

Abstract
An X-linked myopathy was recently associated with mutations in the four-and-a-half-LIM domains 1 (FHL1) gene. We identified a family with late onset, slowly progressive weakness of scapuloperoneal muscles in three brothers and their mother. A novel missense mutation in the LIM2 domain of FHL1 (W122C) co-segregated with disease in the family. The phenotype was less severe than that in other reported families. Muscle biopsy revealed myopathic changes with FHL1 inclusions that were ubiquitin- and desmin-positive. This mutation provides additional evidence for X-linked myopathy caused by a narrow spectrum of mutations in FHL1, mostly in the LIM2 domain. Molecular dynamics (MD) simulations of the newly identified mutation and five previously published missense mutations in the LIM2 domain revealed no major distortions of the protein structure or disruption of zinc binding. There were, however, increases in the nonpolar, solvent-accessible surface area in one or both of two clusters of residues, suggesting that the mutant proteins have a variably increased propensity to aggregate. Review of the literature shows a wide range of phenotypes associated with mutations in FHL1. However, recognizing the typical scapuloperoneal phenotype and X-linked inheritance pattern will help clinicians arrive at the correct diagnosis.
Keywords: X-linked myopathy, scapuloperoneal, FHL1, neurogenetics, muscular dystrophy, genetic diagnosis
Introduction
Inherited myopathies are genetically heterogeneous and can be inherited in autosomal dominant, autosomal recessive or X-linked patterns. Recently, mutations in the four-and-a-half-LIM domains 1 gene (FHL1, OMIM #300163) on Xq27.2 were reported to cause an X-linked myopathy [. FHL1, a 32 kDa protein, is highly expressed in skeletal muscle, where it localizes to sarcomere and sarcolemma, and is expressed at intermediate levels in cardiac muscle. It plays important but poorly understood roles in muscle growth, differentiation, and sarcomere assembly, and acts as a modulator of transcription factors. Structurally, FHL1 comprises four complete LIM domains, which are cysteine-rich double zinc finger motifs, and an N-terminal half LIM domain, which has one zinc finger motif. The reported mutations predominantly cluster in the second and fourth LIM domains, some affecting the same or neighboring residues. However, the clinical features of the associated myopathy vary considerably with respect to age of onset, distribution and severity of muscle weakness, rate of progression, and expression of disease in females (Table). There is also substantial variation in pathologic features. FHL1 immunostaining in muscle was decreased or absent in some cases but was diffusely increased in others. Furthermore, FHL1 accumulations were detected in many but not all cases. By immunoblot, FHL1 ranged from absent to reduced to increased. No clear relationship between the genotype and the clinical and pathologic phenotypes has been recognized. Identification of additional X-linked myopathy families and mutations may help to clarify the genotype-phenotype relationship. We describe a family that harbors a novel FHL1 mutation. In an effort to investigate the effects of the mutations on the structure of the protein we also present results of molecular dynamics simulations of this mutation and a subset of the previously described missense mutations in the LIM2 domain.
Table.
FHL1 mutations and clinical characteristics in X-linked myopathy
| Residuea | LIM | Zincb | Inheritc/sex | Onset age | Wheelchair-bound aged | Phenotype/Affected Musclese | CK | Femalesf | Pathologyg | W. Bloth | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C101F | 2nd | Zn1 | SF | 2 | died at 3 | proximal muscles respiratory failure | ↑ | asymp. | RB, RV, AV, pyknotic Nu. IC. of FHL1, α5-integrin, MyHC | ↓ | [5] |
| 102–104 delKFC | 2nd | Zn1 | SF | 40s | NA | distal muscles | NA | asymp. | RB, RV, IC. of FHL1, α5-integrin, MyHC | ↑ | [5] |
| C104R | 2nd | Zn1 | XLD | 10 | no, current 11 | scapuloperoneal Gowers’, rigid spine | ↑ | o.29 wcb.34 | RB IC. of FHL1, α5-integrin, MyHC | ↓ | [5] |
| 111–229 delG-T | 2nd–4th | Zn1, 2 | XLR | 14 | no, current 35 | scapuloperoneal joints contractures RBBB, respiratory failure | ↑ ↑ | asymp. | non specific myopathic changes | NA | [7] |
| W122C* | 2nd | NBS | XLD | 30s | 40s–50s | scapuloperoneal hypertrophy respiratory insufficiency | ↑ | o.73, mild | RV, necrotic, atrophic fibers, IC. of FHL1, desmin, Ub | NA | This report |
| W122S* | 2nd | NBS | XLD | 20s | 30s | scapuloperoneal hypertrophy respiratory failure | ↑ | o.30s wcb.50s | NA | ↓ | [2] |
| H123Y* | 2nd | Zn1 | SF | <2 | 3 | proximal muscle rigid spine respiratory failure | ↑ | asymp. | RB, IC. of FHL1, desmin, Ub | nl | [1] |
