

Abstract
Connective tissue diseases (CTDs) are a group of clinical disorders that have an underlying autoimmune pathogenesis. These include a diverse set of diseases such as relapsing polychondritis, rheumatoid arthritis, and eosinophilic fasciitis, along with more common entities like Sjogren’s syndrome, dermatomyositis, scleroderma, and lupus erythematosus. The latter three will be the focus of this review, as they constitute the most significant and common CTD with cutaneous manifestations. The cutaneous signs often represent the preliminary stages of disease and the presenting clinical symptoms. Therefore, comprehensive knowledge of CTD manifestations is essential for accurate diagnosis, better assessment of prognosis, and effective management. Although the precise etiologies of CTDs remain obscure, recent advances have allowed for further understanding of their pathogenesis and improved disease classifications. In addition, there have been developments in therapeutic options for CTDs. This review provides an overview of the epidemiology, clinical presentations, and current treatment options of cutaneous lupus erythematous, dermatomyositis and scleroderma.
Keywords: Autoimmunity, connective tissue disease (CTD), cutaneous lupus erythrematous (CLE), dermatomyositis (DM), scleroderma (Scl)
INTRODUCTION
Cutaneous lupus erythematous (LE), dermatomyositis (DM), and scleroderma (Scl) are CTDs with significant cutaneous manfestations that may exhibit widespread systemic dysfunction. The pathogenesis of connective tissue disease (CTD) is quite variable and not completely understood. They may present clinically as localized abnormalities, resulting in only cosmetic deformities, or they may involve debilitating systemic complications. All of the CTDs discussed in this review are polygenic, and can be linked through the concept of autoimmunity and the production of pathogenic autoantibodies [1–6]. There exists strong evidence to suggest that both genetic and environmental factors serve to initiate and promote the autoimmune response. As with many autoimmune diseases, CTDs display a strong predilection for women, ranging from 2:1 up to 15:1 female predominance, and in addition, racial background occasionally plays a role in either the severity or prevalence of disease [7].
Although CTDs are multisystem disorders, the skin is often the presenting sign. Interestingly, the clinical spectrum of presentation for CLE, DM, and Scl can vary from skin only to internal organ only. This offers the clinician a diagnostic challenge and it is thus critical that dermatologists maintain a heightened awareness of non-skin manifestations when working up patients for CTD. Furthermore, the wide array of clinical signs within each disease makes absolute classification of CTDs exceedingly difficult, especially in cases of overlap. Once a diagnosis has been established, the clinician is often disappointed by the lack of curative pharmacological options available for the treatment of CTD. Therefore, clinicians should be aware of the pathogenesis and implications of cutaneous findings in CTDs, as they may be significant to the clinical course and effective management of the disease. Although the precise mechanisms relating to the pathology of CTDs remain obscure, this review aims to elucidate the prevailing knowledge, current clinical assessments, and treatment options of CLE, DM, and Scl.
CUTANEOUS LUPUS ERYTHREMATOUS
Epidemiology & Pathogenetics
Cazenave first coined the term lupus erythematosus (LE) over a century ago [8]. Much progress has been made since then in understanding the complex array of clinical manifestations of LE. LE is a chronic autoimmune disorder that can involve virtually any organ of the body, leading to problems such as arthritis, anemia, nephritis, serositis, and cardiac conduction defects. The prevalence of LE in the United States is estimated at 14–122 per 100,000 persons [9]. In addition, 80–85% of the patients are women who fall into the reproductive age group [10–12]. When LE involves the skin, in the presence or absence of systemic disease, it has been termed cutaneous lupus erythematosus (CLE). Cutaneous forms of LE appear approximately two to three times more often than the systemic variant [13]. More than 75% of patients with LE present with cutaneous lesions during the course of the disease. In an international survey of LE, Vitali et al. found malar rash (40%), alopecia (24%), and oral ulcers (19%) to be the most frequent dermatologic signs [14]. Furthermore, approximately 20% of patients report cutaneous manifestations as the initial symptom of LE [9]. This emphasizes the importance of diagnosis based on cutaneous findings.
CLE can be further subdivided into acute (ACLE), subacute (SCLE), and chronic (CCLE) forms. The clinical features of these 3 variants are summarized in Table 1. Recently, another variant, known as LE tumidus (LET) has been classified as an intermittent subtype of CLE, and the clinical course and prognosis of this subtype is usually more favorable than other forms of CLE [15]. In addition, patients who develop significant scarring with CLE are termed to have discoid LE (DLE), and this is the most prevalent manifestation of CCLE [9]. Vigilance in understanding how to classify and diagnose CLE is necessary as progression to the more grave systemic form, or SLE, occurs at varying frequencies. Genetics appears to convey the greatest risk factor for developing CLE. A strong association exists between specific subtype predispositions and HLA region genes, as well as with complement deficiencies and cytokine abnormalities. ACLE is associated with HLA-DR2 and HLA-DR3, while SCLE is associated with HLA-A1, HLA-B8, HLA-DR2, HLA-DR3, HLA-DRw52, HLA-DRw6, HLA-DQ1, and HLA-DQ2 [9, 16, 17]. Furthermore, SCLE has been strongly linked to deficiencies in MHC class III genes coding for complements C2 and C4 [18]. Interestingly, a polymorphism in the tumor necrosis factor-α (TNF-α) promoter, which is stimulated by ultraviolet (UV) light, has also been found with increased frequency in patients with SCLE [18]. CCLE is associated with HLA-B7, HLA-B8, HLA-Cw7, HLA-DR2, HLA-DR3, and HLA-DQw1, along with decreased levels of complements C2-C5 [9]. MHC class III genes also code for heat shock proteins, which have been previously shown to exacerbate CLE [18].
Table 1.
Cutaneous Lupus Erythematosus (CLE) Variants and Clinical Features
| CLE Variants | Clinical Features |
|---|---|
| Acute CLE |
|
| Subacute CLE |
|
| Chronic CLE |
|
In addition to genetic factors, a number of environmental factors have been postulated to play a role in either the initiation or propagation of the autoimmune response in LE. Ultraviolet light (UV)-induced skin lesions are found in a significant number of patients with CLE, and this phenomenon has been referred to as photosensitivity. The proinflammatory effect of UVB radiation via induction of cytokines such as TNF-α, interleukin-1α (IL-1α), IL-6, IL-8, and IL-10, may be important to CLE [13]. Pathologic photosensitivity is observed in 72% of patients with LET, 63% of patients with SCLE, 60% of patients with ACLE, and 45% of patients with DLE [15, 19]. Patients often do not associate dermatologic flares with sun exposure since the response may be delayed. The presence of Ro/SSA and/or La/SSB autoantibodies, as well as the TNF-α promotor polymorphism, play an important role in the pathogenesis of photosensitivity in SCLE [18]. In addition, a number of medications such as angiotensin-converting enzyme inhibitors (captopril, cilazapril), calcium channel blockers (nifedipine, diltiazem, and verapamil), procainamide, sulfonylureas, and naproxen, amongst others, can induce CLE-like symptoms [18]. Often, these medications are known photosensitizers, and UVA and UVB may be important mediators in this response. Other environmental factors incriminated in the pathogenesis of CLE include smoking, laser-induced thermal injury, and long-term exposure to quartz (silica) [18]. Recently, CD4+CD25+ regulatory T cells (Treg) were found to be deficient in skin lesions of various subtypes of CLE [15]. Since Treg cells are known to be important in the suppression of immune response to self-antigens, a Treg deficiency may play a key role in the pathogenesis of CLE. Furthermore, cytokine abnormalities have been observed in CLE patients. For example, type I interferons (IFNs) are found to be upregulated in nonirradiated skin of CLE patients compared to healthy patients, which may be due to UVB radiation exposure [15].
