

Abstract
The fatty acid ethanolamides are a class of signaling lipids that include agonists at cannabinoid and type-α peroxisome proliferator-activated receptors. In the brain, these compounds are primarily hydrolyzed by the intracellular serine enzyme fatty acid amide hydrolase (FAAH). O-aryl carbamate FAAH inhibitors such as URB597 are being evaluated clinically for the treatment of pain and anxiety, but interactions with carboxylesterases in liver might limit their usefulness. Here we explore two strategies aimed at overcoming this limitation. Lipophilic N-terminal substitutions, which enhance FAAH recognition, yield potent inhibitors but render such compounds both susceptible to attack by broad-spectrum hydrolases and inactive in vivo. By contrast, polar electron-donating O-aryl substituents, which decrease carbamate reactivity, yield compounds, such as URB694, that are highly potent FAAH inhibitors in vivo and less reactive with off-target carboxylesterases. The results suggest that an approach balancing inhibitor reactivity with target recognition produces FAAH inhibitors that display significantly improved drug-likeness.
Keywords: Enzymes, hydrolases, carbamate, FAAH inhibitor, carboxylesterase
Introduction
Fatty acid amide hydrolase (FAAH)[1–4] is a membrane-bound serine enzyme that catalyzes the intracellular hydrolysis of the fatty acid ethanolamide family of signaling lipids, which includes endogenous agonists at cannabinoid CB1 and CB2 receptors[5,6], and type-α peroxisome proliferator-activated receptors[7–9]. Natural FAAH substrates such as the endocannabinoid anandamide[5,6] play important physiological roles, and deficits in the functions of these biomolecules have been implicated in a broad range of disease conditions[10]. Consequently, inhibition of FAAH activity, which is expected to reduce such deficits, has become the focus of intense drug discovery efforts. Among the various classes of FAAH inhibitors identified so far[10–12], two have demonstrated favorable activity and selectivity profiles in vivo: the O-aryl carbamates discovered in our laboratories[13–15] and the piperidine/piperazine ureas developed at Pfizer Inc.[16–17]. The former class comprises N-cyclohexylcarbamic acid O-aryl ester derivatives such as URB524 and URB597 (Figure 1), which feature a unique combination of anxiolytic, antidepressant and analgesic properties[18–21], accompanied by a lack of addiction liability in rodent and primate models[19,21].
Figure 1.

Chemical structures of URB524 (1a) and URB597 (1d).
The mechanism by which O-aryl carbamates inhibit FAAH has been investigated in detail. Quantitative structure-activity relationship (QSAR) and computational studies, aided by the resolution of FAAH’s crystal structure[23], have suggested that these molecules bind to FAAH with their N-terminal moiety positioned within the acyl chain binding (ACB) channel of the enzyme and their O-biphenyl group occupying a large fraction of its cytosolic access (CA) channel[15,24,25]. This orientation allows the catalytic nucleophile, serine 241 (ser241), to attack the compound’s carbonyl group to yield a carbamoylated, inactive enzyme (Figure 2). Consistent with this model, QSAR analyses have shown that aromatically restrained lipophilic substituents with a bent shape, such as a meta-oriented O-biphenyl group, optimally conform to the stereoelectronic requirements of the CA channel[13], while hydrophilic substituents on the distal phenyl ring, such as the 3′-carbamoyl group of URB597, enhance enzyme-inhibitor interactions by forming hydrogen bonds with specific amino acid residues of the same domain[14] (Figure 2). It appears therefore that a combination of covalent binding to catalytic ser241 and noncovalent interactions with specific recognition elements in the active site of FAAH is necessary to achieve high inhibitory potency in this class of compounds. Surprisingly, however, no clear correlation has yet been found between the chemical reactivity of the carbamate group in these molecules and their rank order potency for FAAH inhibition[26]. A plausible explanation for this finding is provided by the unique electronic properties of the serine-serine-lysine catalytic triad of FAAH, which allow ser241 to interact productively with mechanism-based inhibitors of relatively low chemical reactivity, such as the piperidine/piperazine ureas[16,17].
Figure 2.

Representation of URB597 docked into the active site of FAAH. URB597 is oriented with its O-biphenyl moiety positioned within the cytoplasmic access (CA) channel and its N-cyclohexyl group occupying the proximal portion of the acyl-chain binding (ACB) domain of the enzyme.
