

Abstract
Human prion diseases can be sporadic, inherited or acquired by infection and show considerable phenotypic heterogeneity. We describe the clinical, histopathological and pathological prion protein (PrPSc) characteristics of a Dutch family with a novel 7-octapeptide repeat insertion (7-OPRI) in PRNP, the gene encoding the prion protein (PrP). Clinical features were available in four, neuropathological features in three and biochemical characteristics in two members of this family. The clinical phenotype was characterized by slowly progressive cognitive decline, personality change, lethargy, depression with anxiety and panic attacks, apraxia and a hypokinetic-rigid syndrome. Neuropathological findings consisted of numerous multi- and unicentric amyloid plaques throughout the cerebrum and cerebellum with varying degrees of spongiform degeneration. Genetic and molecular studies were performed in two male family members. One of them was homozygous for valine and the other heterozygous for methionine and valine at codon 129 of PRNP. Sequence analysis identified a novel 168 bp insertion [R2–R2–R2–R2–R3g–R2–R2] in the octapeptide repeat region of PRNP. Both patients carried the mutation on the allele with valine at codon 129. Western blot analysis showed type 1 PrPSc in both patients and detected a smaller ~8 kDa PrPSc fragment in the cerebellum in one patient. The features of this Dutch kindred define an unusual neuropathological phenotype and a novel PRNP haplotype among the previously documented 7-OPRI mutations, further expanding the spectrum of genotype–phenotype correlations in inherited prion diseases.
Keywords: Creutzfeldt–Jakob disease, Prion protein, Genetic CJD, Base pair insertion, Neurodegeneration, Amyloidosis, Gerstmann–Sträussler–Scheinker disease
Introduction
Prion diseases, also known as transmissible spongiform encephalopathies, are invariably fatal, neurodegenerative disorders that can be sporadic, inherited or acquired by infection. They can affect both humans and a wide variety of animals, such as cattle, sheep and elk [24]. In humans, inherited prion diseases include familial Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker (GSS) disease and fatal familial insomnia (FFI) [6]. The clinical phenotype in classical GSS disease is characterized by a progressive cerebellar syndrome, accompanied by extrapyramidal and pyramidal signs and cognitive decline, which may evolve into severe dementia [8]. Neuropathological features associated with GSS disease vary substantially, but always include multicentric amyloid plaques in the cerebellum with or without spongiform change.
The central event in prion diseases is thought to be the conversion of the normal prion protein (PrPC) into the pathological isoform, commonly referred to as PrPSc [24]. This process involves a structural rearrangement that results in an increase in β-sheet content, aggregation and partial resistance to proteolytic degradation. The physico-chemical properties of PrPSc can be assessed indirectly by determining the size of the protease-resistant core fragment by Western blotting of proteinase-K (PK) digested homogenates of brain tissue. Abnormal prion protein deposits in affected brain regions of patients with CJD include both full length PrPSc and truncated fragments in the 12–21 kDa range, whereas in GSS disease smaller protease-resistant 7–9 kDa unglycosylated fragments are found [8, 21, 22].
About 10 point mutations in the protein encoding region of the prion protein gene (PRNP) are known to be associated with GSS disease. These are inherited in an autosomal dominant manner and include both missense and nonsense mutations, leading to amino-acid substitutions or a premature stop codon, respectively [8]. In addition, several insertional mutations have been described that occur in the N-terminal octapeptide repeat region of PRNP [14]. The wild-type PRNP octapeptide repeat region is located between codons 51 and 91, and comprises a nonapeptide, designated R1, followed by four copies of an octapeptide, designated R2, R3 and R4 (Table 1). The normal sequence of the entire octapeptide region is R1–R2–R2–R3–R4 [9]. Octapeptide repeat insertions (OPRI) comprise varying numbers (1–9) of 24 base pair multiples. If all insertional mutations are considered together, the disease phenotype is highly variable, often blending features of both CJD and GSS disease or even lacking specific histopathological changes. The molecular basis for this phenotypic heterogeneity remains to be fully elucidated, but appears to depend in part on the size of the base pair insertion and the codon 129 methionine/valine (M/V) polymorphism on the allele carrying the insertion [3, 18, 19].
Table 1.