Diagnosis
Classification of the cutaneous signs of LE were developed by Gilliam, who separated skin lesions into non-specific and specific, with the latter further subdivided into acute, subacute, and chronic [20]. ACLE is characterized by abrupt, usually in the setting of systemic illness, and 100% of ACLE patients develop SLE [9]. The typical ACLE patient is a female in her third decade of life who presents with the classic butterfly rash after sun exposure (Fig. 1). Lesions may be localized or widespread and commonly appear in the face (87%), upper limbs (73%), and the trunk (36%) [9]. Irrespective of location, the rash can range from mild erythema to significant edema, lasting from hours to weeks, and heals without scarring. Accompanying changes include poikiloderma, oral ulceration, scales, and a papular component. There can also be diffuse thinning of scalp hair, as well as loss of the frontal hairline with evidence of hair fragility. Cuticular abnormalities are often observed, such as telengiectasia and erythema in the proximal nail fold. Rare features include bullae on sun-exposed skin, and toxic epidermal necrolysis-like lesions with insidious onset [21, 22]. About 95% of ACLE patients are positive for anti-nuclear antibodies (ANA), including anti-dsDNA, Smith, Ro, or La [18]. The presence of anti-dsDNA antibodies may lead to lupus nephritis, and therefore, these patients should be regularly screened for internal organ disease [23].
Fig. 1.
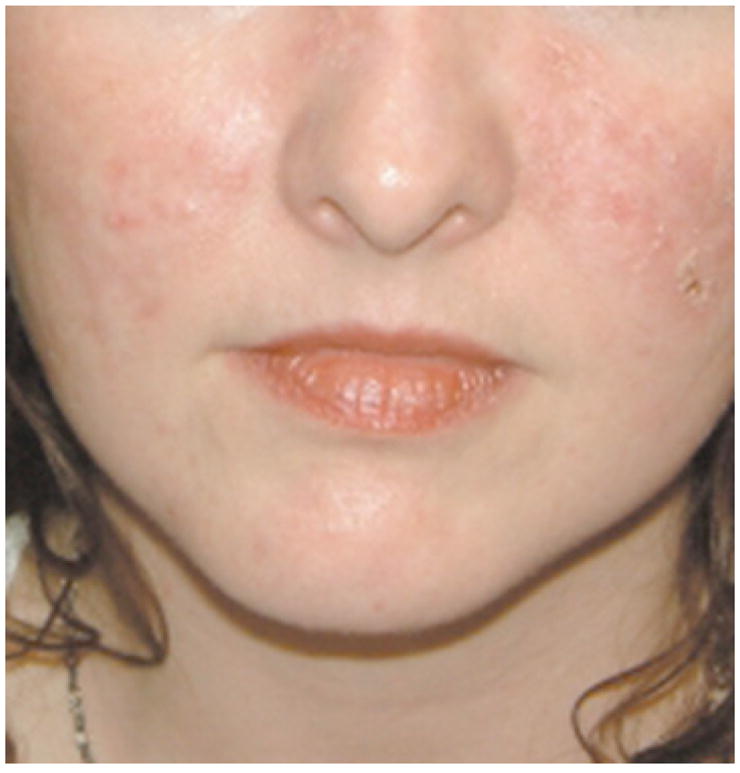
Acute cutaneous lupus. Malar rash with scale. Photo courtesy of David F. Fiorentino, M.D., Ph.D.
In 1979, the term SCLE was coined by Sontheimer, Thomas, and Gilliam to describe a subset of LE patients with a subacute presentation [24]. These patients were also females typically in their third to fourth decade of life with strong correlation to 50% demonstrating photosensitivity. This subtype appears in women 3–4 times more often than men [9]. The typical eruption consists of nonscarring erythematous papules with either an annular (42% of patients) or psoriasiform (39% of patients) pattern [9, 18]. These lesions are commonly distributed on the face, upper trunk, and extensor forearms. Telengiectasia and dyspigmentation are almost always present. Other associated findings include alopecia, malar eruption, perinungal telengiectasia, poikiloderma, livedo reticularis, and rarely a pityriasis-like pattern. In 20% of patients, lesions of the discoid LE type precede the onset of SCLE [24]. Serological testing is useful in diagnosis of SCLE. Patients with SCLE are positive of ANA in 70 to 80% of cases and Ro/SSA in 50 to 71% of cases (especially the annular variant) [9]. In one study, reactivity to anti-dsDNA was detected in only 5% of patients, and therefore, serological testing may often be unreliable for diagnosis of SCLE [18]. It is estimated that approximately 15% of patients will eventually have internal organ involvement [23]. Approximately 50% of patients with SCLE will meet the diagnostic criteria of SLE, however these patients will present with less severe complications [9].
CCLE is the most common type of CLE, and it presents 2–3 times more often than SLE [25]. CCLE can present in multiple forms including discoid LE (DLE), hypertrophic, LE-lichen planus overlap, chilblain, lupus panniculitis (lupus profundus), and lupus tumidus. Some of these clinical entities are not strictly seen in CCLE. The typical patient is a female between the ages of 20 to 40 in the setting of a long-term low grade illness or rarely life threatening SLE [26]. In DLE, a history of Raynaud’s phenomenon can be found in 15% of the cases [18]. Unlike SCLE, no clear association with sun exposure has been established despite a predilection for photosensivity of the face, scalp, and ears. DLE rarely involves the mucosa or regions of the body below the neck (in the absence of lesions above the neck). The findings of DLE are characterized by well demarcated indurated scaly erythematous plaques with adherent scale extending into hair follicles. These plaques heal centrally first, and then, atrophy, scarring, dyspigmentation, and telengiectasia usually follows (Fig. 2). The periphery often is hyperpigmented. Scarring alopecia may result from scalp involvement, and clinicians should distinguish this manifestation from the incidence of alopecia areata in the setting of LE. Often, follicular involvement progresses to the development of keratotic spikes (carpet-tack sign). DLE lesions found exclusively in the head and neck region are classified as localized DLE, and lesions extending both above and below the neck are categorized as generalized DLE [9]. 5% of patients exhibiting localized DLE develop SLE, and 20% of generalized DLE patients develop SLE [9]. Therefore, it seems a transition from localized DLE to a generalized form places the patient at a greater risk for SLE, which should be suspected with the onset of fever, increasing ANAs, leukopenia, hematuria, or albuminuria. Clinicians should be aware of the fact that discoid lesions, the hallmark of DLE, are commonly seen in SLE; therefore, a search for other SLE criteria should be undertaken when evaluating patients with suspected DLE. As in most chronic scarring process, DLE patients are at an increased risk of developing squamous cell carcinoma, most commonly seen on the lower lips of African American patients with hypopigmented scars [18]. Serological screening is somewhat helpful, although testing positive to ANA, dsDNA, Sm, U1RNP, or Ro/SSA antibodies is less common than SCLE or ACLE [16, 27].
Fig. 2.
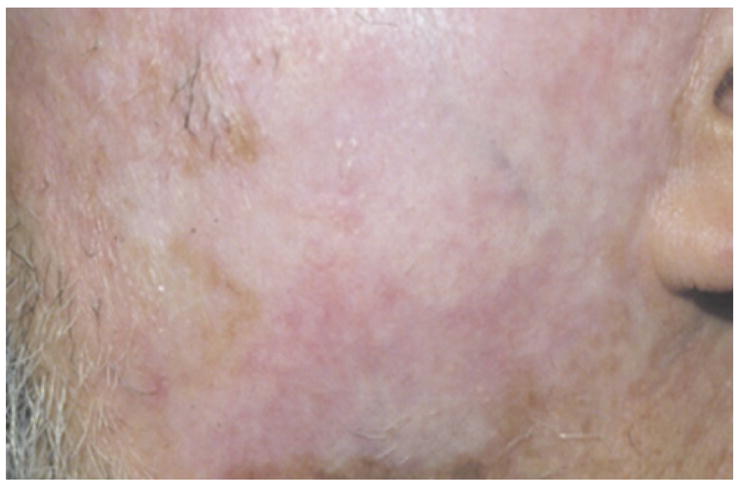
Discoid lesions of chronic cutaneous lupus with significant scarring. Photo courtesy of Amy McMichael, M.D.