URB597, the index compound in the carbamic acid O-aryl ester class of FAAH inhibitors, displays remarkable target selectivity for FAAH both in vitro and in vivo[27–29]. Nevertheless, at concentrations that exceed those needed to fully block FAAH activity, the compound was shown to interact with several broad-spectrum esterases in liver[16,30]. This inhibition may affect the pharmacokinetic behavior of a variety of therapeutic agents that contain ester or amide bonds, and are substrates for liver carboxylesterases[31]. Are these off-target effects inherently linked to the presence of a carbamate moiety, or is it possible to reduce their occurrence without diminishing inhibitory potency? We reported previously that in a set of cyclohexylcarbamic acid aryl esters the electrophilicity of the carbamate influenced its chemical stability and that introduction of small electron donor substituents at conjugated positions of the O-aryl moiety increased overall hydrolytic stability of the carbamate group towards in vitro chemical and enzymatic hydrolysis[32]. In the present study, we have explored two strategies aimed at addressing the above question. The first was to maximize the inhibitors’ complementarity for FAAH by enhancing interactions with unoccupied regions of its active site (Figure 2). The second, similar to what was previously reported,[32] consisted in modulating the reactivity of the carbamate group with the objective of lowering its sensitivity to nucleophilic attack by hydrolases. We selected a focused set of O-aryl carbamates, most of which were previously published[13–15,26,32], that allowed us to test each of these strategies, and investigated the pharmacodynamic and pharmacokinetic properties of these compounds both in vitro and in vivo.
Results
Compounds 1a (URB524)[13], 1b[15], 1c[15], 1d (URB597)[14], 1g (URB694)[26], 1h[26] and 1i[26] were previously published and were synthesized as reported. 4-Phenylbutylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (1e) and 8-phenyloctylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (1f) were obtained by addition of 3′-carbamoylbiphenyl-3-ol to 4-phenylbutylisocyanate (for 1e) or 8-phenyloctylisocyanate (for 1e). 4-Phenylbutylisocyanate was commercially available while 8-phenyloctylisocyanate was synthesized as reported[15].
Effect of lipophilic N-substitutions on inhibitory potency
Previous studies have shown that lipophilic substitutions to the N-fragment of O-aryl carbamate FAAH inhibitors, which likely enhance the complementarity of these molecules for hydrophobic surfaces in the ACB channel, increase their inhibitory potency in vitro[13,24,33]. Consistent with those findings, we found that introducing flexible and hydrophobic N-substituents on the scaffold of URB524 produced compounds that were highly potent at inhibiting FAAH activity in rat brain membranes (Table 1). For example, the N-phenyloctyl derivative 1c inhibited FAAH activity with a median inhibitory concentration (IC50) that was approximately 5 times more potent than parent compound URB524 (Table 1). A similar, albeit slightly less marked, potency shift was also observed with the N-phenylbutyl derivative 1b. Moreover, lipophilic N-substituents produced further effects on potency when combined with small polar substitutions on the distal phenyl ring, as documented by the 3′-carbamoyl derivative 1f, which inhibited FAAH activity with an in vitro potency that was approximately 30-fold higher than that of the index compound, URB597 (Table 1 Figure 3A). Compound 1e did not substantially differ from 1f (Table 1) and was not further examined.
Table 1.
In vitro and in vivo potency and stability of O-aryl carbamate FAAH inhibitors
| Cpd. | Formula | IC50 (nM) | pH 7.4 (%)[a] | Rat Plasma t1/2 (min.) | ID50 (mg-kg−1) |
|---|---|---|---|---|---|
| 1a (URB524) |  |
25.6 ± 2.0 | 41.1 ± 1.1 | 42.7 ± 3.7 | 0.81 |
| 1b | 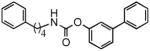 |
9.4 ± 2.8 | 35.9 ± 2.7 | 6.6 ± 0.7 | 0.96 |
| 1c | 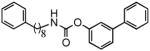 |
5.4 ± 0.5 | 65.9 ± 3.1 | 7.0 ± 0.1 | 105%[b] |
| 1d (URB597) |  |
7.7 ± 1.5 | 40.2 ± 0.6 | 33.0 ± 0.5 | 0.22 |
| 1e |  |
0.33 ± 0.03 | N.D. | N.D. | N.D. |
| 1f | 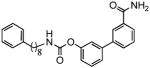 |
0.26 ± 0.02 | 42.8 ± 1.4 | 4.0 ± 0.1 | 65%b |
| 1g (URB694) |  |
30.0 ± 5.8 | 78.0 ± 2.5 | 950 ± 27 | 0.16 |
| 1h | 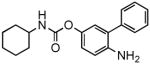 |
27.2 ± 4.8 | 89.0 ± 0.5 | 784 ± 28 | 0.18 |
| 1i | 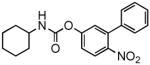 |
> 30,000 | 1.7 ± 0.1[c] | N.D. | N.D. |
Values expressed as percent remaining after 24 h incubation at 37° Celsius.
Percent FAAH activity remaining at 1 mg-kg−1.
Value expressed as t1/2 (min).
Figure 3.