Nucleotide sequences of codons 51–91 in normal, healthy individuals and irregular sequences in patients with insertional mutations in PRNP
| Normal (regular) sequences of codons 51–91 in PRNP | |||||||||
| Codon 51 | |||||||||
| CCT | CAG | GGC | GGT | GGT | GGC | TGG | GGG | CAG | R1 |
| pro | gln | gly | gly | gly | gly | trp | gly | gln | |
| CCT | CAT | GGT | GGT | GGC | TGG | GGG | CAG | R2 | |
| pro | his | gly | gly | gly | trp | gly | gln | ||
| CCT | CAT | GGT | GGT | GGC | TGG | GGG | CAG | R2 | |
| pro | his | gly | gly | gly | trp | gly | gln | ||
| CCC | CAT | GGT | GGT | GGC | TGG | GGA | CAG | R3 | |
| pro | his | gly | gly | gly | trp | gly | gln | ||
| CCT | CAT | GGT | GGT | GGC | TGG | GGT | CAA | R4 | |
| pro | his | gly | gly | gly | trp | gly | gln | ||
| Codon 91 | |||||||||
| Abnormal (irregular) sequence in affected individuals in this report | |||||||||
| CCC | CAT | GGT | GGT | GGC | TGG | GGG | CAG | R3g | |
| pro | his | gly | gly | gly | trp | gly | gln | ||
An important step in clarifying phenotype–genotype correlations is the description of new patients from well-defined kindreds and their comparison with previously described kindreds. In this report, we describe the clinical, neuropathological and biochemical findings in a Dutch family with a novel seven octapeptide repeat insertional mutation (7-OPRI) in PRNP.
Patients and methods
Patients
The pedigree of the kindred examined (Fig. 1) showed a dementing illness in 6 subjects. Clinical details from patients II-2, III-2, III-4 and III-6 were retrieved from available hospital case records. The neuropathological features of subjects III-2, III-4 and III-6 were re-analysed. Additional genetic studies and immunoblotting of affected brain tissue were performed in patients III-4 and III-6.
Fig. 1.

Pedigree of the family described in this manuscript. Circles indicate females; squares correspond to males. Affected family members are shown in shaded symbols, unaffected individuals in open symbols. A diagonal bar in a symbol indicates a deceased family member. The question mark indicates a clinical history suggestive of prion disease, albeit not neuropathologically proven. The numbers refer to the case numbers in the text
Subject II-2
The patient presented in 1962 with forgetfulness, apathy and slurred non-fluent speech at the age of 50 years. She was also emotionally labile and developed anxiety attacks. Her medical history was otherwise unremarkable. There was no known history of iatrogenic prion exposure. On neurological examination, she had a staring gaze and was no longer able to communicate. The extremities were spastic, but tendon and plantar reflexes were normal. Routine blood tests were normal. A pneumo-encephalogram showed severe atrophy and a slightly enlarged ventricular system. An electroencephalogram (EEG) was not recorded. Her condition deteriorated rapidly, complicated by pulmonary infections. She died at the age of 50 years, 8 months after onset of symptoms. She had a family history of neurodegenerative disease. Her mother (I-2) had died at the age of 67 years, shortly after presenting with similar symptoms. Two of the patient’s sisters (II-4 and II-5) had died at the age of 47 and 49 years as a result of an unknown neurological syndrome.
Subject III-2
This patient presented with decreasing driving abilities at the age of 49 years, followed by an inability to complete simple daily tasks, such as dressing. Eleven months later, his family noted that he had become increasingly lethargic. On examination in 1986, 12 months after onset of symptoms, he was unable to write, draw and read with explicit disorders in memory, orientation, planning and judgement. Movements were generally hypokinetic with rigidity in all extremities. No myoclonic jerks were seen. The plantar responses were extensor. Routine blood tests were normal. An EEG showed non-specific changes, but no periodic synchronous wave complexes (PSWC’s). His condition deteriorated thereafter and 12 months later he had become fully dependent on help with daily activities and was disoriented in time and place. He died at the age of 51 years, 36 months after onset of symptoms.
Subject III-4
The patient, brother of subject III-2, presented with progressive gait insecurity, tinnitus and speech disability in 2003 at the age of 59 years. On examination, 1 month after onset of symptoms, he was dysarthric with mild expressive aphasia and saccadic eye movements. He had a mild symmetric action tremor and showed an increased tone of the extremities. The plantar responses were extensor. Three months later his condition had deteriorated and was characterized by a hypokinetic rigid syndrome. Neuropsychological evaluation revealed global aphasia, apraxia, apathy and memory impairment. An EEG showed slowing of background activity with PSWC’s. Magnetic resonance imaging (MRI) showed mild symmetric hyperintense lesions in the basal ganglia, but no atrophy. The last 3 months he became increasingly bedridden and developed myoclonic jerks. He died at the age of 59 years, 7 months after onset of symptoms.