Hypertrophic CCLE lesions constitute about 2% of CLE [18]. In this CCLE variant, a papulonodular often hyperkeratotic eruption is present most commonly on the extensor extremities, but may present of the face, palms, and soles. With diffuse scaling, the hyperkeratotic component gives the skin a chalky dust appearance. Usually, discoid lesions can be detected in other locations of the body, helping secure the diagnosis.
An important clinical challenge lies in the diagnosis of SLE in patients with CLE. Patients who meet at least 4 out of the 11 criteria established by the American Rheumatism Association (ARA), now known as the American College of Rheumatology (ACR), may be diagnosed with SLE [9]. However, mucocutaneous manifestations (malar rash; discoid rash; photosensitivity and oral ulcers) represent 4 of the 11 ACR diagnostic criteria, challenging the reliability of this classification system [9]. More recently, the European Academy of Dermatology and Venereology (EADV) developed criteria aiming to increase the accuracy of diagnosing SLE in CLE patients [28]. In a comparative analysis of the two classification schemes, the ACR criteria exhibited a sensitivity of 88%, a specificity of 79%, a positive predictive value of 56%, and a negative predictive value of 96%, and the EADV criteria demonstrated a sensitivity of 64%, a specificity of 94%, a positive predictive value of 61%, and a negative predictive value of 94% [28]. These findings suggest that the ACR criteria may be unsuitable for diagnosis due to a disproportionately high sensitivity compared to a weak specificity. On the other hand, the EADV criteria are more specific while being less sensitive.
Treatment
Unfortunately in CLE and most autoimmune disorders, most treatments are not curative. Therefore, most therapies aim to prevent disease progression and restore the patient’s normal appearance (Table 2). It is critical to educate patients on heat and sun exposure, as well as the avoidance of particular medications, which may exacerbate the condition. The most important preventive measure is perhaps the use of photoprotection. This can be achieved with various FDA-approved clothing lines, broad spectrum sunscreens, and physical blockers such as zinc oxide or micronized titanium dioxide. The most effective sunscreens contain both UVA (parsol 1789, mexoryl SX, mexoryl XL) and UVB protectants (octocrylene) [29]. A high level of sun protection is necessary to prevent photosensitive eruptions of CLE. Topical or intralesional corticosteroids are sometimes effective in treating localized CCLE as well as CLE involving the scalp [18]. For telengiectasia, the pulse dye laser has proven to be a safe treatment with few side effects and a clearance rate approaching 70% [18]. Scarring of CCLE can be addressed using a carbon-dioxide laser; however, this should be avoided in patients who have been treated with isotretinoin in the past 1–2 year [18].
Table 2.
Summary of Connective Tissue Diseases
| Disease | Genetic Associations/Etiologies | Auto-Antibodies | Prevalent Complications | Standard Treatment (Refs.)1 |
|---|---|---|---|---|
| CLE | HLA-DR2 | anti-nuclear | Raynaud’s phenomenon | UVA and UVB sunscreens [29] |
| HLA-DR3 | dsDNA | progression to SLE | topical corticosteroids [18] | |
| HLA-B7 | Smith | glomerulonephritis | intralesional corticosteroids [18] | |
| HLA-B8 | Ro | pulmonary fibrosis | chloroquine [14] | |
| C2–C5 deficiency | La | serositis | hydroxycholoroquine [14] | |
| quinacrine [14] | ||||
| DM | ↑ C5b-C9 levels | anti-jo-1 | interstitial lung disease | UVA and UVB sunscreens [32] |
| MxA protein | Mi-2 | arthralgias | Corticosteroids [41,42] | |
| Th1 activation | SRP | dyspnea | Methotrexate [39, 41] | |
| TNF receptor | anti-U1RNP | dysphagia | IVIG2 [39, 41] | |
| MHC-I | Ro | arrhythmia | cyclophosphamide [39, 41] | |
| IL-1α | PM | dysphonia | cyclosporine [39, 41] | |
| malignancy (ovarian, stomach, lymphoma) | mycophenolate mofetil [39, 41] | |||
| Scl | oxidative stress | anti-nuclear | arthritis | Corticosteroids [45] |
| aberrant free radicals | dsDNA | Raynaud’s phenomenon | Sulfasalazine [45] | |
| ↓ ascorbic acid, | histone | neurological abnormalities | pencillamine [45] | |
| ↓ selenium, | topoisomerase centromere | vascular changes | λ-interferon [45] | |
| ↓ vitamin E | gastrointestinal conditions | methotrexate [45] | ||
| ↓ carotene | respiratory disorders | phototherapy with UVA light [45] | ||
| other autoimmune disorders (eg. thyroiditis) | calcipotriene [39] | |||
| stem cell transplantation [60] | ||||
Refs.: reference number.
IVIG: intravenous immunoglobulin.
In the absence of systemic involvement, the anti-malarial drug hydroxychloroquine (400 mg/d) is used as first-line treatment for widespread CLE. This treatment has a slow onset, and hence, patients should be forewarned that improvement may not surface until 6–8 weeks after treatment. Quinacrine (100mg/d) may be supplemented in refractory cases [14]. Chlorquine phosphate may be appropriate for patients who fail the combination of hydroxycholorquine and quinacrine. However, this treatment poses risk of ocular toxicity and therefore should be dosed at less than 3.5mg/kg/d and followed by an ophthalmologist visit every six months. Furthermore, patients on anti-malarial medications should be advised to avoid smoking, which diminishes the efficacy of these drugs. Other longstanding treatment options include mycophenolate mofetil, thalidomide, dapsone, clofazamine, retinoids, and auranofin [14]. However, recent evidence on significant toxicity should be considered. For example thalidomide, in a recent study, was found to successfully treat 81% of patients with LE exhibiting skin involvement, but, other studies have found that the incidence of irreversible neuropathy associated with treatment is observed in greater than 50% of patients [24]. Overall, existing treatment options for CLE are not efficacious. In recent years, promising results were observed in photosensitive variants of CLE treated with low-dose UVA1 therapy [14]. The efficacy of this treatment is believed to lie in the ability of UVA1 radiation to induct IL-12, which plays a role in DNA repair (14). Emerging therapeutic strategies are aimed at exploiting the immunopathogenesis of CLE, since immune dysregulation, involving antibody-secreting B cells and aberrant T cells, is thought to be responsible for the development of LE [9]. One novel therapeutic target is B cell-dependent T cell activation. CTLA-4, which is expressed by activated T cells, serves to diminish T cell activation via fusion of co-stimulatory molecules CD28 (expressed on T cells) and B7 (expressed on B cells) [9]. These interactions served as the basis for the development of fusion proteins, which bind to CTLA-4 and result in down regulation of co-stimulation of T cells by B7-CD28 interactions. Fusion proteins have shown promising results in treating lupus in murine models and are currently in human trials for lupus patients [9]. Other potential therapies are aimed at targeting B and T cells directly as well as cytokines such as IL-1, TNF-α, and IFN-α, which are upregulated in lupus [9]. Further understanding of the pathogenesis of CLE will lead to improved diagnosis and treatment options for patients.
DERMATOMYOSITIS
Epidemiology & Pathogenetics
DM is an idiopathic inflammatory myopathy of autoimmune etiology characterized by a cutaneous eruption in the setting of symmetric proximal, extensor myopathy. Often, the cutaneous manifestations are the most difficult to treat and result in significant morbidity among patients. The mean age of onset of adult DM is 52 years and there is a strong female predilection, as well as an increased risk of malignancy [30]. Generally, the cutaneous signs precede the development of cancer and dermatologists play an integral role in the early detection of internal organ involvement. The pathogenesis remains unknown, although supporting evidence is building for a model of immune-mediated destruction of muscle in association with apoptotic reactions leading to skin lesions. Recent evidence has implicated complement components in the development of vascular pathology associated with DM [22]. Elevated C5b-9 deposition in vessels was found in biopsies from DM patients, suggesting the possibility of humoral and complement-mediated damage of endothelium and kertinocytes in DM [22]. Wenzel and colleagues have observed increased expression of M×A protein, along with cytokine profile suggestive of enhanced Th1 activation in DM patients [31]. This finding supports that innate immune responses along with adaptive immune responses may be associated with the etiology of DM. Other studies have found unusual levels of nitric oxide, upregulation of circulating TNF receptors, increased soluble CD40 expression, and upregulation of MHC I genes and IL-1α within the muscle of patients with DM [32].