Pharmacodynamic and pharmacokinetic profiles of compounds URB597, 1f and 1c. (a) Effects of URB597 (squares) and 1f (circles) on rat brain FAAH activity in vitro. Results are expressed as the mean ± SEM of 3–5 experiments, each performed in triplicate. (b) Brain tissue and serum (inset) concentrations of URB597 and 1f over a 2 h time-course following a single administration in mice (1 mg-kg−1 i.p.). (c) Time-course of brain FAAH inhibition by URB597 and 1f measured ex vivo after a single drug injection in mice (1 mg-kg−1 i.p.). **P < 0.01 vs. 0 time, ANOVA, followed by Student-Newman-Keuls post-test. (d) Dose-dependent inhibition of brain FAAH activity by administration of URB597, 1f and 1c (triangles) in mice (i.p). FAAH activity was measured ex vivo as in panel (c).
Effect of N-substitutions on metabolic stability
N-Substituted derivatives of URB524 and URB597, such as compounds 1b, 1c, 1e, and 1f are very potent FAAH inhibitors, but are much less stable than their parent molecules when incubated in rat plasma in vitro (Table 1). For example, the N-phenylbutyl derivative 1b displayed a median life-time (t1/2) in plasma that was approximately 6-fold shorter than that of URB524 (Table 1). A comparable loss in metabolic stability was observed with compound 1c (Table 1). This instability likely resulted from increased sensitivity to hydrolysis by plasma esterases, because both 1b and 1c were as stable as parent URB524 in aqueous buffer at neutral pH (Table 1).
Effects of N-substituted inhibitors in vivo
The metabolic lability of O-aryl carbamates with hydrophobic N-substituents suggests that the effectiveness of these compounds in vivo might also be suboptimal. To test this idea, we examined the pharmacokinetic and pharmacodynamic properties of the most potent inhibitor in the series, 1f, comparing them to those of URB597. Administration of a maximally efficacious dose of URB597 to mice (1 mg-kg−1, intraperitoneal, i.p.)[34] was followed by a rapid distribution of the drug in serum and brain tissue (Figure 3B). URB597 and 1f were measured by liquid chromatography/mass spectrometry (LC/MS), using the structural analog N-cyclohexyl biphenyl-3-ylacetamide as an internal standard. The limit of quantification in the assays was 0.4 pmol/sample. Brain concentrations of URB597 reached maximal levels (Cmax) approximately 15 min following administration and rapidly declined afterwards (Figure 3B). Consistent with this time-course of distribution, the drug produced a complete blockade of brain FAAH activity, measured by an ex vivo assay, within 10–15 min of injection (Figure 3C). Finally, URB597 inhibited mouse brain FAAH with a median effective dose (ID50) (Table 1, Figure 3D) comparable to the ID50 value previously reported in rats (0.15 mg-kg−1)[18]. In contrast with the results obtained with URB597, administration of compound 1f produced little or no increase in drug levels in serum and brain (Figure 3B) or inhibition of brain FAAH activity (Figure 3C and 3D). A similar dissociation between in vitro and in vivo inhibitory potencies was observed with compounds 1b and 1c (Table 1). The latter, in particular, was approximately 5 times more potent than parent URB524 in vitro (Table 1), yet was completely inactive in vivo when injected at doses as high as 1 mg-kg−1 (Figure 3D).
The results outlined above confirm prior observations suggesting that lipophilic phenylbutyl or phenyloctyl N-substituents, which enhance hydrophobic interactions with the ACB pocket of FAAH, improve the potency of O-aryl carbamate inhibitors in vitro[15]. The findings also indicate, however, that the same structural modifications markedly reduce the compounds’ stability thus impairing their ability to reach effective concentrations in vivo.
Modulation of carbamate reactivity increases metabolic stability in vitro
We hypothesized that reducing the reactivity of the carbonyl carbon would yield compounds that might escape nucleophilic attack by broad-spectrum esterases in plasma and liver while retaining the ability to interact with FAAH. Consistent with this prediction, insertion of an electron-donating substituent, such as a hydroxyl or amino group, in the para position of the proximal phenyl ring of URB524 did not significantly affect inhibitory potency in vitro (1g and 1h, Table 1 and Figure 4A). The modifications caused, however, a marked increase in the compounds’ stability in plasma, in comparison to other molecules in the series (Table 1). For example, compound 1g (URB694) displayed an average t1/2 in plasma that was approximately 29-fold greater than that of URB597 (Table 1). By contrast, para substitution with the strong electron-withdrawing nitro group (1i) negatively impacted potency, likely due to the very low chemical stability of the compound in water (Table 1).
Figure 4.

Pharmacodynamic and pharmacokinetic profile of URB694. (a) Effects of URB524 (squares) and URB694 (circles) on rat brain FAAH activity in vitro. Results are expressed as the mean ± SEM of 3 experiments, each performed in triplicate. (b) Brain tissue concentrations of URB524 and URB694 over a 2 h time-course following a single drug injection in mice (1 mg-kg−1 i.p.). (c) Time-course of brain FAAH inhibition by URB524 and URB694 measured ex vivo 1 h after a single drug injection in mice (1 mg-kg −1 i.p.). *P < 0.05 vs. 0 time, **P < 0.01 vs. 0 time, ***P < 0.001 vs. 0 time, ANOVA, followed by Student-Newman-Keuls post-test. (d) Dose-dependent inhibition of FAAH activity by systemic administration of URB524 and URB694 in mice. FAAH activity was measured ex vivo as in panel (c).