Subject III-6
The patient, brother of III-2 and III-4, presented at the age of 51 years with balance insecurity, forgetfulness, depression and panic attacks. He suffered from tinnitus and sometimes noted numbness in fingers and toes. On examination, 12 months after onset of symptoms, he was mildly dysarthric and hypokinetic and showed increased tone of all extremities. Tendon reflexes were brisk, but plantar responses were extensor. Neuropsychological examination revealed decreased concentration and impaired memory. An EEG showed slowing of background activity, but no PSWC’s. Over the next 3 years depression and panic attacks continued, which improved with the use of anti-depressive medication. On re-examination, five years after onset of symptoms, his condition had suddenly deteriorated with increasing bradyphrenia, hypokinetic rigidity, apraxia and myoclonic jerks. In addition, he suffered from visual hallucinations. Tendon reflexes were normal. MRI showed cerebral atrophy, but no hyperintense lesions in the basal ganglia. The final stages of his disease course resembled the rapid neurodegenerative decline of his family members. He died at the age of 57 years, 65 months after onset of symptoms.
PRNP analysis
DNA was extracted from brain tissue of patients III-4 and III-6. Full sequence analysis of the prion protein gene was performed using established methods [13]. In addition the genotype (methionine/valine) at the polymorphic codon 129 of PRNP was determined by restriction fragment length analysis.
Neuropathology
Histopathological examination was performed on 5-μm thick sections of formalin-fixed and paraffin-embedded brain tissue blocks, after decontamination for 1 h in concentrated formic acid. Sections were stained with haematoxylin–eosin and combined Luxol-periodic acid-Schiff (PAS) stains according to standard procedures. Congo red stains were performed on sections from tissues untreated with formic acid. The monoclonal antibody 3F4 (1:400, overnight at 4°C, Signet Laboratories, Dedham, MA, USA) was used for PrP immunohistochemistry. Pretreatment protocols for PrP staining involved antigen retrieval by autoclaving in citric acid buffer pH 6.0 at 121°C for 10 min followed by incubation with Proteinase K (10 μg/mL, for 5 min at room temperature). Other immunohistochemical stains included antibodies against Aβ (Aβ1–17, 1:400, Dako, Glostrup, Denmark), phosphorylated tau (AT8 1:250, Innogenetics, Gent, Belgium), Glial Fibrillary Acidic Protein (GFAP, 1:800, Dako, Glostrup, Denmark), HLA-DRα (1:80, Dako, Glostrup, Denmark) and ubiquitin (1:500, Dako, Glostrup, Denmark).
Western blotting
Frozen tissues from the temporal, occipital and cerebellar cortices (patient III-4 and III-6) were homogenized and analysed for the presence and type of protease-resistant PrPSc by Western blotting, using an established method with minor modifications [12]. The homogenates were immunoblotted with mAb 3F4 (1:1000, Dako, UK). The isoform of PrPSc was classified as type 1 (unglycosylated PrPres of M r ~ 21 kD) or type 2 (unglycosylated PrPres of M r ~ 19 kD) based on the classification system by Parchi et al. [23]. Samples from both patients were run alongside brain homogenates from sporadic and variant Creutzfeldt–Jakob disease, a case of GSS (P102L-129M) and a case of protease sensitive prionopathy [11] for comparison of the relative mobility of the smaller fragment sizes. PrPres glycoform ratios were analysed using densitometry using a GS-800 scanning densitometer (Bio-Rad, UK).
Results
Genetic analysis
Patient III-4 was found to be heterozygous for methionine and valine at codon 129 of PRNP, whereas patient III-6 was homozygous for valine. Amplification of the octapeptide repeat region of the PRNP by PCR resulted in two fragments, one of which was larger than the expected size, suggesting the presence of an insertional mutation in one of the PRNP alleles. Sequencing of this PCR product demonstrated a 168-bp insertion in the octapeptide repeat region [R2–R2–R2–R2–R3g–R2–R2] (Table 1). Both patients carried the mutation on the codon 129 valine allele.
Neuropathology
Gross examination of the brain of patient II-2 (weight 970 g) showed severe atrophy with frontal predominance. The autopsy report mentioned extensive spongiform changes in the cortex, accompanied by gliosis and neuronal loss, consistent with prion disease. The presence of plaques was not reported. Unfortunately, neither the slides nor the tissue blocks could be retrieved for this study.