| H123Y | 2nd | Zn1 | SF | <1.5 | no, current 2.5 | proximal muscle, neck Gowers’ | mild ↑ | asymp. | RB, RV, IC. of FHL1, pericentrin, tubulin | NA | [6] |
| H123L | 2nd | Zn1 | SF | 4 | no, current 7 | generalized hypotrophy scapular Gowers’, scoliosis | nl. | asymp. | RB, RV, IC. of FHL1, pericentrin, tubulin | NA | [6] |
| H123Q* | 2nd | Zn1 | SF | 4 | 10 | proximal and axial scapular, scoliosis | ↑ | asymp. | RB, RV, IC. of FHL1, pericentrin, tubulin | NA | [6] |
| H123Q | 2nd | Zn1 | SF | 3 | 8 | neck, axial Gowers’, scoliosis respiratory insufficiency | ↑ ↑ | asymp. | RB, RV, IC. of FHL1, pericentrin, tubulin | NA | [6] |
| H123Q | 2nd | Zn1 | SM | 1 | 2 | proximal, scoliosis cardiomyopathy, respiratory insufficiency | ↑ ↑ | asymp. | RB, RV, IC. of FHL1, pericentrin, tubulin | NA | [6] |
| K124Rfs X6 | 2nd | Zn1, 2 | XLD | 10 | no, current 26 | humeral, peroneal, facial JC, RBBB, AFb | ↑ | NA | dystrophic | ↓ | [7] |
| 127–128in sFT | 2nd | NBS | XLR | 30s–40s | 50s | postural, hypertrophy respiratory failure | ↑ ↑ | asymp. | Atrophy and necrotic fibers. Variation in fiber size | NA | [3] |
| C132F* | 2nd | Zn1 | SF | 2 | 4.5, died at 6.5 | generalized hypotrophy respiratory failure | ↑ | asymp. | RB, IC. of FHL1, desmin, Ub | ↑ | [1] |
| C150Y* | 2nd | Zn2 | SF | 2.5 | died at 4.5 | proximal muscles respiratory failure | mild ↑ | asymp. | RB, RV, AV, pyknotic Nu. | NA | [5] |
| 151–153 delVTC | 2nd | Zn2 | SM? | 13 | no, current 16 | scapuloperoneal, rigid spine, Gowers’ | mild ↑ | asymp. | RB, RV, increased FHL1staining | ↓ | [4] |
| C153R | 2nd | Zn2 | XLD | 5 | 8 | proximal, cardiomyopathy respiratory insufficiency | ↑ ↑ | o.30s wcb.40s | RB | NA | [1] |
| C153Y | 2nd | Zn2 | XLD | 10 | 16 | proximal muscles rigid spine | ↑ | o.30s | RB | NA | [1] |
| K157Vfs X36 | 2nd | Zn1, 2 | XLD | 4 | no, current 23 | scapuloperoneal, facial, JC, heart hypertrophy | ↑ | cardiac disease | non specific myopathic changes | ↓ | [7] |
| A168Gfs X27 | 2nd | Zn1, 2 | XLD | 20s | NA | rigid spine, sinus valsalvae aneurysm | ↑ ↑ | o. 50s | protein body, loss of FHL1 staining | absent | [8] |
| C209R | 3rd | Zn2 | XLD | 11 | no, current 41 | scapular, pelvic, JC arrhythmias | ↑ ↑ | o.10to69 | dystrophic changes | ↓ | [7] |
| C224W | 4th | Zn1 | XLR | 30s | died at 45–72 | postural, scapulo-axio-peroneal, hypertrophy cardiomyopathy respiratory failure | ↑ or ↑ ↑ | asymp. | core-like in NADH, AV FHL1 absent or reduced staining | absent | [3] |
| C224W | 4th | Zn1 | XLR/XLD | 6–45 | NA | rigid spine, hypertrophy scapuloperoneal respiratory failure | ↑ | asymp. | absent FHL1 staining | absent | [8] |
| H246Y | 4th | Zn1 | SM? | 17 | NA | rigid spine, scapuloperoneal | mild ↑ | asymp. | hypertrophy, FHL1 no staining | NA | [8] |
| C273Lfs X11 | 4th | Zn2 | XLD | 6 | no, current 30 | scapular, pelvic hypertrophy, JC | ↑ | cardiac disease | non specific myopathic changes | NA | [7] |
| C276Y | 4th | Zn2 | XLR | 48 | no, current 54 | axial, pelvic, hypertrophy JC, AFb, AFI | ↑ | asymp | dystrophic/myopathic reduced FHL1 staining | ↓ | [7] |
| V280M | 4th | NBS | XLR | 8 | NA | rigid spine, scapuloperoneal | ↑ | asymp | absent FHL1 staining | absent | [8] |
| X281E | 4th | NBS | XLR | 10 | no, current 18 | Scapuloperoneal, JC septal hypertrophy | ↑ | asymp | dystrophic reduced FHL1 staining | ↓ | [7] |
Notes:
* = mutations studied in molecular dynamics simulations; ins = in-frame nucleotide insertion; del = in-frame nucleotide deletion; fsX = frameshifting mutation, with the following number indicating the number of new amino acids downstream of the mutation preceding introduction of a premature stop codon.
Binding to the first or second Zn of the domain; NBS = not a binding site.
Inheritance pattern: SF = sporadic female; SM = sporadic male; XLD = X-linked dominant; XLR = X-linked recessive.
Age (years) at which patient became wheel-chair bound or died, or current age if still ambulatory; NA=not available
JC = joint contractures; RBBB = right bundle branch block; AFb = atrial fibrillation; AFI = atrial flutter
Myopathy symptoms in female members: o = age of onset; wcb = age at which wheelchair bound; asymp = asymptomatic.
RB = reducing bodies; RV = rimmed vacuoles; AV = autophagy vacuoles; Nu = nucleus; IC = inclusion; Ub = ubiquitin;
FHL1 expression level by western blot; nl = normal.
Patients and Methods
Subjects
We identified family MD15 of Northern European ethnic background with eight members affected by myopathy (Figure 1). Under a protocol approved by the University of Washington’s Institutional Review Board, clinical evaluations were performed and samples were obtained from eight individuals from two generations. Genomic DNA was isolated from leukocytes by standard methods.
Figure 1.