Diagnosis
DM can present with a wide spectrum of clinical findings and can be subtyped as amyopathic DM, clinically amyopathic DM, or classic DM [33]. Amyopathic DM refers to the presence of characteristic cutaneous features of DM without muscle involvement, as confirmed by muscle biopsy, electrography, and magnetic resonance imaging [34]. This diagnosis cannot be established if patients have undergone recent immunosuppressive therapy of 2 months or greater duration within the first 6 months of onset of cutaneous disease [33]. Coincidental appearance of classic skin findings with use of medications known to generate DM-like cutaneous changes (hydroxyurea or the statin class of lipid lowering agents) also precludes a diagnosis of amyopathic DM [33, 34]. Patients, who on more rigorous muscle testing (laboratory, electrophysiologic, or radiologic evaluation), are found to have positive test results representing subclinical myositis are best classified as hypomyopathic DM [33]. The term clinically amyopathic DM includes both amyopathic DM as well as hypomyopathic DM, since cutaneous lesions represent the primary clinical finding in both entities. In classic DM, patients present with hallmark skin findings of DM as well as both clinical and objective evidence of muscle inflammation [33].
The two characteristic and perhaps pathognomonic cutaneous manifestations of DM are the heliotrope rash and Gottron’s papules [32]. The heliotrope rash is characterized by a violeceous erythema in the periorbital region. In some cases, the heliotrope rash is represented solely by dilated veins in the eyelids, which is believed to result from inflammation of striated muscle [32]. Also, affected regions undergo scaling and desquamation. Clinicians should be aware that the heliotrope rash may range from a slight discoloration surrounding the margin of the eyelid to profuse edema. Gottron’s paules are indurated, violaceous papules and plaques found on the metacarpophalangeal joints, the proximal interphalangeal joints, and/or the distal interphalangeal joints (Fig. 3). Often, telangiectasia occurs in the lesions, which may appear similar to lesions of LE or papulosquamous disorders such as psoriasis or lichen planus [32]. Hence, frequent histologic analysis may be needed in differential diagnosis. In addition, to the heliotrope rash and Gottron’s papules, DM patients exhibit other cutaneous manifestations such as malar erythema, poikiloderma (photosensitive distribution), violaceous erythema on extensor regions, periungal and cuticular alterations, and alopecia. Poikiloderma classically appears on the extensor regions of the arm, the “V” of the neck or the upper back (shawl sign), and/or the lateral aspects of the upper hips and thigh (holster sign). Furthermore, DM patients often exhibit psoriasiform dermatitis in the scalp, and this often necessitates histological differentiation from seborrheic dermatitis or psoriasis [32]. Uncommon cuteaneous findings in DM patients include vesiculobullous lesions, an eruption that induces pityriasis rubra pilaris, vasculitits, erosive lesions, and exfoliatiative erythroderma; there is speculation that these findings are observed more commonly in patients with malignancy [32].
Fig. 3.
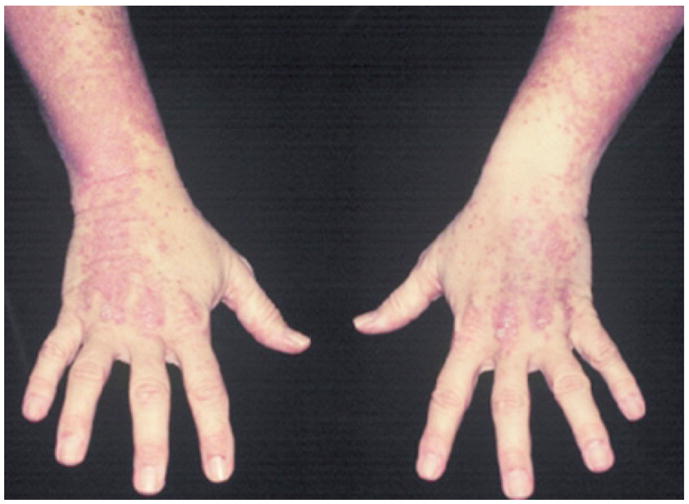
Dermatomyositis of the dorsal hands and forearms. Photo courtesy of Amy McMichael, M.D.
DM is a multisystem autoimmune disease and can lead to significant internal organ involvement. The two most clinically important associations with dermatomyositis are malignancy and pulmonary disease. The increased risk of malignancy (from 1.7 to 6.5 fold) is greatest in the first 1 to 2 years post-diagnosis but remains elevated for many years [36]. The occurrence of malignancy in adult DM patients is estimated to be 6% to 60%, with large population-based cohort studies predicting incidence in 20% to 25% of cases [35]. The most often reported malignancies are gynecological, especially ovarian carcinoma; however, others associated with DM include lung, pancreatic, stomach, colorectal, and non-Hodgkin’s lymphoma [35]. Ovarian cancers are usually found in female patients while stomach and lymphoma prevail in men. In addition, In addition, malignancy is more frequent in older patients [31]. Pulmonary disease is observed in 15% to 65% of patients [36, 37]. Interstitial pneumonitis is the most prevalent lung complication associated with DM, and it also appears to affect patients with the amyopathic variant as well. In a retrospective study at Mayo Clinic of 70 patients with DM and interstitial lung disease, it was found that only 15 patients presented with simultaneous involvement, suggesting that patients may more commonly experience symptoms of lung disease and myositis separately [38]. Patients with interstitial lung disease usually present with a non-productive cough and dyspnea on exertion. Pulmonary function tests in these patients exhibit a restrictive pattern. Other systemic manifestations include proximal muscle weakness, dyspnea, dyphagia, arrhythmia, and dyphonia [21]. Diagnosis is validated through laboratory tests revealing increased muscle enzymes such as creatine kinase, aldolase, aspartate aminotransferase, and/or lactate dehydrogenase [22]. Arthralgias are found in nearly 25% of patients exhibiting inflammatory myopathy and usually present with morning joint stiffness [32]. Esophageal disease, characterized by dysphagia is found in 15% to 50% of patients, and distal dysphagia correlates with a worse prognosis than proximal [32]. Although rare, cardiac abnormalities also may occur in DM patients, with conduction defects and arrhythmias being the most common [32].
Juvenile DM is classified as disease onset prior to the age of 16, and the peak incidence occurs between the ages of 5 and 10 [34]. The mean age at diagnosis is 10.8 years, and there is approximately an equal ratio of male and female patients [39]. The juvenile form exhibits a greater frequency of calcinosis, vascular inflammation, insulin resistant-lipidystrophy than the adult DM [32, 39]. Contrary to adult DM, there is no significant risk of malignancy associated with the juvenile form. Symptomatic treatment of cutaneous manifestations and careful management of clinical course is correlated with good prognosis [39].
As with other CTDs, DM is characterized by autoanti-bodies to different cellular constituents. Autoantibodies that are specific for DM are known as myositis-specific autoantibodies and include antisynthetases (anti-jo-1 being the most common), Mi-2, and SRP [40]. Autoantibodies associated with DM, but share overlap with other pathological conditions of myositis, are U1RNP, Ro/SSA, and PM/Scl. Autoantibodies specific for DM have been grouped into myositis-specific (antisynthetases, Mi-2, and SRP) and myositis-associated (PM/Scl, Ro/SSA, U1RNP) autoantibodies [40]. Anti-synthetase antibodies such as anti-Jo-1 have been associated with a clinical manifestation known as Anti-synthetase syndrome, which is characterized by myositis, interstitial pneumonia, polyarthritis, mechanic’s hand, Raynaud’s phenomenon, and fever [40]. The clinical usefulness of these autoantibodies as diagnostic screening tools is questionable in the face of their low frequency. In a past European study of 181 classic DM patients, no autoantibodies were found in more than 50% of patients [30]. However, recent research has found two additional autoantibodies associated with DM: anti-CADM-140 antibody and anti-p155 antibody [40]. In a small study, anti-CADM-140 was found in more than 50% of patients with amyopathic DM, and in a separate study, anti-p155 was found in 20% of DM patients, with strong correlation to cancer-associated DM [40]. The discovery of these novel antibodies increases the significance of serological testing and offers further insight into the pathology of DM.