Gross examination of the brains of subjects III-2 (weight unknown), III-4 (weight 1,295 g) and III-6 (weight 1,235 g) showed mild atrophy of the cerebellar cortex. The results of microscopic examination (subjects III-4 and III-6) are summarized in Table 2. Microscopic examination revealed variable spongiform degeneration, astrocytic gliosis and neuronal loss in all areas of the cerebral cortex (Fig. 2a, b), basal ganglia (Fig. 2e, f) and thalamus. The cerebellum was also affected to a variable degree, characterized by mild to moderate spongiosis of the molecular layer (Fig. 2g, h). Furthermore, many PrPSc-positive and congophilic multicentric and unicentric plaques were present (Fig. 2c, d, j, k). These showed characteristic green birefringence under polarized light, confirming the presence of amyloid. The plaques were most numerous in the molecular layer of the cerebellar cortex (Fig. 2j, k), but were also found in large numbers in the cerebral cortex, basal ganglia, thalamus and hippocampus. On higher magnification, many plaques seemed to consist of smaller eosinophilic globules, resembling the deposits reported in an individual with a 144 bp insertion [7]. Elongated cerebellar deposits of PrPSc with perpendicular orientation to the meningeal outline were not seen. The brainstem and hippocampus were relatively spared. The spongiform degeneration was more severe in the brain of patient III-6, whereas the brain of patient III-4 showed less spongiosis, but more amyloid plaques. Immunohistochemical staining with anti-PrP antibodies showed positive staining of all plaques, against a background of diffuse synaptic staining in all cortical areas, basal ganglia, thalamus and the molecular layer of the cerebellum. Fine ubiquitin deposits were observed around some of the plaques, accompanied by increased numbers of microglia in the HLA-DRα stain. There were moderate numbers of diffuse plaques and few cored Aβ plaques in the frontal cortex, but less in other cortical areas. Neuritic plaques were not seen. Immunohistochemical staining with AT8 showed diffuse punctuate staining with sparse neuropil threads in all areas examined, including the striatum and molecular layer of the cerebellum. In addition, few neurofibrillary tangles were focally noted in the hippocampus, frontal cortex and temporal cortex of subject III-6, whereas they were absent in subject III-4. Together, these changes were regarded as Alzheimer changes, corresponding to Braak stage 1B. Microscopic examination of the remaining regions of the brain was unremarkable.
Table 2.
Regional distribution of spongiosis, neuronal loss/gliosis and multicentric plaque density
| FC | PC | TC | OC | Hipp | STR | Th | BRST | CE | |
|---|---|---|---|---|---|---|---|---|---|
| III-4 | |||||||||
| Spongiosis | ++ | + | + | ++ | − | ++ | + | − | + |
| Gliosis/neuronal loss | ++ | + | ++ | ++ | + | ++ | + | − | − |
| Multicentric plaques | ++ | + | + | ++ | + | +++ | + | − | +++ |
| III-6 | |||||||||
| Spongiosis | +++ | ++ | ++ | ++ | + | ++ | ++ | −/+ | ++ |
| Gliosis/neuronal loss | ++ | ++ | ++ | ++ | + | ++ | ++ | −/+ | ++ |
| Multicentric plaques | + | + | + | + | + | + | + | − | ++ |
Each lesion was scored semi-quantitatively as not detectable (−), mild (+), moderate (++) and severe (+++). The number of PrP-positive multicentric plaques were counted per HPF and scored as not detectable (−), 1–4 plaques (+), 5–9 plaques (++) and 10–15 plaques (+++)
FC frontal cortex, PC parietal cortex, TC temporal cortex, OC occipital cortex, Hipp hippocampal formation, STR striatum, Th thalamus, BRST brainstem, CE cerebellum
Fig. 2.
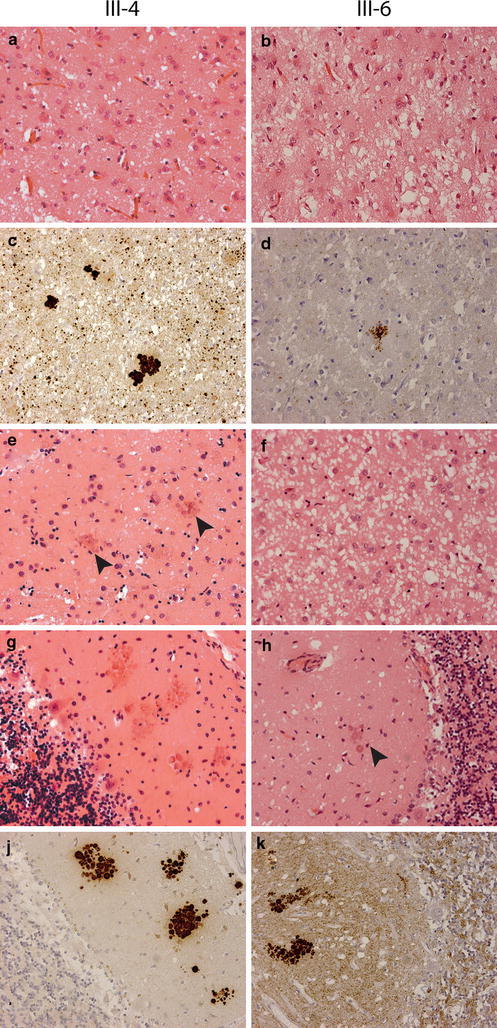
Histopathological findings in two family members (III-4 and III-6) with 7-OPRI. Sections are stained with haematoxylin–eosin (a, b, e, f, g and h) and the 3F4 anti-PrP antibody (c, d, j and k), original magnification ×200. Mild (a) and severe (b) spongiform change in the frontal cortex; c, d multicentric and unicentric plaques in the frontal cortex; e mild spongiform change with multicentric plaques (arrows) in the caudate nucleus; f severe spongiform change in the caudate nucleus; g, j many multicentric plaques in the molecular layer of the cerebellum; h, k moderate spongiform change with multicentric plaques (arrow) in the molecular layer of the cerebellum
Western blotting
Western blot analysis of protease-resistant prion protein (PrPres) in brain homogenates of patients III-4 and III-6 demonstrated the presence of type 1 PrPres. In addition, patient III-4 had a smaller PrPres fragment seen consistently, but only in the cerebellar specimen (Fig. 3a). Densitometric analysis of the 20–30 kDa PrPres bands showed a glycoform ratio similar to that found in sporadic CJD, being dominated by the monoglycosylated (middle) band seen on Western blots, however this pattern was found to be modified towards over-representation of non-glycosylated forms in the cerebellar specimen from patient III-4, in which the small fragment was also evident (Fig. 3b). Comparison of the cerebellar specimen from patient III-4 with samples from other prion diseases in which low-molecular weight PrPres fragments are also found, showed that the PrPres fragment in this 7-OPRI case was of the same general mobility as the major low-molecular weight fragments found in cases of GSS and PSPr (Fig. 3c).