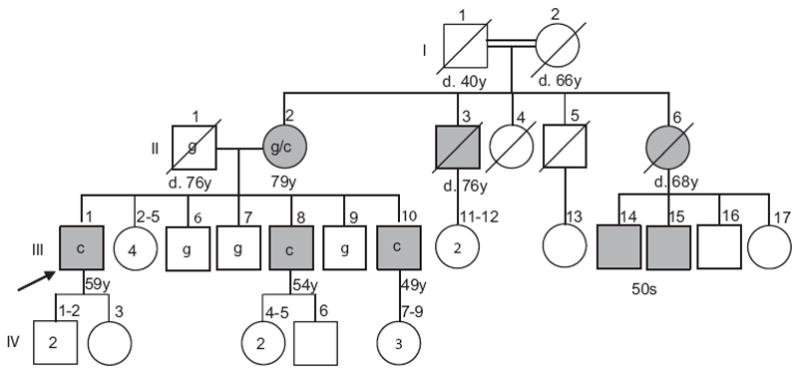
Pedigree of the family segregating an X-linked scapuloperoneal myopathy. Affected individuals are shaded. Only the branch on the left side of the figure was available for evaluation; remaining affected persons are known only through family report. The FHL1 allele at nt 366 is shown within the pedigree symbol for all eight tested individuals (c = mutant nt). Current age or age of death is indicated. Arrow indicates index case.
Linkage analysis targeted to the FHL1 genomic region
Genotyping of markers flanking FHL1 was performed using fluorescently-labeled polymorphic markers DXS1001, DXS1047, DXS1062, and DXS1227 (from ABI Linkage Mapping Set V2.5, ABI, Foster City, CA) in the eight available members in MD15. PCR amplifications were carried out using FastStart DNA Taq polymerase (Roche Applied Science, Indianapolis, IN) under the conditions of initial activation at 95°C for 4min, 30 cycles of denaturation at 94°C for 45sec, annealing at 60°C for 45sec, and extension at 72°C for 1min. The products were analyzed on an ABI Prism 3130xl Genetic Analyzer using GeneMapper version 4.0 software.
Mutation detection
Fragments encompassing each of the six coding exons (exon 2 to exon 7) of FHL1 and their corresponding splice junctions were amplified using the following forward and reverse primer pairs given in 5′ to 3′ direction:
exon 2 ATGAGGGAGCCAGATCCAC and AGACATTGCCCCCGAGAG (486 bp);
exon 3 GTCCTCAGACCCCATGGAC and CTCAAGGTGGCTGCAGTG (415 bp);
exon 4 CCCAGGTGTAAGGGACTGTG and ACGTTAGCCCCACCATGA (387 bp);
exon 5 CATCCCCTCACCTCTGGA and TACGTCACAGGGCTGTCGT (382 bp);
exon 6 CCCATGGCTGTTGTGGAG and CAATAGCAGGGGGAGAAGG (441 bp);
exon 7-1 GCTTGTCGGTCTGTGAGTGG and ACTTTGCTGCAGGGTTGC (484 bp);
exon 7-2 AGGGGAAGAGTGGTCCTTCC and AGGGCAGAGTTCTGATGAGG (426 bp).
Amplifications were performed with FastStart Taq polymerase as described, with optimized annealing temperatures of 62°C for exon 5 and 58°C for the remaining exons. Direct DNA sequencing was performed as previously described. Briefly, sequencing reactions were carried out using BigDye V3.1 Cycle Sequencing Kit (Applied Biosystems) with the same forward primers as in the PCR amplification and sequencing products were electrophoresed on an ABI PRISM 3130xl Genetic Analyzer. To confirm the mutation, exon 5 was also sequenced in reverse.
TaqMan SNP genotyping assays were performed to screen for the 366g→c mutation in the available family members and 228 unrelated unaffected individuals (118 males and 110 females for a total of 338 control × chromosomes). Exon 5 fragments were amplified in TaqMan Genotyping Master Mix (Applied Biosystems), using the protocol provided in the manual, in a total volume of 10 ml. The sequences of primers are: forward - AGGAGATCAAAACGTGGAGTACAAG; and reverse - CTTGCAGTTACTAACAGGTGAAGCA. The sequences of the fluorescent dye-labeled TaqMan hybridization probes, in the reverse direction complementary strands, are: wild-type allele G - CTTTGTGCCAGACGGT; and mutant allele C - TTTGTGGCAGACGGT. PCR amplification was carried out with an ABI 7500 Real-Time PCR system under the condition of an initial denaturation step at 95°C for 10 min, 40 cycles of 95°C for 15 s, 60°C 1 min, and a final extension for 1min.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalin-fixed, paraffin-embedded sections of an anterior tibial muscle. Sections were deparaffinized and hydrated in gradated xylene and alcohol. All IHC was performed following antigen retrieval (15 min at 100°C in phosphate buffer at pH 6.0) and quenching of endogenous peroxidase (3% H2O2 for 15 min). Primary antibodies FHL-1 (AbCam Inc, Cambridge, UK at 1:1,000 dilution), Ubiquitin (Millipore, Billerica, MA at 1:50,000 dilution) and Desmin (D-EU-10, Sigma, St. Louis, MO at 1:2,500 dilution) were incubated for 1 hr. Biotinylated anti-mouse secondary antibodies were subsequently incubated for 40 min and ABC reagent (Vector Laboratories, Burlingame, CA) for 1 hr. Immunoreactivity was developed using Diaminobenzidine/H2O2. Hematoxylin and eosin (H&E) and modified Gomori trichrome staining were performed by routine methods on frozen tissues.
Molecular dynamics studies of the FHL1 LIM2 domain
Models for molecular dynamics (MD) simulations were constructed from the consensus NMR structure of the second LIM domain of human skeletal muscle FHL1 (PDB ID: 1×63). The protein was trimmed to remove unstructured regions at either end, leaving residues 98 through 158 and the two zinc atoms. Mutations were introduced individually by a procedure that optimized the initial conformations of the new side-chains while holding the rest of the structure constant. The protein was embedded in water to fill a sphere with a radius of 23.5 Å, and the energy of the system was minimized locally by an all-atom MD trajectory of 4 ps at 30 K, using a slightly modified version of the ENZYMIX force field. The temperature then was raised to 310 K, and MD trajectories were propagated for a 15-ns equilibration period, during which relatively stiff force constants for stretching and bending of the Zn-ligand bonds were maintained. At the end of this period, the Zn-ligand force constants were decreased to values just sufficient to keep the Zn-S and Zn-N distances in the wild-type protein close to those in the NMR structure and crystal structures of other proteins with similar zinc centers (see Supplementary Information for additional details on this and other aspects of the simulations). MD trajectories with the relaxed force constants then were propagated for 12.5 ns at 310 K. All the MD results presented below pertain to this last set of trajectories. Structures were saved every 20 ps for analysis.