Treatment
The prognosis of DM has been significantly influenced by the development of treatments (Table 2). Factors influencing prognosis are patient’s age, severity of myositis, occurrence of dysphagia, cardiopulmonary complications, DM-associated malignancy, and the response to corticosteroid therapy [32]. Mortality ranges from 8.9% to 52% due to variation of complications in patients [41]. One-year, 5-year, and 9-year or greater survival rates range from 83% to 95%, 63% to 95%, and 53% to 100%, respectively [41]. These survival rates reflect a much better prognosis than the 50% mortality rate associated with the progression of the natural course of DM without corticosteroid therapy [42]. Juvenile DM has a significantly lower mortality rate [39]. Corticosteroid therapy has diminished the mortality rate of juvenile DM from 34% to 1.5% or 0%, depending on the study [41, 42]. These results demarcate why immunosuppression with corticosteroids remains the gold standard therapy in DM. Early treatment may be life saving in DM patients. Therapy should be catered to the manifestations of DM, the patient’s responsiveness to treatment, and the effect on a patient’s quality of life. Methotrexate is a second-line treatment for patients with severe DM or those whose conditioned has not improved with corticosteroids [39]. Other second-line and third-line treatments include intravenous immunoglobulin, cyclophosphamide, cyclosporine, and mycophenolate mofetil [41]. Since most cutaneous lesions are photosensitive, patients with cutaneous disease should follow strict UV avoidance measures and daily use of sunscreens with broad spectrum activity is recommended [32]. Cutaneous calcinosis is often a severe complication of DM in children, and research has attributed it to delays in diagnosis and treatment [41]. Case reports have detailed successful treatments with varying treatments including diltiazem, probenecid, and alendronate, however surgical excision may be the best option for large deposits [41]. Ongoing research is investigating the potential for other immunomodulatory treatments for DM. A recent pilot study has found that B-cell depletion via the anti-CD20 monoclonal antibody rituximab resulted in increased muscle strength from baseline by 36% to 113% in DM patients, possibly implicating B cells in pathogenesis [43]). Another study found promising results with the use of tumor necrosis factor-α as monotherapy for DM [41]. These immune-targeting therapies may represent the future of DM treatment. Astute clinical diagnosis of DM remains critical to the prognosis of DM.
SCLERODERMA
Epidemiology & Pathogenetics
Scleroderma is a disorder characterized by excessive fibrosis, which may be systemic or localized to skin. When fibrosis is limited to the skin and subcutaneous tissue, the disorder is classified as localized scleroderma or morphea [44]. Systemic sclerosis refers to a multi-system disorder involving fibrosis of skin, blood vessels, and internal organs [45, 46]. In spite of significant research efforts, the precise pathological origin of scleroderma remains obscure. Recent research supports the hypothesis that scleroderma may be due to oxidative stress and an aberrant activation of damaging free radicals [47]. Scleroderma patients were found to have reduced serum levels of antioxidants such as ascorbic acid, selenium, vitamin E, and carotene [48, 49]. Furthermore, elevated levels of nitric oxide, a reactive oxygen molecule involved in inflammatory injury, were found in scleroderma patients, along with autoantibodies against peroxiredoxin I, an antioxidant enzyme [47]. The generation of free radicals may lead to the development of fibrosis through their link to cytokines and growth factors such as tumor growth factor-β and platelet derived growth factor, which have been implicated as mediators of fibrosis in scleroderma [50]. In addition, recent evidence has implicated endothelin-I as an important mediator in the pathogenesis of scleroderma [51]. Endothelin-I has been found in early lesions, is released from damaged endothelial cells, and mediates effects associated with scleroderma, such as inducing extracelluar matrix biosynthesis and inhibiting matrix-degrading enzymes [51]. It is challenging to diagnose scleroderma, as there are various other disorders that exhibit cutaneous sclerosis, such as sclerederma, myxedema, scleromyxedema, nephrogenic fibrosing dermatopathy and eosinophilic fasciitis. The clinical course of scleroderma often makes accurate diagnosis very challenging. Various genetic disorders (Progeria), metabolic diseases (porphyria, lipodystrophy) and paraneoplastic syndromes may display significant cutaneous sclerosis. In addition, toxins (rapseed oil), medications (bleomycin) and even external trauma (vibrations) can results in scleroderma-like signs. Therefore, knowledge of the broad range of clinical manifestations is essential to accurate diagnosis and effective clinical management of the disorder.
Diagnosis
True scleroderma occurs in two distinct categories – localized or systemic. Localized scleroderma, or morphea, although limited to the skin and subcutaneous tissue, can produce lesions that cause severe functional and aesthetic defects. Tuffanelli and Winkelmann classified localized scleroderma into the following three categories, which are commonly used today: morphea, linear morphea, and generalized morphea [46]. Patients with morphea usually present with an insidious onset of an indurated plaque with an ivory-colored center and violoaceous halo (Fig. 4) [50]. Often a violaceous or erythmatous border, commonly referred to as the lilac ring, appears in the early course of the disorder. This halo indicates active inflammation, and it is replaced by sclerosis with central atrophy as the disease progresses, providing an ivory appearance to the skin. “Superficial morphea” refers to lesions in which collagen thickening is limited to the upper dermis, and theses lesions may be hyper-pigmented without underlying sclerosis. Punctate morphea is variant of localized morphea and is characterized by small plaque complexes [50] Linear scleroderma, which is the most common type observed in pediatric cases, appears as a linear streak of fibrotic lesions that may extend from the dermis to the underlying bone [45]. When linear morphea afflicts the face or scalp, it is known as en coup de sabre [45]. A severe manifestation of en coup de sabre is known as Parry Romberg Syndrome, and this form presents as hemi-facial atrophy of skin and subcutaneous tissue below the forehead [45]. Generalized morphea is the most severe type of localized scleroderma, and it is characterized by extensive full-thickness involvement of the skin along with plaques, hyperpigmentation, and muscle atrophy [50]. Recent research has implicated generalized morphea in the development of squamous cell carcinoma [52].
Fig. 4.
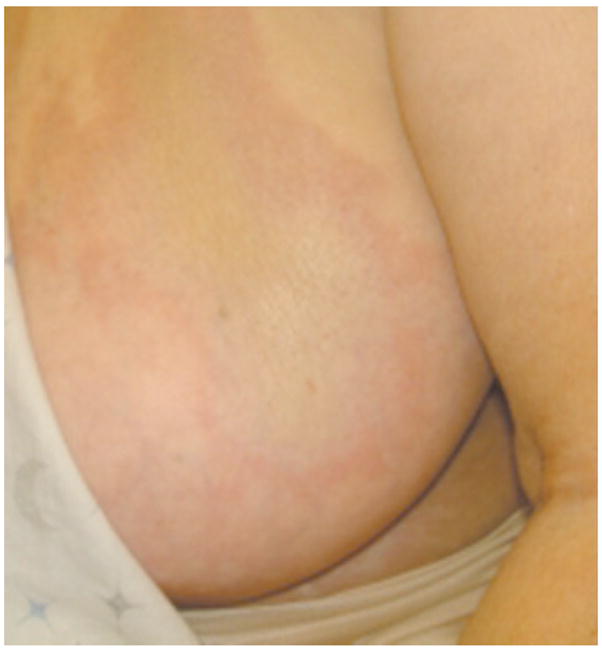
Morphea. A well demarcated sclerotic plaque with central yellowish color and peripheral violaceous erythema. Photo courtesy of David F. Fiorentino, M.D., Ph.D.