Fig. 3.
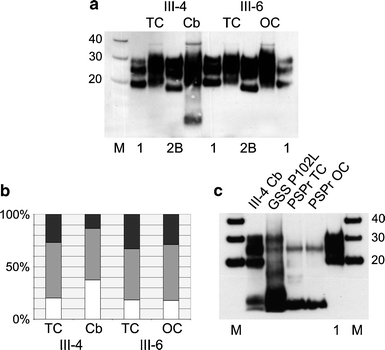
Western blot analysis of two family members (III-4 and III-6) with 7-OPRI. In a protease-resistant prion protein (PrPres) from specimens of temporal cortex (TC) and cerebellum (Cb) from one patient (III-4) and from specimens of temporal cortex (TC) and occipital cortex (OC) from the other patient (III-6) are shown flanked by reference standards of frontal cortex from sporadic CJD of the MM1 subtype (1) and frontal cortex from variant CJD (2B). In b the relative abundance of the three different glycoforms of PrPres is shown, calculated on the basis of densitometric analysis of three independent samples from the available frozen tissues shown in a. The relative abundance of the upper (diglycosylated) band is shown in black, the middle (monoglycosylated) band is shown in grey and the lower (non-glycosylated) band is shown in white. In c the PrPres profile from the cerebellum in patient III-4 (III-4 Cb) is compared with those found in a patient with Gerstmann–Sträussler–Scheinker disease carrying a proline to leucine substitution at position 102 (GSS P102L), and samples from the temporal cortex and occipital cortex of a Dutch case of protease sensitive prionopathy (PSPr TC and PSPr OC). M denotes molecular weight markers with their M r indicated in kDa. The loading of individual samples on Western blots (volume of 10% weight/volume brain homogenate) was adjusted to allow easy comparison of the PrPres profiles obtained. All densitometric readings for glycoform analysis were within the linear range for each band in every sample
Analysis of combined data
Clinical and neuropathological features in this Dutch kindred may be combined with those of other families and patients with 7-OPRI (Table 3). Marked phenotypic heterogeneity is a consistent feature in these reports. Age at onset was known for four patients in our family, ranging from 49 to 59 years (mean 52.2 years, SD = 4.6 years). The disease duration ranged from 0.6 to 5.4 years (mean 2.4 years; SD = 2.3 years). In contrast, the age at onset was known in 9 previously reported patients, carrying similar 7-OPRI, ranging from 18 to 44 years (mean 28.9 years; SD = 7.3 years). The disease duration in these patients ranged from 4 to 16 years (mean 10.3 years; SD = 3.4). Whereas the insertional mutation in our patient was coupled to valine on codon 129 of PRNP, it occurred on a methionine background in all previously reported patients. Therefore, haplotype defined by direct physical linkage of the insertion to valine at position 129 appears to be associated with later age at disease onset (independent samples t test with assumption of equal variances, mean difference 23.4 years, p < 0.001) and shorter disease duration (independent samples t test with assumption of equal variances, mean difference 7.9 years, p = 0.001). It is also noteworthy that of the two siblings (III-4 and III-6), the patient with valine at codon 129 of the wild-type allele (in addition to valine linked to the mutation) had an earlier onset and a substantially longer disease duration. Therefore, PRNP codon 129 appears to exert a modifying effect on the age at onset and disease duration in 7-OPRI, although these findings must be interpreted with caution, being limited to single cases only. A similar modifying effect of codon 129 on age at onset was seen in 5-OPRI and 6-OPRI cases [18, 19], although there was no effect on disease duration.