Results
Pedigree and clinical features
The pedigree of family MD15 is shown in Figure 1. The parents in generation I were first cousins. Our study focused on II-2 and her children. A more distant branch of the family was not available for evaluation. Disease in this family is characterized by a progressive scapuloperoneal myopathy syndrome. The age of onset ranged from 32 to 36 years with progression to wheelchair use in the 40–50s. Life span did not appear to be appreciably decreased.
The proband (III-1 in Fig. 1) is a 59 year-old man who developed weakness of his legs at approximately age 36. Previously he had been athletic and had an occupation requiring frequent long distance walking. In retrospect he believes his physical strength became difficult to maintain at age 18–20, but he was on the wrestling team in college and played softball without difficulty to age 36. At age 36 he noted falling several times while walking and weakness of his legs while walking uphill or climbing stairs. Examination at age 37 showed winging of the right scapula and mild weakness of the extensors of his toes and feet. Moderate calf hypertrophy was noted. A serum creatine kinase (CK) was mildly elevated at 210 units (upper limit of normal 186 units in that laboratory at that time) and electromyography (EMG) showed diffuse denervation potentials in both proximal and distal muscles of his lower limbs. A muscle biopsy at age 39 was reported to show myopathic changes with rimmed vacuoles. His weakness progressed slowly over the next 20 years and he began using a wheelchair at age 48 and required a motorized wheelchair by age 50. Repeat EMG was reported as consistent with a myopathic process. Repeat serum CKs ranged from 750 to 1020 units (normal <200). Examination at age 52 showed bilateral winging of the scapulae, weakness and atrophy of the triceps and biceps muscles, marked weakness of both proximal and distal muscles in both lower limbs. Cranial nerves were normal. Facial strength was normal. His strongest muscles were in his forearms and hands. Tendon reflexes were hypoactive. Sensation was intact. His initial diagnosis of possible spinal muscular atrophy was later changed to a scapuloperoneal syndrome. At age 59 he had normal facial muscle strength, some residual motor function of hand/finger flexors, knee flexors and ankle flexors, but was otherwise profoundly weak in all other muscle groups. Respiratory function was compromised with an FVC 27% of predicted, use of mouthpiece ventilator during the day and BIPAP at night. Genetic tests for the common mitochondrial DNA mutation (m. 8344A>G) for MERRF, dystrophin deletions and facioscapulohumeral (FSH) muscular dystrophy were normal.
The proband has two similarly affected younger brothers. His 54-year old brother (III-8 in Fig. 1) was very physically active and played football in college. At age 32 he was asymptomatic but had an elevated serum CK (400 units) obtained when his older brother had become symptomatic. At age 36 this man noted a decreased ability to play basketball. He first noted weakness in his feet and ankles, followed later by weakness of his shoulder muscles. In his early 40s he began using bilateral ankle foot orthotics (AFO). Examination at age 48 showed a waddling lordotic gait with bilateral foot drop. He had evidence of both proximal and distal weakness in his lower limbs that was more prominent distally. Facial strength was normal. He had bilateral winging of the scapulae and weakness of shoulder elevation, biceps and pectoralis muscles. Forearm and hand muscles were strong. Tendon reflexes were depressed. No calf hypertrophy was noted. Sensation was intact and plantar reflexes were down. His disease continued to progress and he required a wheelchair at age 52.
The youngest brother is now 49 years old (III-10 in Fig. 1). He had been athletic and played football in high school. At approximately age 32 he noticed leg weakness while running and playing softball. At age 40 an examination revealed mild weakness of shoulder muscles and prominent weakness of foot extensors. Facial strength was normal. He had bilateral calf hypertrophy. Tendon reflexes were absent. An EMG was compatible with myopathy and a serum CK was 766 units. At age 43 he was wearing bilateral AFOs and was noted to have bilateral winging of the scapulae, bilateral foot drop, mild proximal weakness of his legs with normal sensation and down-going plantar reflexes. By age 47 he was using a motorized scooter.
The mother (II-2 in Fig. 1) is presently 79 years old and was examined at age 73. Her only complaint at that time was mild chronic diffuse pain and limited range of motion of her right shoulder. Her only findings on physical examination were mild winging of the right scapula, bilateral large calves and depressed ankle tendon reflexes. An EMG demonstrated low amplitude, short duration motor unit action potentials consistent with a myopathy. Presently she uses a walker for ambulation.
The family by report has two similarly affected male maternal first cousins (III-14 and III-15 in Fig. 1) whom we have not examined. Their mother (II-6) used a walker in her 60s, had weakness of neck extensors and died of a stroke at age 68. A maternal uncle (II-3) was also reported to be affected. No affected members of this family have had complaints or clinical signs suggestive of cardiac disease, in contrast to the families described recently by Gueneau et al. [7].
Genotype Analyses
Although autosomal dominant inheritance could not be excluded, the pedigree was consistent with X-linked inheritance of disease as there were no male-to-male transmissions. Recently, mutations in the FHL1 gene have been associated with a form of X-linked myopathy (Table). To investigate if FHL1 might be responsible for disease in MD15, we performed a targeted linkage study using all eight available subjects in the family and four markers spanning the critical region of FHL1 on Xq27.2. Genotyping and manually constructed haplotypes of the region bordered by markers DXS1001 (75.79cM) and DXS1227 (88.33cM) revealed that the three affected sons shared a disease-associated haplotype inherited from their mother, while the three unaffected sons inherited the other X-chromosome haplotype (data not shown). This result is consistent with linkage of disease in MD15 to the FHL1 gene locus.