In addition to cutaneous findings, localized scleroderma has been linked to systemic complications, such as arthritis, neurological abnormalities, other autoimmune conditions (ex. thyroiditis), vascular changes, GI conditions, and respiratory disorders [45]. Of these, arthritis appears to be most common extracutaneous complication of localized scleroderma [45]. Localized scleroderma also results in an elevation of autoantibodies in many patients. Antinuclear antibodies are found in 73% of patients, more specifically, anti-double-stranded (50%), anti-histone (47%), anti-topoisomerase (76%), and anti-centromere (12%) antibodies are found [50]. In addition, plasma rheumatoid factor is found in 60% of the patients [50]. The link between the prevalence of these autoantibodies and the pathology of scleroderma remains unknown. Eosinophilia has been reported in patients with linear and generalized morphea, and may correlate with disease severity. Autoantibodies markers should be viewed skeptically because patients with localized scleroderma have minimal risk of systemic disease. Prognosis of skin disease is favorable, with lesions regressing spontaneously over several years; however, linear and generalized morphea may be chronic and progressive.
Treatment
Management of localized scleroderma remains a clinical challenge. The subjective nature of clinical assessment of localized scleroderma often poses difficulties in established skin scoring methods. Recent technological advances have allowed for improved evaluation of localized scleroderma. Infrared thermography has been used to detect active lesions [53]. Also, laser Doppler flowmetry, a non-invasive tool to evaluate cutaneous microcirculation, was found successful in evaluating active en coup de sabre lesions, which lack external signs of inflammation [54].
Treatment of localized scleroderma has included topical and systemic drugs, including corticosteroids, sulfasalazine, penicillamine, gamma-interferon, methotrexate, and phototherapy with UVA light (Table 2) [45]. Pruritus may be managed by dry skin care and topical (pramoxine, menthol) or oral antihistamine therapy. Intralesional corticosteroids may be used for localized lesions. Since the effects of localized scleroderma are usually limited to aesthetic deformities, therapies with low therapeutic indices are usually not recommended. For example, although phototherapy with ultraviolet light has shown some promise in managing localized scleroderma, the potential side effects with long-term use, such as accelerated skin aging and carcinogenesis, should be considered before therapy initiation [45]. For debilitating cases of localized scleroderma, systemic treatment involving a combination of methotrexate and a corticosteroid such as prednisone is a potential option. Recently, calcipotriene, a vitamin D3 derivative, has been reported to regulate fibrosis associated with localized scleroderma through inhibiting fibroblast proliferation [39].
Diagnosis and Treatment of Systemic Sclerosis
Systemic sclerosis is a chronic connective tissue disorder characterized by symmetrical fibrotic change in the skin, with visceral organ involvement. Denton and Black classified the pathology of systemic sclerosis with three key clinical manifestations [55]. The first main facet of the disorder is endothelial cell injury, which is observed in all patients who experience the disorder. This component manifests clinically as Raynaud’s phenomenon. The second component is inflammation, resulting in mononuclear cell infiltration. The last manifestion is fibrosis, characterized by irreversible damage of normal tissue and organs, and this component is responsible for the morbidity of systemic sclerosis. Systemic sclerosis is further divided based the nature and extent of the sclerosis. Patients in whom sclerosis is limited to involvement of the forearms, hands, regions proximal to the neck and perioral area are deemed to have limited systemic sclerosis [55, 56]. This form was formerly known as CREST syndrome, which characterized mainly by the pentad of calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclero-dactyl (Fig. 5), and telangiectasias [55]. Limited systemic sclerosis patients are at a high risk for isolated pulmonary hypertension, but they are at low risk (2%) for the development of scleroderma renal crisis, which remains a major cause of morbidity associated with scleroderma [55]. Patients who present with widespread sclerosis involving regions proximal to the neck and proximal limbs are deemed to have diffuse systemic sclerosis. The most common systemic complication involves the gastrointestinal tract, which can lead to altered motility, bacterial overgrowth, and malabsorption [55]. Diffuse systemic sclerosis can also present with fibrosis in other visceral organs such as the lung (30%), heart (10%), and kidney (12%) [55].
Fig. 5.
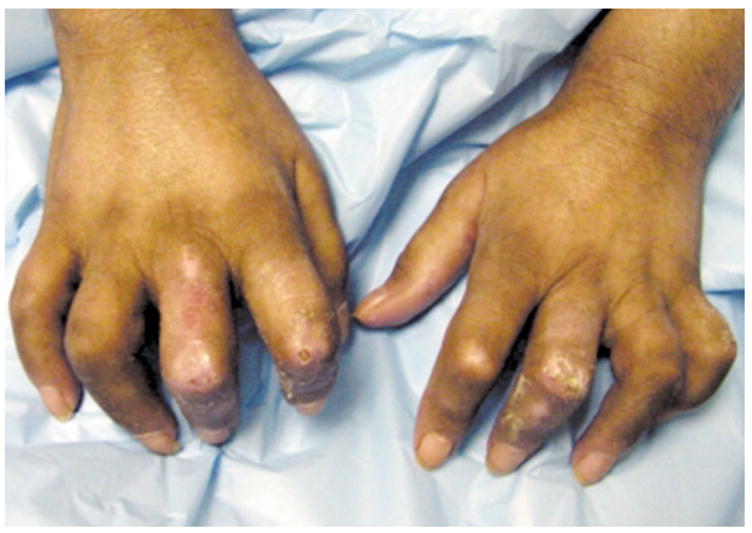
Sclerodacytly of systemic sclerosis. Note claw-like hands and digital ischemic ulcers. Photo courtesy of David F. Fiorentino, M.D., Ph.D.
As with other autoimmune disorders, there is an elevation in plasma autoantibodies commonly observed in patients with scleroderma. Autoantibodies are found in more than 95% of the patients with the disorder [56]. Furthermore, elevation of certain antibodies is associated with specific organ complications. Pulmonary arterial hypertension is found almost exclusively in patients positive for anti-centromere antibodies [56]. Patients with anti-RNA (POL3) appear to suffer the most severe skin fibrosis and have the highest frequency of renal complications, but these patients do not experience severe interstitial fibrosis [57, 58]. Patients with anti-topoisomerase classically present with renal crisis and severe lung disease, and these patients usually experience digital ulcers, gangrene and acroosteolysis more commonly than other patients with systemic sclerosis [56, 59]. The nucleolar antibody, U3-ANP, is found in patients have the most severe visceral organ involvement and have the worst prognosis among scleroderma patients [56]. Due to the existence of a strong correlation between specific autoantibodies and organ complications, clinicians can have insight into prognosis and management of scleroderma after serological analysis. Furthermore, clinical manifestations and forms of scleroderma have been associated with nationalities and ethnicities of patients, suggesting a genetic basis for the development of disease [56].
Management of complications is usually organ specific. Angiotensin converting enzyme inhibitors have significantly improved the prognosis of renal crisis associated with scleroderma (Table 2) [55]. Results from early case-control studies show a decrease in 12-month mortality from 76% to below 15% [56]. Cardiac complications are currently treated with prostacyclin analogues, and in cases of severe conduction abnormalities, pacemakers are implanted [56]. Cyclophosphamide and corticosteroids are used to treat chronic interstitial lung disease, and gastrointestinal involvement is managed with dietary alterations, antispasmodics, and antibiotics [56]. Recent advances in research and technology are exploring alternative options in therapy. Two current phase 3 clinical trials, the Autologous Stem Cell Transplantation International Scleroderma trial in Europe and the Scleroderma Cyclophosphamide or Transplant trial in the US, have generated promising results in scleroderma patients after autologous stem cell transplantations [60]. In these trails, 70% of patients exhibited an improvement in skin hardness, and 33% of patients displayed prolonged remission after transplantion with CD34+ hematopoietic stem cells [60]. The success of these trails is believed to attributed to the fact that scleroderma patients have a deficiency in circulating bone marrow-derived endothelial progenitor cells, and hence the ability of stem cells to increase the number of circulating progenitor cells may improve blood vessel repair and and neovascularization in scleroderma patients [60]. Also, early trials show an efficacy of intravenous immunoglobulin therapy in treating patients with systemic sclerosis, however, the precise mechanism of pharmacological action is not completely known [61]. Imatinib (STI571, Gleevec/Glivec) has shown potent anti-fibrotic effects in patients with chronic myelogenous leukemia and is currently being investigated for treatment of scleroderma [61].