Table 3.
Clinical, genetic and neuropathological features of all known 14 patients carrying 7-OPRI
| Country | Mutation | Gender | Family history | Age at onset (years) | Duration (years) | Dementia | Ataxia | Myoclonus | Pyramidal | Extrapyramidal | Epileptic seizures | Psychiatric disturbances | Codon 129 | Immunoblot | Neuropathology | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | USA | R2c–R3–R2–R3–R2–R3–R2–R3g | F | Positive | 31 | 11 | Y | Y | Y | Abnormal behaviour | Met | Spongiosis, gliosis, neuronal loss | [1, 9] | ||||
| 2 | USA | F | 23 | 10 | Y | Y | Y | Y | Depression/mood change | Met | Mild neuronal loss and gliosis, no spongiosis | ||||||
| 3 | USA | F | 28 | 13 | Y | Y | Y | Y | Euphoria | Met | n.a. | ||||||
| 4 | Japan | R3–R2–R2–R2–R3g–R2–R2 | n.a. | Positive | n.a. | 7 | Y | n.a. | n.a. | [25] | |||||||
| 5 | Belgium | n.a. | n.a. | Positive | 32 | 7 | Y | Ya | Met | n.a. | [4] | ||||||
| 6 | Belgium | n.a. | 24 | 11 | Y | Y | Y | Y | Psychosis | Met | Spongiosis, gliosis, neuronal loss, elongated cerebellar plaques cerebellar cortex | ||||||
| 7 | Belgium | n.a. | 31 | 11 | Y | Y | Y | Depression/mood change | Met/Met | Spongiosis, gliosis, neuronal loss, elongated cerebellar plaques cerebellar cortex | |||||||
| 8 | Australia | R3–R2–R3–R2–R2–R2–R2 | n.a. | Positive | 29 | 16 | Y | Y | Y | Abnormal behaviour | Met/Val | Type 1 | Spongiosis, gliosis, neuronal loss, non-linear granular deposits cerebellar cortex | [16] | |||
| 9 | China | R2–R2–R2–R3g–R2–R3g–R2a | F | Positive | 44 | 4 | Y | Y | Y | Y | Depression/mood change | Met/Met | Type 1 | Spongiosis, gliosis, neuronal loss | [10, 27] | ||
| 10 | Italy | R3–R2–R2–R3g–R2–R2–R2 | n.a. | Negative/de novo | 18 | 10 | Y | Y | Depression, psychosis, bipolar disorder | Met/Met | n.a. | [2, 17] | |||||
| 11 | Netherlands | n.a. | F | Positive | 50 | 0.7 | Y | Y | Emotional lability, anxiety attacks | n.a. | n.a. | This report | |||||
| 12 | Netherlands | n.a. | M | 49 | 3 | Y | Y | Emotional lability, apathetic | n.a. | Spongiosis +++, gliosis, neuronal loss, multicentric plaques (cerebellum ++, cortex +) | |||||||
| 13 | Netherlands | R2–R2–R2–R2–R3g–R2–R2 | M | 59 | 0.6 | Y | Y | Y | Y | Apathetic | Met/Val | Type 1 | Spongiosis +, gliosis, neuronal loss, multicentric plaques (cerebellum +++, cortex ++) | ||||
| 14 | Netherlands | R2–R2–R2–R2–R3g–R2–R2 | M | 51 | 5.4 | Y | Y | Y | Y | Depression, panick attacks | Val/Val | Type 1 and 8 kDa | Spongiosis +++, gliosis, neuronal loss, multicentric plaques (cerebellum ++, cortex +) |
aDescribed as motor problems
M male, F female, Met methionine, Val valine, Y present, n.a. not available/insufficient information, + mild, ++ moderate, +++ severe
Discussion
In this report, we describe a Dutch family with inherited prion disease and a novel 7-octapeptide-repeat-insertion (7-OPRI). This kindred represents the sixth reported pedigree to harbour a 7-OPRI in PRNP. In our family, the 7-OPRI is carried on the valine allele of codon 129 of PRNP, and the sequence and arrangement of the 168 bp insertion differs from those previously described in the literature. In all 10 patients with 7-OPRI reported in the literature so far, the mutation was located on the methionine allele of codon 129 of PRNP [1, 2, 4, 9, 10, 16, 17, 25, 27]. The median age at onset in these patients was 28.9 years (range 18–44 years) and the median disease duration 10.0 years (range 4–16 years). Histopathological features usually comprise varying degrees of spongiosis, neuronal loss and gliosis with characteristic linear deposits of PrP in the cerebellar cortex in two patients [4]. Interestingly, no amyloid plaques were reported. In contrast, the clinical phenotype in our patients was characterized by a relatively late age at onset (mean 52.3 years; range 49–59 years), progressive cognitive decline, extrapyramidal symptoms, psychiatric disorders and a mean disease duration of 2.4 years (range 0.6–5.4 years). Neuropathological findings included many multicentric plaques, consistent with a GSS-like phenotype, with moderate to severe spongiosis, neuronal loss and gliosis. Although it is tempting to relate the shorter disease duration observed in this family to the presence of valine at codon 129 in the mutated PRNP allele, there is also a strong effect of age on disease duration in inherited prion diseases (i.e. longer disease duration being associated with younger age at onset), which may confound the comparison. Interestingly, the two patients in this family with the longer disease duration showed more severe spongiform change, whereas a shorter disease duration seemed to be associated with more abundant multicentric amyloid plaques and less severe spongiform change. This observation is unexpected, since among the known prion protein amyloidoses, the number of amyloid plaques usually increases with longer disease duration. At the moment, we do not have a suitable explanation for this apparent discrepancy, although the PRNP codon 129 polymorphism in the non-mutated allele appears to have played a role since the presence of valine at codon 129 in both the wild-type and mutant alleles was associated with an earlier age at onset, a longer disease duration, more spongiosis and a lower number of amyloid plaques. Of course, the interpretation of these findings should be performed with caution because of the limited number of cases.