Identification of an FHL1 mutation
After confirming linkage of myopathy in MD15 to the FHL1 locus, we screened subjects in the family for FHL1 mutations. The six protein-coding exons in FHL1 and their splice junctions were PCR-amplified and sequenced in the proband (III-1 in Fig. 1) and his mother (II-2 in Fig. 1) in MD15. A hemizygous G to C transversion was identified in nucleotide 366 (366g→c) (Fig. 2) in the proband and in heterozygous state in the mother. This sequence change predicts substitution of cysteine for tryptophan in residue 122 (W122C). Tryptophan 122 is evolutionarily conserved in all mammals and invertebrates. We performed SNP genotyping with probes for wild-type G and mutant C alleles to screen the other family members. Both affected siblings of the proband (III-8 and III-10 in Fig. 1) carried the hemizygous mutation of G to C nucleotide change and all three at-risk unaffected members had the wild-type sequence. This nucleotide change was not detected in 338 control X-chromosomes. Given the variability in phenotype in FHL1-related myopathy families reported to date, we then screened the FHL1 coding region in two other families with myopathy that, consistent with X-linked inheritance, did not have instances of male-to-male transmission; no pathologic nucleotide changes were identified in those families.
Figure 2.
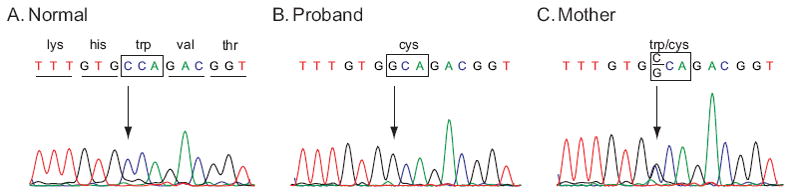
Sequence chromatograms for a portion of exon 5 of FHL1 showing a mutation in the family with X-linked scapuloperoneal myopathy compared to a control with the wild-type nucleotide c at nt 366 (A). A hemizygous g to c transversion in an affected subject (B) and heterozygous g to c change in his mother (C) that predicts substitution of tryptophan for cysteine in residue 122 (W122C). W122C co-segregated with disease in the family and was not present in 338 normal control X-chromosomes.
FHL1 accumulation in muscle
H&E staining of an anterior tibial muscle biopsy from the proband at age 39 revealed myopathic changes with rimmed vacuoles (Fig. 3A and F). Muscle fiber size was markedly variable with numerous small rounded atrophic fibers and enlarged hypertrophic fibers and fiber splitting. Fascicles had increased endomysial fibrosis. Central nuclei were increased in number and some cells contained multiple central nuclei. Scattered necrotic and atrophic fibers were observed. Scattered intrasarcoplasmic eosinophilic inclusions were present in some fibers. Significant inflammation or neurogenic changes were not seen. Modified Gomori trichrome staining demonstrated similar findings (Fig. 3E). To study mutant FHL1 expression and localization, IHC analysis of FHL1 was performed in paraffin-embedded muscle from the proband and an age-matched normal control. In contrast to the control muscle, which was negative for FHL1 immunostaining, in the proband, subsarcolemmal and intracytoplasmic immunoreactivity to FHL1 was observed in some atrophic fibers (Fig. 3B). The inclusion bodies were also positive for desmin (Fig. 3C) and ubiquitin (Fig. 3D). The quality of morphology with IHC staining is compromised by the PFA fixation and paraffin embedding, whereas structural abnormalities are clearly seen with H&E (Fig. 3F) and modified Goromi trichrome staining (Fig. 3E) previously performed on frozen tissues. Unfortunately, frozen tissue is no longer available from the patient for IHC study or menadione_ nitroblue_ tetrazolium (menadione-NBT) staining.
Figure 3.
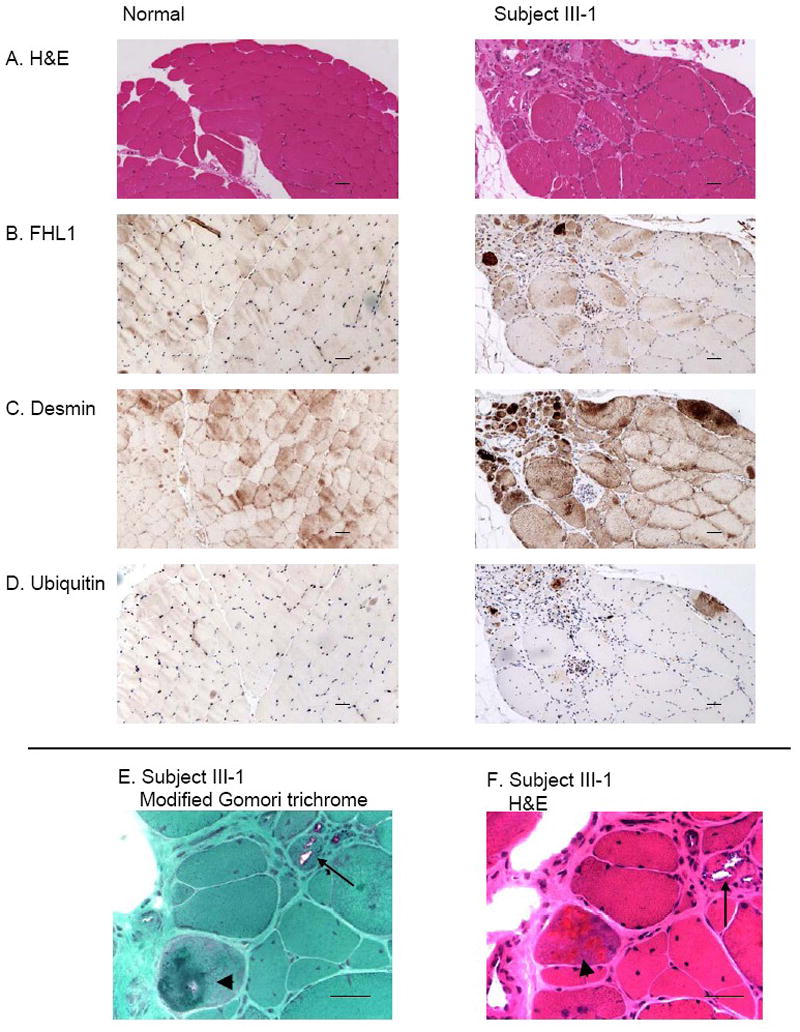
Muscle biopsy from a subject III-1 in the family with FHL1 mutation revealing myopathy and FHL1 inclusions that are desmin and ubiquitin-positive. In muscle from the proband, H&E staining (A and F) reveals that muscle fiber size is markedly variable with numerous small rounded atrophic fibers and enlarged hypertrophic fibers. Central nuclei are increased in number and some fibers contain multiple central nuclei. Scattered intrasarcoplasmic eosinophilic inclusions are present in some fibers. In muscle from a normal control, muscle fiber size is generally equal and nuclei are peripherally placed and central nuclei are uncommon. There is no increase in fibrous connective tissue. Immunohistochemistry for FHL1 (B), desmin (C) and ubiquitin (D) shows abnormal dense staining present in some atrophic fibers. In a normal control, normal desmin-positive striations are present; no ubiquitin or FHL1 staining is observed. Rimmed vacuoles (arrow) and eosinophilic inclusions (arrowhead) are revealed more clearly at higher magnification in E (modified Gomori trichrome stain) and F (H&E). Scale bars: 100 μm.
