ, Van Duyne RP. Anal. Bioanal. Chem . 2004;379:920–930. doi: 10.1007/s00216-004-2708-9. [DOI] [PubMed] [Google Scholar]
- 56.Im H, Lindquist NC, Lesuffleur A, Oh SH. ACS Nano. 2010;4:947–954. doi: 10.1021/nn901842r. [DOI] [PubMed] [Google Scholar]
- 57.Anderson TH, Min Y, Weirich KL, Zeng H, Fygenson D, Israelachvili JN. Langmuir. 2009;25:6997–7005. doi: 10.1021/la900181c. [DOI] [PubMed] [Google Scholar]
- 58.Schonherr H, Johnson JM, Lenz P, Frank CW, Boxer SG. Langmuir. 2004;20:11600–11606. doi: 10.1021/la049302v. [DOI] [PubMed] [Google Scholar]
- 59.Lingler S, Rubinstein I, Knoll W, Offenhausser A. Langmuir. 1997;13:7085–7091. [Google Scholar]
- 60.Soumpasis DM. Biophys. J. 1983;41:95–97. doi: 10.1016/S0006-3495(83)84410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Worsfold O, Voelcker NH, Nishiya T. Langmuir. 2006;22:7078–7083. doi: 10.1021/la060121y. [DOI] [PubMed] [Google Scholar]
- 62.Starr TE, Thompson NL. Langmuir. 2000;16:10301–10308. [Google Scholar]
- 63.Mager MD, Almquist B, Melosh NA. Langmuir. 2008;24:12734–12737. doi: 10.1021/la802726u. [DOI] [PubMed] [Google Scholar]
- 64.Jonsson P, Jonsson MP, Höök F. Nano Lett. 2010;10:1900–1906. doi: 10.1021/nl100779k. [DOI] [PubMed] [Google Scholar]
- 65.Studer A, Han X, Winkler FK, Tiefenauer LX. Colloids Surf. B. 2009;73:325–331. doi: 10.1016/j.colsurfb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 66.Oshannessy DJ, Brighamburke M, Soneson KK, Hensley P, Brooks I. Anal. Biochem. 1993;212:457–468. doi: 10.1006/abio.1993.1355. [DOI] [PubMed] [Google Scholar]
- 67.Ragle BE, Wardenburg JB. Infect. Immun. 2009;77:2712–2718. doi: 10.1128/IAI.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bakás L, Ostolaza H, Vaz WLC, Goñi FM. Biophys. J. 1996;71:1869–1876. doi: 10.1016/S0006-3495(96)79386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee SH, Bantz KC, Lindquist NC, Oh SH, Haynes CL. Langmuir. 2009;25:13685–13693. doi: 10.1021/la9020614. [DOI] [PubMed] [Google Scholar]
- 70.Nagpal P, Lindquist NC, Oh SH, Norris DJ. Science. 2009;325:594–597. doi: 10.1126/science.1174655. [DOI] [PubMed] [Google Scholar]
- 71.Johnson PB, Christy RW. Phys. Rev. B. 1972;6:4370. [Google Scholar]
- 72.Johnson PB, Christy RW. Phys. Rev. B. 1974;9:5056. [Google Scholar]
- 73.Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Biophy. J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.