CONCLUSION
In summary, this review has addressed the current clinical assessment and therapeutic options for CLE, DM, and Scl. CTDs remain challenging disorders to diagnose and treat effectively. Current research aims to elucidate the unknown elements involved in the pathogenesis of these disorders. New molecular techniques, such as high-throughput gene and protein analysis have advanced the prevailing concepts of etiologies of autoimmune dysfunctions. In addition, ongoing clinical trials of novel therapies may contribute to the emergence of improved therapeutic options in the near future. The extensive efforts in basic and applied science research are rapidly illuminating our comprehension of CTDs. Such knowledge will serve to enhance evaluation of prognosis and delivery of treatment in patients with CTDs.
ABBREVIATIONS
- ACLE
Acute cutaneous lupus erythrematosus
- ACR
American College of Rheumatology
- ANA
Anti-nuclear antibodies
- ARA
American Rheumatism Association
- CCLE
Chronic cutaneous lupus erythrematosus
- CTD
Connective tissue disease
- CLE
Cutaneous lupus erythrematosus
- DM
Dermatomyositis
- DLE
Discoid LE
- EADV
European Academy of Dermatology and Venereology
- IFN
Interferon
- IL
Interleukin
- LE
Lupus erythematosus
- LET
Lupus erythematosus tumidus
- Treg
Regulatory T cells
- Scl
Scleroderma
- SCLE
Subacute cutaneous lupus erythrematosus
- SLE
Systemic lupus erythrematosus
- TNF-α
Tumor necrosis factor alpha
- UV
Ultraviolet light
Footnotes
This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
References
- 1.Gaffney PM, Ortmann WA, Selby SA, et al. Genome screening in human systemic lupus erythematosus: results from a second Minnesota cohort and combined analyses of 187 sib-pair families. Am J Hum Genet. 2000;66:547–56. doi: 10.1086/302767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray-McGuire C, Moser KL, Gaffney PM, et al. Genome scan of human systemic lupus erythematosus by regression modeling: evidence of linkage and epistasis at 4p16–15.2. Am J Hum Genet. 2000;67:1460–9. doi: 10.1086/316891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valentini G, Improta RDG, Reese M, et al. Antinuclear antibodies in first degree relatives of patients with polymyositis/dermatomyositis: analysis of the relationship with HLA haplotypes. Br J Rheumatol. 1991;30:429–32. doi: 10.1093/rheumatology/30.6.429. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein R, Duvic M, Targoff IN, et al. HLA-D region genes associated with autoantibody responses to histidyl-trans RNA synthetase (Jo-1) and other translation-related factors in myositis. Arthritis Rheum. 1990;33:1240–8. doi: 10.1002/art.1780330826. [DOI] [PubMed] [Google Scholar]
- 5.Condemi JJ. The autoimmune diseases. Progressive systemic sclerosis (scleroderma) JAMA. 1992;268:2882–92. [PubMed] [Google Scholar]
- 6.Steen VD, Powell DL, Medsger TA., Jr Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 1988;31:196–203. doi: 10.1002/art.1780310207. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Tan FK, Wang N, et al. Genome-wide association study for regions of systemic sclerosis susceptibility in a Choctaw Indian population with high disease prevalence. Arthritis Rheum. 2003;48:2585. doi: 10.1002/art.11220. [DOI] [PubMed] [Google Scholar]
- 8.Cazenave PLA. Lupus erythemateux (erythema centrifuge) Ann Lad Peau Syph. 1850;3:297–9. [Google Scholar]
- 9.Lee HJ, Sinha AA. Cutaneous lupus erythematous: understanding of clinical features, genetic basis, and pathobiology of disease guides therapeutic strategies. Autoimmunity. 2006;39:433–44. doi: 10.1080/08916930600886851. [DOI] [PubMed] [Google Scholar]
- 10.McCarty DJ, Manzi S, Jr, Medsger TA. Incidence of systemic lupus erythrematosus: race and gender differences. Arthritis Rheum. 1995;38:1260–70. doi: 10.1002/art.1780380914. [DOI] [PubMed] [Google Scholar]
- 11.Gudmundsson S, Stein K. Systemic lupus erythrematosus in Iceland 1975–1984: a nationwide in an epidemiological study in an unselected population. J Rheumatol. 1990;17:1162–7. [PubMed] [Google Scholar]
- 12.Hopkinson ND, Doherty M, Powell RJ. The prevalence and incidence of systemic lupus erythrematosus in Nottingham, UK, 1989–1990. Br J Rheumatol. 1993;32:110–15. doi: 10.1093/rheumatology/32.2.110. [DOI] [PubMed] [Google Scholar]
- 13.Werth VP. Cutaneous Lupus: insights into pathogenesis and disease classification. Bull NYU Hosp Jt Dis. 2007;65:200–4. [PubMed] [Google Scholar]
- 14.Vitali C, Doria A, Tincani A, et al. International survey on the management of patients with SLE. I. General data on the participating centers and the results of a questionnaire regarding mucocutaneous involvement. Clin Exp Rheumatol. 1996;14:S17–22. [PubMed] [Google Scholar]
- 15.Kuhn A, Bijl M. Pathogenesis of cutaneous lupus erythrematosus. Lupus. 2008;17:389–93. doi: 10.1177/0961203308090019. [DOI] [PubMed] [Google Scholar]
- 16.McCauliffe DP. Cutaneous lupus erythematosus. Semin Cutan Med Surg. 2001;20:14–26. doi: 10.1053/sder.2001.23091. [DOI] [PubMed] [Google Scholar]
- 17.Werth VP, Dutz JP, Sontheimer RD. Pathogenetic mechanisms and treatment of cutaneous lupus erythematosus. Curr Opin Rheumatol. 1997;9:400–9. doi: 10.1097/00002281-199709000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin. 2002;20:373–85. doi: 10.1016/s0733-8635(02)00016-5. [DOI] [PubMed] [Google Scholar]
- 19.Hansan T, Nyberg F, Stephansson E, et al. Photosensitivity in lupus erythematosus, UV photoprotection results compared with history of photosensitivity and clinical findings. Br J Dermatol. 2000;136:699–705. [PubMed] [Google Scholar]
- 20.Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4:471–5. doi: 10.1016/s0190-9622(81)80261-7. [DOI] [PubMed] [Google Scholar]
- 21.Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions. Arch Dermatol. 1963;88:176–80. doi: 10.1001/archderm.1963.01590200064012. [DOI] [PubMed] [Google Scholar]
- 22.Costner MI, Grau RH. Update on connective tissue diseases in dermatology. Semin Cutan Med Surg. 2006;5:207–20. doi: 10.1016/j.sder.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Sontheimer RD, Provost TT. Cutaneous Manifestation of Rheumatic Diseases. In: Sontheimer RD, Provost TT, editors. Lupus Erythematous: Lipincott Williams & Wilkins. 2. 2004. pp. 104–58. [Google Scholar]
- 24.Sontheimer RD, Thomas JR, Gilliam JN. Subacute cutaneous lupus erythematosus: a cutaneous marker for a distinct lupus erythematosus subset. Arch Dermatol. 1979;115:1409–15. [PubMed] [Google Scholar]
- 25.Tebbe B, Orfanos CE. Epidemiology and socioeconomic impact of skin disease in lupus erythematosus. Lupus. 1997;6:96–104. doi: 10.1177/096120339700600204. [DOI] [PubMed] [Google Scholar]