The functional significance of the octapeptide repeat region in PRNP still remains controversial, although it was shown to bind copper and may play a role in neuroprotective function [20]. It may also be related to amyloid formation, as patients with 8 or 9 extra repeats often show a GSS disease phenotype with numerous multicentric plaques [5, 9, 15, 26], whereas patients with OPRI mutations consisting of less than 5 extra repeats present in a way similar to the typical presentation of CJD. Nevertheless, the exact mechanism of amyloid formation in prion diseases still remains to be elucidated. Data obtained from the study of purified amyloid fractions in patients with several forms of GSS disease indicate that the minimal PrPSc segment essential for amyloidogenesis is an N- and C-terminally truncated fragment spanning residues 88–146 [8]. This corresponds to the 7–9 kDa unglycosylated fragments typically seen on Western blotting. Interestingly, one of our patients showed a similar ~8-kDa fragment in the cerebellum. Another interesting finding was the simultaneous presence of 20–30 kDa PrPSc fragments, corresponding to type 1 PrPSc. This PrPSc isoform was originally described in individuals with sCJD, which is characterized by the presence of spongiform degeneration [22]. Therefore, the presence of both type 1 PrPSc isoforms and smaller fragments of PrP may account for the neuropathological phenotype seen in our patients, consisting of spongiform degeneration and the presence of amyloid plaques. Smaller fragments were not described in two other cases with 7-OPRI reported in the literature [16, 27], which is quite in agreement with the fact that they lacked amyloid plaques. It is not exactly clear what caused the failure to detect smaller PrP fragments in our second patient (III-6), but the (difference in) size, quality and/or precise origin of the frozen specimens taken at autopsy for further molecular analysis might have contributed to this failure. In addition, the lower amyloid burden in this patient may have played a role as well.
The 7-OPRI mutation in this pedigree was inherited in an autosomal dominant pattern. However, it is important to note that a negative family history does not exclude the possibility of inherited prion disease, exemplified by a recently reported Italian patient with a de novo 7-OPRI [2, 17]. Although genetic cases of prion disease may be identified well on the basis of a positive family history or unusual clinical and neuropathological features alone, pre-mortem PRNP sequencing should be considered in all patients with a clinical history of atypical dementia.