Predicted structural effects of FHL1 LIM2 missense mutations
Molecular dynamics (MD) simulations of the second LIM domain were carried out with wild-type (wt) FHL1, the W122C mutant described here, and five of the mutants that have been described previously: W122S, H123Q, H123Y, C132F and C153Y. Histidine 123 is one of the four ligands of the first Zn atom in the domain, whereas cysteines 132 and 153 are ligands of the second Zn. Patients with these mutations present phenotypes ranging from milder disease with onset ages in the 40s (in W122C) to very severe myopathy beginning by age 2 (in H123Q, H123Y and C132F). We undertook the MD studies to explore whether predictable structural differences resulting from the mutations might account for the variable severity of the clinical phenotypes. Figure 4 shows the locations of the mutations in representations of the mean structures during 12.5-ns MD simulations following a 15-ns equilibration period. Plots of the deviations of the Cα carbons from their original positions, the fluctuations of the coordinates about the means, and other quantities derived from the simulations are given in Supplementary Data (Figures S2–S8). All the models remained well folded, with both zinc atoms bound to the ligands that had not been mutated. Water stayed outside the first shell of ligands in the intact zinc sites, and moved in to replace the missing ligand in the mutated sites (Figure S4). The distance between the two Zn atoms changed by less than ±0.8 Å (Fig. S5). The root-mean-square fluctuations of the Cα coordinates about their means also were similar in the wt and all the mutant proteins (Fig. S3). The mean distance between the Cα carbons of the residues at the ends of the two β-hairpins increased by 2.1 Å in W122S, but decreased by 1.2 Å in W122C (Fig. S5). Other minor structural differences between the wild-type and mutant proteins are evident in Fig. 4, but these are comparable to the structural fluctuations seen in many proteins and none of them correlated well with the variations in clinical phenotype among the six mutations.
Figure 4.

Structures of the second LIM domain from MD simulations of wild-type FHL1 and the W122C, W122S, H123Y, H123Q, C132F and C153Y mutants. Each structure shown is the mean of 625 structures saved at 20-ps intervals during the trajectory. A tube representation of the wild-type protein is shown in green in each panel, with the indicated mutant structure superimposed in blue. The mutant sidechain and zinc atoms are shown in space-filling (van der Waals) representations colored by atom type. The zinc atoms and the N- and C-termini of the model are labeled in the W122C structure. Over all, the mutant proteins remain folded in essentially the native conformation, with their Zn sites almost fully intact. A few subtle changes are indicated by arrows. The figures were made with VMD 1.8.7 [24].
Probably the most significant effects seen in the MD simulations were changes in the amount or distribution of non-polar surface exposed to the solvent. Both the total solvent-accessible surface area (SASA) and the non-polar SASA increased in most of the mutants relative to the wt protein (Fig. S6). These changes occurred predominantly in two clusters of residues, one near the N-terminal end of the models (C101, F105, A107–A110) and the other at the base of the second β-hairpin (S140–P143) (Figs. 5, S7 and S8). The non-polar SASA in the first cluster increased in the W122S, S122C and H123Y mutants; that in the second cluster increased in W122S, H123Q, C132F and C153Y (Fig. S8).
Figure 5.