Modulation of carbamate reactivity improves activity in vivo
The enhanced stability of URB694 in vitro was accompanied by an improved distribution of the inhibitor in vivo. Thus, after a single systemic administration (1 mg-kg−1, i.p.), URB694 reached a Cmax in brain tissue that was 6-fold and 2-fold higher than those achieved by URB524 and URB597, respectively (Cmax in pmol/g: URB524, 74 ± 3; URB597, 226 ± 29; URB694, 466 ± 96; n = 3 per timepoint) (Figure 4B). Additionally, time-course experiments revealed that the area under the curve (AUC) for URB694 in the brain, a measure of tissue exposure to the drug, was substantially greater in comparison with URB524 or URB597 (AUC in arbitrary units: URB524, 2589 ± 423; URB597, 6036 ± 899; URB694, 30314 ± 4374; n = 3) (Figure 4B). As expected, increased drug exposure resulted in a marked enhancement of inhibitory potency in vivo (Table 1, Figure 4D). Importantly, a similar effect on in vivo potency was observed with the p-amino derivative 1h (Table 1).
Effects on liver carboxylesterases
It has been suggested that O-aryl carbamate FAAH inhibitors react with several broad-spectrum carboxylesterases in liver[16,30]. We reasoned that lowering the reactivity of these compounds might not only reduce their interaction with plasma hydrolases, but also limit their interactions with liver esterases. Consistent with this prediction, URB694 and compound 1h had reduced effects on liver carboxylesterase activities in rat microsome preparations, compared to those exerted by URB597 (Figure 5). Together, the results suggest that appropriate modulation of reactivity in a series of O-aryl carbamate FAAH inhibitors yields new pharmacological agents endowed with markedly improved stability and selectivity for FAAH.
Figure 5.

Effect of FAAH inhibitors on liver carboxylesterase activity. In vitro carboxylesterase activities were measured in rat liver microsomes in the presence of URB597 (squares), URB694 (circles) and 1h (triangles). Results, in percent of control activity measured in the presence of vehicle, are expressed as the mean ± SEM of 6 experiments, **P < 0.01 URB694 or 1h vs URB597, ***P < 0.001 URB694 or 1h vs URB597, ANOVA, followed by Student-Newman-Keuls post-test.
Discussion
Clinical evidence indicates that Δ9-tetrahydrocannabinol (Δ9-THC), the active constituent of cannabis, alleviates neuropathic pain[35,36], improves muscle spasticity in multiple sclerosis[37] and reduces chemotherapy-induced nausea[38]. Because the psychotropic effects of this drug limit its therapeutic usefulness, an alternative approach might be to develop agents that amplify endocannabinoid signaling in the brain and other tissues. Animal studies indicate indeed that inhibitors of FAAH-mediated anandamide degradation are potent at relieving symptoms of pain, anxiety, depression and nausea[18–21]. Additionally, evidence suggests that FAAH inhibitors lack reinforcing properties in rodent and primate models[19,22], a therapeutic advantage that distinguishes this class of drugs from direct-acting cannabinoid agonists such as Δ9-THC.
Although current FAAH inhibitors incorporate chemical scaffolds of diverse structures, carbamate-based compounds remain important for three reasons. First, a number of potent and selective carbamate FAAH inhibitors have been reported in the scientific and patent literature[10–12]. Second, such inhibitors – and particularly O-aryl carbamates such as URB597 – are widely used experimentally and cross-validated data on their pharmacological actions are now becoming available. Finally, preclinical studies show that URB597 has favorable target selectivity and safety profiles, providing an early indication for the ‘druggability’ of this class of agents[27,29]. Despite these favorable properties, reports suggest that URB597 may interact with carboxylesterases in liver and possibly other tissues[16,30]. In the present study, we evaluated two strategies to improve the drug-likeness of O-aryl carbamate inhibitors while reducing their untoward effects. The first strategy was directed at enhancing inhibitor recognition by FAAH. We reasoned that adding flexible lipophilic groups onto the N-portion of the compounds might increase their interactions with FAAH enhancing target specificity and inhibitory potency in vivo. The second strategy was aimed at modulating the reactivity of the carbamate group through substitution of small polar groups on the para position of the proximal phenyl ring. We hypothesized that these changes would increase the compounds’ stability without significantly affecting their ability to inhibit FAAH.