Acknowledgments
This work was supported in part by the Rijksinstituut voor Volksgezondheid en Milieu.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Brown P, Goldfarb LG, McCombie WR, et al. Atypical Creutzfeldt-Jakob disease in an American family with an insert mutation in the PRNP amyloid precursor gene. Neurology. 1992;42:422–427. doi: 10.1212/wnl.42.2.422. [DOI] [PubMed] [Google Scholar]
- 2.Cannella M, Martino T, Simonelli M, et al. De novo seven extra repeat expanded mutation in the PRNP gene in an Italian patient with early onset dementia. J Neurol Neurosurg Psychiatry. 2007;78:1411–1413. doi: 10.1136/jnnp.2007.121137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capellari S, Vital C, Parchi P, et al. Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology. 1997;49:133–141. doi: 10.1212/wnl.49.1.133. [DOI] [PubMed] [Google Scholar]
- 4.Dermaut B, Cruts M, Backhovens H, et al. Familial Creutzfeldt–Jakob disease in a patient carrying both a presenilin 1 missense substitution and a prion protein gene insertion. J Neurol. 2000;247:364–368. doi: 10.1007/s004150050603. [DOI] [PubMed] [Google Scholar]
- 5.Duchen LW, Poulter M, Harding AE. Dementia associated with a 216 base pair insertion in the prion protein gene. Clinical and neuropathological features. Brain. 1993;116(Pt 3):555–567. doi: 10.1093/brain/116.3.555. [DOI] [PubMed] [Google Scholar]
- 6.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 7.Gelpi E, Kovacs GG, Strobel T, et al. Prion disease with a 144 base pair insertion: unusual cerebellar prion protein immunoreactivity. Acta Neuropathol. 2005;110:513–519. doi: 10.1007/s00401-005-1073-x. [DOI] [PubMed] [Google Scholar]
- 8.Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P. Hereditary prion protein amyloidoses. Clin Lab Med. 2003;23:65–85. doi: 10.1016/S0272-2712(02)00064-1. [DOI] [PubMed] [Google Scholar]
- 9.Goldfarb LG, Brown P, McCombie WR, et al. Transmissible familial Creutzfeldt–Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci USA. 1991;88:10926–10930. doi: 10.1073/pnas.88.23.10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo YJ, Wang XF, Han J, et al. A patient with Creutzfeldt-Jakob disease with an insertion of 7 octa-repeats in the PRNP gene: molecular characteristics and clinical features. Am J Med Sci. 2008;336:519–523. doi: 10.1097/MAJ.0b013e3181643e50. [DOI] [PubMed] [Google Scholar]
- 11.Head MW, Knight R, Zeidler M, Yull H, Barlow A, Ironside JW. A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol. 2009;35:628–632. doi: 10.1111/j.1365-2990.2009.01040.x. [DOI] [PubMed] [Google Scholar]
- 12.Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt–Jakob disease. Histopathology. 2000;37:1–9. doi: 10.1046/j.1365-2559.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- 13.Kovacs GG, Puopolo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118:166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- 14.Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002;249:1567–1582. doi: 10.1007/s00415-002-0896-9. [DOI] [PubMed] [Google Scholar]
- 15.Laplanche JL, Hachimi KH, Durieux I, et al. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain. 1999;122(Pt 12):2375–2386. doi: 10.1093/brain/122.12.2375. [DOI] [PubMed] [Google Scholar]
- 16.Lewis V, Collins S, Hill AF, et al. Novel prion protein insert mutation associated with prolonged neurodegenerative illness. Neurology. 2003;60:1620–1624. doi: 10.1212/01.wnl.0000065887.14609.0e. [DOI] [PubMed] [Google Scholar]
- 17.Mauro C, Giaccone G, Piscosquito G, et al. A novel insertional mutation in the prion protein gene: clinical and bio-molecular findings. J Neurol Neurosurg Psychiatry. 2008;79:1395–1398. doi: 10.1136/jnnp.2007.142976. [DOI] [PubMed] [Google Scholar]
- 18.Mead S, Poulter M, Beck J, et al. Inherited prion disease with six octapeptide repeat insertional mutation–molecular analysis of phenotypic heterogeneity. Brain. 2006;129:2297–2317. doi: 10.1093/brain/awl226. [DOI] [PubMed] [Google Scholar]
- 19.Mead S, Webb TE, Campbell TA, et al. Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology. 2007;69:730–738. doi: 10.1212/01.wnl.0000267642.41594.9d. [DOI] [PubMed] [Google Scholar]
- 20.Mitteregger G, Vosko M, Krebs B, et al. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007;17:174–183. doi: 10.1111/j.1750-3639.2007.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notari S, Strammiello R, Capellari S, et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt–Jakob disease. J Biol Chem. 2008;283:30557–30565. doi: 10.1074/jbc.M801877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parchi P, Chen SG, Brown P, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann–Straussler–Scheinker disease. Proc Natl Acad Sci USA. 1998;95:8322–8327. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 24.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tateishi J. Recent advances in the research of Creutzfeldt-Jakob disease (CJD) and Gerstmann–Sträussler syndrome (GSS) Rinsho Shinkeigaku. 1991;31:1306–1308. [PubMed] [Google Scholar]
- 26.van Gool WA, Hensels GW, Hoogerwaard EM, Wiezer JH, Wesseling P, Bolhuis PA. Hypokinesia and presenile dementia in a Dutch family with a novel insertion in the prion protein gene. Brain. 1995;118(Pt 6):1565–1571. doi: 10.1093/brain/118.6.1565. [DOI] [PubMed] [Google Scholar]
- 27.Wang XF, Guo YJ, Zhang BY, et al. Creutzfeldt-Jakob disease in a Chinese patient with a novel seven extra-repeat insertion in PRNP. J Neurol Neurosurg Psychiatry. 2007;78:201–203. doi: 10.1136/jnnp.2006.09433. [DOI] [PMC free article] [PubMed] [Google Scholar]