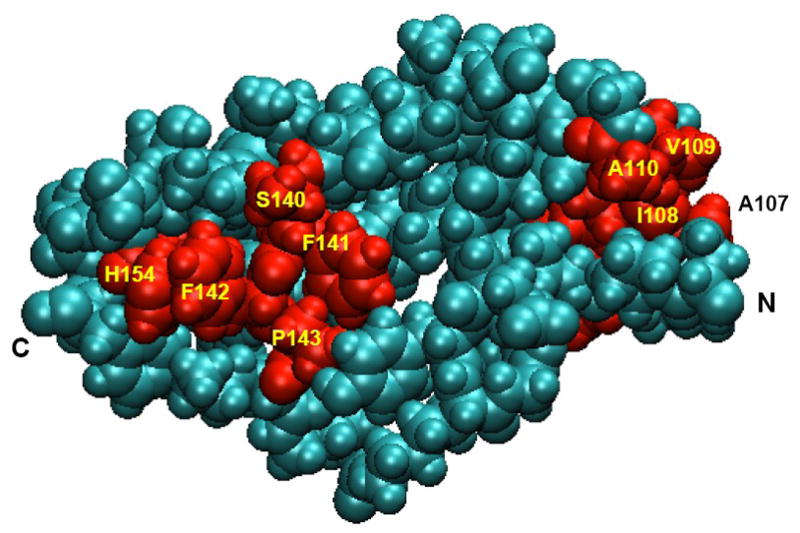
A space-filling representation of the 2nd LIM domain of wt FHL1, with the residues color-coded to indicate their mean values of EXNS, a measure of excess, solvent-accessible nonpolar surface for the protein atoms in and surrounding the residue (see Supplementary Data, eq. 1). EXNS was calculated with a cutoff radius of 8 Å and was averaged over the 625 structures saved during the MD trajectory. In this image, residues that had mean values of EXNS > 0.1 are colored red and other residues cyan. The viewpoint is rotated 180° about a vertical axis with respect to the viewpoints in Fig. 4. Key residues and the N- and C-termini are labeled. The figure was made with VMD 1.8.7 [24].
Discussion
Mutations in FHL1 now define a distinct but phenotypically variable X-linked myopathy. To date, 26 different mutations, including missense mutations, a splice-site mutation, deletions, insertions and a duplication, have been identified. Among them, 18 mutations are in LIM2, 1 in LIM3 and 7 in LIM4. Within LIM2 there are mutational clusters at residues 122, 123 and 153 (Table). In most of the cases, a cysteine or histidine residue that provides a zinc ligand is directly altered by a missense mutation. In addition, some frame-shift mutations truncate the protein upstream of one or more of these residues. The rare exceptions are V280M and X281E, which are located at the C terminus of the protein, and W122S and W122C (reported in this study), which modify a residue adjacent to a histidyl ligand in LIM2. This limited spectrum of mutations likely reflects the particular importance of the LIM domains, and the LIM2 and LIM4 domains in particular, for proper function.
Despite the clustering of mutations, the clinical phenotypes can vary substantially (Table). The onset ages range from 1 year-old to the 40s, but most develop symptoms by age 20. Accordingly, the age at which patients become wheel-chair bound also varies considerably; the youngest was 2 years old and some currently remain ambulant as old as 54 years. Three toddlers progressed quickly to death between one and four years after the onset [1, 5]. However, the course in some early-onset patients was slow and they remained ambulant after more than 20 years of disease [7]. Most female carriers are asymptomatic and the few exceptions (7 in 29 families) have later onset and mild myopathy. Notably, the clinical phenotypes can vary even between different amino acid substitutions at the same residue. The family with substitution of cysteine for tryptophan at residue 122 reported in this study has a relatively mild initial clinical phenotype, with onset delayed until the fourth decade, followed by slow progression to use of a wheelchair late in the fifth to sixth decades. There is much milder disease in the one examined female. In contrast, substitution of serine at this same residue (W122S) is associated with onset in the 20s, loss of ambulation in the 30s, more prominent muscle hypertrophy, and moderate disease in females. In six cases, mutations of the adjacent codon 123, which affect the zinc-coordinating histidine residue (H123L, H123Q, H123Y), result in very severe myopathy with onset in infancy to toddler age, rapid loss of ambulation and, in some cases, respiratory insufficiency. Overall, mutations that replace cysteine or histidine at one of the Zn binding sites in domain LIM2, in comparison to those in LIM4, result in more severe phenotypes with very early onset, rapid progression and loss of ambulation at an early age. Interestingly, mutations (K124RfsX6, K157VfsX36, A168GfsX27, C273LfsX11) that presumably produce a truncated protein are associated with less severe clinical phenotypes ([7, 8], Table).
There is considerable variation in the effects of mutations on FHL1 concentration and intracellular localization, although not all mutations have been studied in this regard (Table). As shown by western blotting and IHC analyses, the C224W and V280M mutations in LIM4 result in total or near total loss of FHL1 protein. In our family with a W122C mutation IHC studies showed muscle sequestration of FHL1 in inclusion bodies containing ubiquitin and desmin. Similar inclusions were seen in the majority of patients with the other mutations in LIM2 reported to date. In addition, most LIM2 mutations were associated with inclusion bodies that stained abnormally by menadione-NBT and these patients therefore were described as having a reducing body myopathy (RBM). RBM is a rare muscle disorder with progressive muscular weakness and atrophy, characterized by the histopathologic findings in myofibers of intracytoplasmic inclusion bodies that reduce NBT and thus stain strongly with menadione-NBT. The identification of FHL1 mutations in all RBM cases that have been screened confirms that FHL1 is the causative gene for this disorder. The common clinical phenotype shared with RBM, the presence of an FHL1 mutation, and the intracytoplasmic FHL1 aggregates in our patient’s muscle fibers suggests that disease in this family is highly similar to RBM, but unfortunately no frozen muscle was available from the family in the current report to evaluate for NBT reducing bodies. FHL1-positive structures in myofibers previously have been reported to be identical to the menadione-NBT positive reducing bodies, but we have not been able to document this in the present family [6]. Mutations in LIM4 and out-of-frame mutations appear to be associated with lack of both reducing bodies and FHL1 aggregates [7, 8] (see Table). As noted above, they are also associated with moderately severe clinical courses. These features suggest that more severe myopathy caused by missense mutations in LIM2 may reflect toxic effects of aggregated protein, rather than loss of protein function.