- 26.O’Quinn S. Problems of disability in patients with chronic skin disease. Arch Dermatol. 1972;105:35–41. [PubMed] [Google Scholar]
- 27.Ng PP, Tan SH, Koh ET, et al. Epidemiology of cutaneous lupus erythematosus in a tertiary referral center in Singapore. Australas J Dermatol. 2000;41:229–33. doi: 10.1046/j.1440-0960.2000.00443.x. [DOI] [PubMed] [Google Scholar]
- 28.Parodi A, Rebora A. ARA and EADV criteria for classification of systemic lupus erythematosus in patients with cutaneous lupus erythematosus. Dermatology. 1997;194:217–20. doi: 10.1159/000246105. [DOI] [PubMed] [Google Scholar]
- 29.Callen J. Therapy for cutaneous lupus. Dermatol Ther. 2001;14:61–9. [Google Scholar]
- 30.Dawkins MA, Jorizzo JL, Walker Fo, et al. Dermatomyositis: a dermatological-based case series. J Am Acad Dermatol. 1998;38:397–404. doi: 10.1016/s0190-9622(98)70496-7. [DOI] [PubMed] [Google Scholar]
- 31.Wenzel J, Scheler M, Bieber T, Tuting T. Evidence for a role of type I interferons in the pathogenesis of dermatomyositis. Br J Dermatol. 2005;153:462–3. doi: 10.1111/j.1365-2133.2005.06786.x. [DOI] [PubMed] [Google Scholar]
- 32.Callen JP, Wortmann RL. Dermatomyositis. Clin Dermtol. 2006;24:363–73. doi: 10.1016/j.clindermatol.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 33.Sontheimer RD. Dermatomyositis: an overview of recent progress with emphasis on dermatologic aspects. Dermatol Clin. 2002;20:387–408. doi: 10.1016/s0733-8635(02)00021-9. [DOI] [PubMed] [Google Scholar]
- 34.Quian RD, Werth VP. Management of cutaenous dermatomyositis: current therapeutic options. Am J Clin Dermatol. 2006;7:341–51. doi: 10.2165/00128071-200607060-00002. [DOI] [PubMed] [Google Scholar]
- 35.Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96–100. doi: 10.1016/S0140-6736(00)03540-6. [DOI] [PubMed] [Google Scholar]
- 36.Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositits. Rheumatology. 2005;44:1282–86. doi: 10.1093/rheumatology/keh723. [DOI] [PubMed] [Google Scholar]
- 37.Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis, dermatomyositits and dermatomysositis. Ann Rheum Dis. 2004;63:297–301. doi: 10.1136/ard.2003.006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Douglas WW, Tazezlaer HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. AM J Respir Crit Care Med. 2001;164:1182–5. doi: 10.1164/ajrccm.164.7.2103110. [DOI] [PubMed] [Google Scholar]
- 39.Gamboa DG, Sugarman JL. An update on selected connective tissue diseases in adolescents. Curr Opin Pediatr. 2008;20:413–418. doi: 10.1097/MOP.0b013e328306eba1. [DOI] [PubMed] [Google Scholar]
- 40.Mimori T, Imura Y, Nakashima R, et al. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and patho-physiological significance. Curr Opin Rheumatol. 2007;19:523–9. doi: 10.1097/BOR.0b013e3282f01a8c. [DOI] [PubMed] [Google Scholar]
- 41.Iorizzo LJ, Jorizzo JL. The treatment and prognosis of dermatomyositis: an updated review. J Am Acad Dermatol. 2008;59:99–112. doi: 10.1016/j.jaad.2008.02.043. [DOI] [PubMed] [Google Scholar]
- 42.Bohan A, Peter JB. Polymyositis and dermatomyositis (two-part article) N Engl J Med. 1975;292:344–8. 403–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 43.Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601–7. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- 44.Gupta RA, Fiorentino D. Localized scleroderma and systemic sclerosis: is there a connection? Best Pract Res Clin Rhematol. 2007;21:1025–36. doi: 10.1016/j.berh.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Zulian F. Systemic sclerosis and localized scleroderma in childhood. Rheum Dis Clin N Am. 2008;34:239–55. doi: 10.1016/j.rdc.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Tufarnelli DL. Systemic scleroderma. Med Clin North AM. 1989;5:1167–80. doi: 10.1016/s0025-7125(16)30626-5. [DOI] [PubMed] [Google Scholar]
- 47.Gabrielli A, Svegliati S, Moroncini G, et al. Oxidative stress and the pathogenesis of scleroderma: the Murrell’s hypothesis revisited. Semin Immunopathol. 2008;3:329–37. doi: 10.1007/s00281-008-0125-4. [DOI] [PubMed] [Google Scholar]
- 48.Herrick AL, Rieley F, Schofield D, et al. Micronutrient antioxidant status in patients with primary Raynaud’s phenomenon and systemic sclerosis. J Rheumatol. 1994;21:1477–83. [PubMed] [Google Scholar]
- 49.Lundberg AC, Akesson A, Akesson B. Dietary intake and nutritional status in patients with systemic sclerosis. Ann Rheum Dis. 1992;51:1143–8. doi: 10.1136/ard.51.10.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takehara K, Sato S. Localized scleroderma is an autoimmune disorder. Rheumatology. 2005;44:274–9. doi: 10.1093/rheumatology/keh487. [DOI] [PubMed] [Google Scholar]
- 51.Mayes MD. Endothelin and endothelin receptor antagonists in systemic rheumatic disease. Arthritis Rheum. 2003;48:1190–9. doi: 10.1002/art.10895. [DOI] [PubMed] [Google Scholar]
- 52.Parodi PC, Riberti C, Draganic Stinco D, et al. Squamous cell carcinoma arising in a patient with long-standing pansclerotic morphea. Br J Dermatol. 2001;2:417–9. doi: 10.1046/j.1365-2133.2001.04041.x. [DOI] [PubMed] [Google Scholar]
- 53.Martini G, Murray KJ, Howell KJ, et al. Juvenile-onset localized scleroderma activity detection by infrared thermography. Rheumatology (Oxford) 2002;41:1178–82. doi: 10.1093/rheumatology/41.10.1178. [DOI] [PubMed] [Google Scholar]
- 54.Wiebel L, Howell KJ, Visentin MT, et al. Laser Doppler flowmetry for assessing localized scleroderma in children. Arthritis Rheumol. 2007;56:3489–95. doi: 10.1002/art.22920. [DOI] [PubMed] [Google Scholar]
- 55.Denton CP, Black CM. Scleroderma-clinical and pathological advances. Bes Prac Res Clin Rheumatol. 2004;18:271–90. doi: 10.1016/j.berh.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Steen VD. The many faces of scleroderma. Rheum Dis Clin N Am. 2008;34:1–15. doi: 10.1016/j.rdc.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 57.Okano Y, Steen VD, Jr, Medsger TA, et al. Autoantibody reactive with RNA polymerase III in systemic sclerosis. Ann Intern Med. 1993;119:1005–13. doi: 10.7326/0003-4819-119-10-199311150-00007. [DOI] [PubMed] [Google Scholar]
- 58.Santiago M, Baron M, Hudson M, et al. Antibodies to RNA Polymerase III in systemic sclerosis detected by ELISA. J Rheumatol. 2007;34:1528–34. [PubMed] [Google Scholar]
- 59.Steen VD, Conte C, Owens GR, et al. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994;37:1283–9. doi: 10.1002/art.1780370903. [DOI] [PubMed] [Google Scholar]
- 60.Gomer RH. Circulating progenitor cells and scleroderma. Curr Rheumatol Rep. 2008;10:183–8. doi: 10.1007/s11926-008-0031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Distler J, Distler O. Novel treatment approaches in scleroderma. Rheum Dis Clin N Am. 2008;34:145–59. doi: 10.1016/j.rdc.2007.12.003. [DOI] [PubMed] [Google Scholar]