The results reported in the present study show that an approach aimed at enhancing the interactions of O-aryl carbamate FAAH inhibitors with FAAH may be less effective than one directed at balancing inhibitor recognition with lower carbamate reactivity. We found that analogs of the compound URB524, which contained lipophilic phenylalkyl substituents (compounds 1b and 1c), were more potent in vitro, but considerably less stable in rat plasma. This lack of metabolic stability resulted in a loss of activity in vivo (Table 1). For example, compound 1f was 27 times more potent than URB597 at inhibiting FAAH in vitro yet it failed to reach the brain and inhibit FAAH in vivo. Based on these results, we conclude that addition of flexible lipophilic N-substituents to the O-aryl carbamate scaffold enhances FAAH inhibitory potency in vitro, but creates molecules that are more susceptible to metabolism in plasma and less pharmacologically active in vivo.
Our second approach was aimed at managing the high reactivity inherent to the carbamate group, a property that likely underlies the off-target interactions of URB597. We have previously shown that small electron-donating substitutions at the para position of the proximal phenyl ring of URB524, which are expected to reduce the partial positive charge on the carbonyl group, yield inhibitors of significant in vitro potency[26]. Two factors might explain this result. First, the strong nucleophilic nature of catalytic ser241 may allow FAAH to react with substrates and inhibitors of widely different electrophilicity[39]. Second, modeling studies suggest that hydrophilic substituents of appropriate size are sterically tolerated within the CA channel, where they may engage in favorable electrostatic interactions with neighboring amino-acid residues[26].
To modulate the reactivity of the carbamate group we introduced hydroxyl (URB694) or amino (compound 1h) groups onto the para position of the proximal phenyl ring of URB524. We expected that these electron-donating substitutions would increase the electron density around the carbonyl carbon of the molecules thereby reducing their electrophilicity and increasing their chemical and metabolic stability. Indeed, URB694 and 1h had in vitro potencies that were similar to that of URB524, but were more stable than URB524 both in neutral buffer and rat plasma (Table 1). Accordingly, URB694 achieved superior brain tissue penetration and was 5-fold more potent in vitro than parent compound URB524 (Figure 4). Finally, URB694 and 1h were significantly less reactive than URB597 toward liver carboxylesterases (Figure 5).
In conclusion, the O-aryl carbamate URB694 demonstrated greater plasma stability, prolonged life-span in vivo, and decreased activity toward liver carboxylesterases in comparison to URB597. Thus, URB694 might complement URB597 as an experimental tool and offer a starting point for new FAAH inhibitors with improved drug-likeness. More generally, our findings highlight the flexibility of carbamate-based agents as enzyme inhibitors, and underscore the value of integrating target recognition and reactivity to achieve optimal effectiveness with this class of agents.
Experimental Section
Chemistry
Anandamide was prepared as previously described[40]. [3H]-Anandamide was purchased from American Radiolabeled Chemicals (St. Louis, MO). Other compounds were synthesized as outlined below. All reagents were purchased from Sigma-Aldrich or Lancaster in the highest quality commercially available. Solvents were RP grade. Chromatographic separations were performed on silica gel columns by flash chromatography (Kieselgel 60, 0.040–0.063 mm, Merck). TLC analyses were performed on precoated silica gel on aluminum sheets (Kieselgel 60 F254, Merck). Melting points were determined on a Büchi SMP-510 capillary melting point apparatus. EI-MS analyses (70 eV) were recorded with a Fisons Trio 1000 spectrometer; only molecular ions (M+) and base peaks are given. 1H NMR and 13C NMR spectra were recorded on a Bruker AC 200 or 50, respectively, spectrometer and analyzed using the WIN-NMR software package; chemical shifts were measured by using the central peak of the solvent. IR spectra were obtained on a Nicolet Avatar 360 spectrometer. Elemental analyses were performed on a Carlo Erba analyzer.
Synthesis of alkylcarbamic acid biphenyl-3-yl esters 1e and 1f: The opportune isocyanate (2.2 mmol) and Et3N (12 mg, 0.017 mL, 0.12 mmol) were added to a stirred solution of 3′-carbamoylbiphenyl-3-ol (426 mg, 2 mmol) in toluene (10 mL). After the reactants were refluxed [24 h in the case of 1f; 5 h, then 3 h after the addition of a further amount of 4-phenylbutylisocyanate (771 mg, 4.4 mmol) in the case of 1e], the mixture was cooled and concentrated. Purification of the residue by column chromatography (cyclohexane:EtOAc, 3:7) and recrystallization gave the desired compounds.