The variable phenotypes caused by mutations that cluster in the same domain, and sometimes involve the same or adjacent residues, raise the question of how these mutations affect the structure of the protein. None of the mutations we considered resulted in gross unfolding or major distortions of the structure during MD simulations that continued for 12.5 ns beyond a 15-ns equilibration period at 310 K (Figs. 4 and S2). The Zn binding sites remained largely intact. Since the MD simulations began with the structure of the native protein, it is possible that kinetic barriers prevented them from reaching the equilibrium structures of the mutant proteins. More severe structural changes might occur if the simulations were continued for longer times or were run at higher temperatures. However, all the mutant proteins displayed a larger amount of non-polar surface in one or both of two clusters of residues (Figs. 5 and S6–S8). Such hydrophobic patches can lead to protein aggregation [17–21], and thus might account for the intracellular aggregates of FHL1 seen in many patients with FHL1 mutations. Variations in the size or shape of these patches would affect the rate of aggregation, and also might affect specific interactions of FHL1 with other proteins, as Quinzii et al. have noted in connection with the W122S mutation. Mutations that cause subtle changes in the exposure of β-strands have been linked to aggregation of several proteins [22].
It is curious that FHL1 mutations are associated with seemingly different inheritance patterns, despite the marked clustering and the propensity to affect cysteine residues. Families with C104R, W122C, W122S, C153R or C153Y manifested an apparently X-linked dominant inheritance pattern, while those with 127–128insFT or C224W manifest an apparently X-linked recessive inheritance pattern. However, the majority of mutations in the LIM2 domain have been reported in sporadic cases, predominantly young females. These observations support the suggestion by Dobyns that inheritance of X-chromosome-related traits should not be defined as dominant or recessive, but rather generally as X-linked. Although a rare disease, mutations in FHL1 should be considered as the cause for scapuloperoneal myopathy in families in which X-linked inheritance cannot be ruled out, even in isolated affected females (that is, families such as the one described here without male to male transmission). Clinicians must be alert to the possibility of this disease because of the phenotypic overlap with other scapuloperoneal syndromes, FSH and Becker muscular dystrophies.
In conclusion, the notable clustering of mutations in the LIM2 domain suggests that this region has special importance for maintenance of the structural integrity of muscles, or that mutations in this domain result in formation of aggregates with particularly deleterious properties. Mutations at the same or adjacent residues cause phenotypes that differ markedly with respect to the muscle groups affected, the presence or absence of hypertrophy, the age of onset, the rate of progression and the likelihood that females will manifest disease, possibly because they increase the nonpolar solvent-accessible surface area in ways that have variable effects on the propensity of FHL1 to aggregate.
Supplementary Material
Acknowledgments
This research was supported by Veterans Affairs Medical Research funds (TDB, HL, WHR, JW), National Institutes of Health grant NS069719 (WHR), and National Science Foundation grant MCB-0641640 (WWP). Randy Small, Department of Pathology, University of Washington, provided technical assistance for the histopathology studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–912. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–213. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Windpassinger C, Schoser B, Straub V, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet. 2008;82:88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shalaby S, Hayashi YK, Goto K, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1) Neuromuscul Disord. 2008;18:959–961. doi: 10.1016/j.nmd.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology. 2009;72:375–376. doi: 10.1212/01.wnl.0000341311.84347.a0. [DOI] [PubMed] [Google Scholar]
- 6.Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132:452–464. doi: 10.1093/brain/awn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gueneau L, Bertrand AT, Jais J-P, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–353. doi: 10.1016/j.ajhg.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73:543–551. doi: 10.1212/WNL.0b013e3181b2a4b3. [DOI] [PubMed] [Google Scholar]
- 9.McGrath MJ, Cottle DL, Nguyen MA, et al. Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem. 2006;281:7666–7683. doi: 10.1074/jbc.M512552200. [DOI] [PubMed] [Google Scholar]
- 10.Loughna PT, Mason P, Bayol S, Brownson C. The LIM-domain protein FHL1 (SLIM 1) exhibits functional regulation in skeletal muscle. Mol Cell Biol Res Commun. 2000;3:136–140. doi: 10.1006/mcbr.2000.0206. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi Y, Furukawa T, Tun T, Han H, Honjo T. LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol Cell Biol. 1998;18:644–54. doi: 10.1128/mcb.18.1.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen DH, Matsushita M, Rainier S, et al. Presence of alanine-to-valine substitutions in myofibrillogenesis regulator 1 in paroxysmal nonkinesigenic dyskinesia: confirmation in 2 kindreds. Arch Neurol. 2005;62:597–600. doi: 10.1001/archneur.62.4.597. [DOI] [PubMed] [Google Scholar]
- 13.Qin XR, Nagashima T, Hayashi F, Yokoyama S. Solution structure of the second LIM domain of skeletal muscle LIM protein 1 [database on the Internet] 2005 Available from: http://www.pdb.org/pdb/explore.do?structureId=1X63.
- 14.Alden RG, Parson WW, Chu ZT, Warshel A. Orientation of the OH dipole of tyrosine (M)210 and its effect on electrostatic energies in photosynthetic reaction centers. J Phys Chem. 1996;100:16761–16770. [Google Scholar]
- 15.Lee FS, Chu ZT, Warshel A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J Comp Chem. 1993;14:161–185. [Google Scholar]
- 16.Parson WW, Warshel A. Calculations of electrostatic energies in proteins using microscopic, semimicroscopic and macroscopic models and free-energy perturbation approaches. In: Aartsma TJ, Matysik J, editors. Biophysical Techniques in Photosynthesis. Dordrecht: Springer; 2008. pp. 401–420. [Google Scholar]
- 17.Young L, Jernigan RL, Covell DG. A role for surface hydrophobicity in protein-protein recognition. Protein Sci. 1994;3:717–729. doi: 10.1002/pro.5560030501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai C, Lin SL, Wolfson HJ, Nussinov R. Studies of protein-protein interfaces: a statistical analysis of the hydrophobic effect. Protein Sci. 1997;6:53–64. doi: 10.1002/pro.5560060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caflish A. Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol. 2006;10:437–444. doi: 10.1016/j.cbpa.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci USA. 2009;106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 23.Dobyns WB, Filauro A, Tomson BN, et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A. 2004;129A:136–143. doi: 10.1002/ajmg.a.30123. [DOI] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD- Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.