4-Phenylbutylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (1e)
white crystals (691 mg, 89%); mp: 168–170 °C (EtOH); 1H NMR (200 MHz, CDCl3): δ = 1.57–1.81 (m, 4H), 2.64–2.71 (t, 2H), 3.27–3.36 (q, 2H), 5.09 (br t, 1H), 5.76 (s, 1H), 6.30 (s, 1H), 7.09–7.55 (m, 10H), 7.72–7.81 (m, 2H), 8.02 (m, 1H) ppm; 13C NMR (50 MHz, CDCl3): δ = 28.5, 29.4, 35.5, 41.2, 120.5, 121.0, 124.1, 125.9, 126.2, 126.5, 128.4 (2xC), 129.1, 129.7, 130.6, 134.0, 140.7, 141.6, 142.0, 151.5, 154.7, 169.5 ppm; IR (Nujol): ν = 3349, 3156, 1712 cm−1; MS (EI): m/z 388 (M+), 91 (100); Anal. calcd for C24H24N2O3: C 74.21, H 6.23, N 7.21, found: C 74.42, H 6.19, N 7.09.
8-Phenyloctylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (1f)
white crystals (196 mg, 22%) (calculated from the corresponding carboxylic acid derivative); mp: 140–141 °C (EtOH); 1H NMR (200 MHz, CDCl3): δ = 1.26–1.74 (m, 12H), 2.57–2.65 (t, 2H), 3.23–3.33 (q, 2H), 5.08 (br t, 1H), 5.76 (s, 1H), 6.30 (s, 1H), 7.12–7.55 (m, 10H), 7.73–7.80 (m, 2H), 8.02 (m, 1H) ppm; 13C NMR (50 MHz, CDCl3): δ = 26.8, 29.2 (2xC), 29.4, 29.8, 31.5, 36.0, 41.3, 120.6, 121.0, 124.0, 125.6, 126.2, 126.5, 128.2, 128.4, 129.1, 129.7, 130.6, 134.0, 140.8, 141.5, 142.8, 151.5, 154.6, 169.4 ppm; IR (Nujol): ν = 3354, 3186, 1712 cm−1; MS (EI): m/z 444 (M+), 91 (100); Anal. calcd for C28H32N2O3: C 75.65, H 7.26, N 6.30, found: C 75.83, H 7.10, N 6.26.
Cyclohexylcarbamic acid 6-aminobiphenyl-3-yl ester (1h)
white-coloured crystals; mp: 126 °C (EtOH); 13C NMR (50 MHz, CDCl3): δ = 24.7, 25.5, 33.3, 50.1, 108.7, 111.8, 125.0, 127.2, 128.8, 129.1, 131.1, 138.9, 144.2, 151.3, 153.7 ppm; 1H NMR, IR and MS (EI) spectra are according to the literature[26].
Cyclohexylcarbamic acid 6-nitrobiphenyl-3-yl ester (1i)
off-white needles; mp: 132–134 °C (EtOH). 13C NMR (50 MHz, CDCl3): δ = 24.7, 25.4, 33.2, 50.4, 117.6, 125.7, 128.0, 128.2, 128.7, 132.5, 133.1, 137.0, 149.1, 150.3, 152.6 ppm; 1H NMR, IR and MS (EI) spectra are according to the literature[26].
Animals
Male Swiss Webster mice (20–35 g) and male Wistar rats (250–300 g) were group-housed in standard cages at room temperature on a 12 h light/dark cycle. Water and standard chow pellets were available ad libitum. Experiments at the University of California, Irvine (UCI), were performed in an AAALAC-accredited facility, met the National Institutes of Health guidelines for the care and use of laboratory animals, and were approved by the Institutional Animal Care and Use Committee of UCI. Experiments performed at the University of Parma were in compliance with the European Community Council Directive 86 (609) EEC, and the experimental protocol was carried out in compliance with Italian regulations (DL 116/92) and with local ethical committee guidelines for animal research.
Brain Homogenate and Membrane Preparation
Animals were anesthetized with isoflurane and decapitated. The brains were removed and immediately frozen in liquid N2. For ex vivo determination of FAAH activity, frozen brains were thawed and homogenized in 10 vol of ice-cold Tris buffer (50 mM, pH 7.5) containing 0.32 M sucrose. The homogenates were centrifuged at 1000 x g for 10 min at 4 °C, and FAAH activity was measured as described below. For in vitro FAAH activity assays, the supernatants from the 1000 x g spin were further centrifuged at 27,000 x g for 30 min at 4 °C. Pellets were suspended in 20% of the original homogenization volume in Tris buffer (50 mM, pH 7.5). Homogenate and membrane protein concentrations were determined with the BCA protein assay kit (Thermo Scientific, Rockford, Il).
Stability Measurements in Buffer and Plasma
Compounds (final concentration: 1 μM; 1% dimethylsulfoxide, DMSO) were incubated at 37 °C in 10 mM phosphate buffered saline (PBS) at pH 7.4. Samples were withdrawn after various time intervals, and injected into a Shimadzu LC system (Shimadzu Europe, Duisburg, Germany) for quantification. Calibration curves were built for each compound by spiking PBS with known amounts of compounds. For plasma stability experiments, whole blood was drawn from rats via cardiac puncture and transferred to heparinized tubes. The plasma was separated by centrifugation at 1,900 x g and diluted to 80% (vol/vol) with PBS (100 mM, pH 7.4) to buffer the pH. Test compounds were dissolved in DMSO at a concentration of 100 μM. 5 μL of the stock solution were added to 0.5 mL of pooled rat plasma in PBS at pH 7.4. Samples were allowed to incubate at 37 °C in a shaking water bath and, at various time points, 50 μL samples were withdrawn, added to two volumes of ice-cold acetonitrile and centrifuged at 8000 x g for 5 min at 4 °C. The supernatants (20 μL) were fractionated on a reversed-phase Supelcosil LC-18-DB, 5 μm, 150 × 4.6 mm column (Supelco, Bellefonte, PA) with a linear gradient of acetonitrile/0.1% formic acid in water at a flow rate of 1 ml-min−1. Detection was accomplished at the absorbance maximum of each compound with a Shimadzu SPD-10A UV-VIS detector. Calibration curves were built for each compound by spiking rat plasma with known amounts of compounds in the tested concentration range. Apparent half-life times (t1/2) for the disappearance of test compounds were calculated from the pseudo first-order rate constants obtained by linear regression of the log compound concentration versus time plots.
FAAH Activity
Rat brain membranes and mouse brain homogenates were used in the in vitro and ex vivo experiments, respectively. FAAH activity was measured at 37°C for 30 min in 0.5 mL Tris buffer (50 mM, pH 7.5) containing fatty acid-free bovine serum albumin (BSA) (0.05%, weight/vol), 50 μg of protein from total brain homogenates or membranes, 10 μM anandamide and [3H]-anandamide (10,000 cpm, specific activity 60 Ci/mmol). The reactions were stopped with 1 mL chloroform/methanol (1:1) and centrifugation at 2,000 x g for 10 min at 4 °C. Radioactivity in the aqueous layer was measured by liquid scintillation counting. For in vitro experiments, the drugs were dissolved in DMSO and assayed in the reaction without pre- incubation. Final DMSO concentration was 1%.
Carboxylesterase Activity
Rat liver microsomes (BD Biosciences, Bedford, MA) were used as a source of liver carboxylesterases. Inhibition assays were conducted as reported elsewhere[41] with minor modifications. Briefly, 2.5 μg of liver microsomes were incubated with different concentrations of test compounds (30, 100 and 300 nM) for 30 min at room temperature. Substrate p-nitrophenylacetate (Sigma, Gallarate, Italy) was added to a final concentration of 1 mM. 20 min after substrate addition, UV absorbance was measured at 405 nm for the appearance of the p-nitrophenol product.
Sample preparation for LC/MS
Brain
Mice were sacrificed with isoflurane and tissues were collected and immediately frozen in liquid N2. Frozen tissues were weighed and homogenized in methanol (1 mL) containing N- cyclohexyl biphenyl-3-ylacetamide as internal standard. Drugs were extracted from tissue with chloroform (2 vol) and washed with water (1 vol). Organic phases were collected and dried under N2. The pellet was resuspended in 50 μL methanol for LC/MS analysis.
Serum
Trunk blood was collected from decapitated animals, allowed to clot and placed on ice. The clotted blood was immediately centrifuged at 18,000 x g for 10 min at 4 °C and the serum was transferred to glass vials and brought up to 1 mL with water. Proteins were precipitated with ice-cold acetone (1 mL) containing N-cyclohexyl biphenyl-3-ylacetamide as an internal standard, and the precipitate was removed by centrifugation at 3000 x g for 10 min at 4 °C. The samples were dried under N2 to remove the acetone and extracted with chloroform/methanol as described above.
LC/MS analysis
Tissue levels of compounds 1a, 1d, 1f, and 1g were measured using an 1100-LC system coupled to a 1946A-MS detector (Agilent Technologies, Inc., Palo Alto, CA) equipped with an electrospray ionization interface. Drugs were separated using a XDB Eclipse C18 column (50 X 4.6-mm inner diameter, 1.8 μm, Zorbax). 1a, 1d, and 1g were eluted using a linear gradient of 60% to 100% of A in B over 3 min at a flow rate of 1.0 mL-min−1. Mobile phase A consisted of methanol containing 0.25% acetic acid, 5 mM ammonium acetate; mobile phase B consisted of water containing 0.25% acetic acid, 5 mM ammonium acetate. 1f was eluted with a gradient of methanol in water (from 85% to 90% methanol in 2.5 min) at a flow rate of 0.5 mL-min−1. Column temperature was kept at 40 °C. MS detection was in the positive ionization mode, capillary voltage was set at 3 kV, and fragmentor voltage was varied from 120 to 140 V. N2 was used as drying gas at a flow rate of 13 L-min−1 and a temperature of 350 °C. Nebulizer pressure was set at 60 psi. Quantitations were conducted using N-cyclohexyl biphenyl-3-ylacetamide (m/z = 294) as an internal standard. The protonated ions ([M+H+]) for 1a, 1g, and the Na+ adduct ([M+Na+]) for 1d and 1f were monitored in the selective ion-monitoring mode. Limit of quantification was 0.4 pmol.
Statistical Analyses
Results are expressed as the mean ± SEM. Statistical significance was evaluated using one-way ANOVA followed by Student-Newman-Keuls post hoc test. P ≤ 0.05 was considered significant (SigmaStat v. 3.2).
Acknowledgments
This work was supported by the National Institute on Drug Abuse (DA012413) and by the Italian Ministry of Instruction, University and Research (2005032713_002) and University of Urbino “Carlo Bo”. DP, AD, AT, GT, and MM are inventors in a patent disclosing FAAH inhibitors, owned by the University of California, Irvine, the University of Urbino, ‘Carlo Bo’ Italy, and the University of Parma, Italy. The patent was licensed to Organon Biosciences, now a unit of Schering-Plough.
References
- 1.Désarnaud F, Cadas H, Piomelli D. J Biol Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 2.Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. J Biol Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- 3.Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Biochim Biophys Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- 4.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 5.Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 6.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 7.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez de Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 8.Astarita G, Di Giacomo B, Gaetani S, Oveisi F, Compton TR, Rivara S, Tarzia G, Mor M, Piomelli D. J Pharmacol Exp Ther. 2006;318:563–570. doi: 10.1124/jpet.106.105221. [DOI] [PubMed] [Google Scholar]
- 9.LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 10.Di Marzo V. Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 11.Minkkilä A, Saario SM, Käsnänen H, Leppänen J, Poso A, Nevalainen T. J Med Chem. 2008;51:7057–7060. doi: 10.1021/jm801051t. [DOI] [PubMed] [Google Scholar]
- 12.Seierstad M, Breitenbucher JG. J Med Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 13.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 14.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 15.Mor M, Lodola A, Rivara S, Vacondio F, Duranti A, Tontini A, Sanchini S, Piersanti G, Clapper JR, King AR, Tarzia G, Piomelli D. J Med Chem. 2008;51:3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 17.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Becelaere KV, Stevens RC, Cravatt BF. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 19.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2006;103:2465. [Google Scholar]
- 20.Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Biol Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Russo R, LoVerme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 22.Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, Goldberg SR. Biol Psychiatry. 2008;64:930–937. doi: 10.1016/j.biopsych.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 24.Alexander JP, Cravatt BF. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Chem Commun. 2008;38:214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]
- 26.Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]
- 27.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lichtman AH, Leung D, Shelton C, Saghatelian A, Hardouin C, Boger D, Cravatt BF. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- 29.Clapper JR, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Pharmacol Res. 2006;54:341–344. doi: 10.1016/j.phrs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 30.Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, Brioni JD, Surowy CS. Neuropharmacology. 2007;52:1095–1105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Satoh T, Hosokawa M. Annu Rev Pharmacol Toxicol. 1998;38:257–288. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 32.Vacondio F, Silva C, Lodola A, Fioni A, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Mor M, Tarzia G. ChemMedChem. 2009 doi: 10.1002/cmdc.200900120. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minkkilä A, Myllymäki MJ, Saario SM, Castillo-Melendez JA, Koskinen AM, Fowler CJ, Leppänen J, Nevalainen T. Eur J Med Chem. 2009;44:2994–3008. doi: 10.1016/j.ejmech.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 35.Ellis RJ, Toperoff W, Vaida F, van den Brande G, Gonzales J, Gouaux B, Bentley H, Atkinson JH. Neuropsychopharmacology. 2009;34:672–680. doi: 10.1038/npp.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abrams DI, Jay CA, Shade SB, Vizoso H, Reda H, Press S, Kelly ME, Rowbotham MC, Petersen KL. Neurology. 2007;68:515–521. doi: 10.1212/01.wnl.0000253187.66183.9c. [DOI] [PubMed] [Google Scholar]
- 37.Rog DJ, Nurmikko TJ, Friede T, Young CA. Neurology. 2005;65:812–819. doi: 10.1212/01.wnl.0000176753.45410.8b. [DOI] [PubMed] [Google Scholar]
- 38.Davis MP. Exp Opin Investig Drugs. 2008;17:85–95. doi: 10.1517/13543784.17.1.85. [DOI] [PubMed] [Google Scholar]
- 39.McKinney MK, Cravatt BF. J Biol Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]
- 40.Astarita G, Ahmed F, Piomelli D. J Lipid Res. 2008;49:48–57. doi: 10.1194/jlr.M700354-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Wheelock CE, Severson TF, Hammock BD. Chem Res Toxicol. 2001;14:1563–1572. doi: 10.1021/tx015508+. [DOI] [PubMed] [Google Scholar]