

Abstract
Pomegranate from the plant Punica granatum possesses strong antioxidant and anti-inflammatory properties. Recently, we have demonstrated that treatment of normal human epidermal keratinocytes with pomegranate fruit extract (PFE) inhibited ultraviolet B (UVB)-mediated activation of nuclear factor kappa B (NF-κB) and mitogen activated protein kinases (MAPK) pathways. Here, we evaluated the effect of PFE on early biomarkers of photocarcinogenesis employing SKH-1 hairless mice. PFE was provided in drinking water (0.2%, wt/vol) to SKH-1 hairless mice for 14 days before a single UVB (180 mJ/cm2) irradiation. We found that oral feeding of PFE inhibited UVB-induced: (i) skin edema, (ii) hyperplasia, (iii) infiltration of leukocytes, (iv) lipid peroxidation, (v) hydrogen peroxide generation, (vi) ODC activity, and (vii) ODC, COX-2 and PCNA protein expression. Oral feeding of PFE enhanced repair of UVB-mediated formation of cyclobutane pyrimidine dimers (CPDs) and 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG). Importantly, PFE treatment further enhanced UVB-mediated increase in tumor suppressor p53 and cyclin kinase inhibitor p21. Furthermore, oral feeding of PFE inhibited UVB-mediated: (i) nuclear translocation of NF-κB, (ii) activation of IKKα, and (iii) phosphorylation and degradation of IκBα. Taken together, we provide evidence that oral feeding of PFE to mice affords substantial protection from the adverse effects of UVB radiation via modulation in early biomarkers of photocarcinogenesis and provide suggestion for its photochemopreventive potential.
Introduction
Nonmelanoma skin cancer accounts for more than 1 million cases of human malignancies annually in the United States and the incidence continues to rise (1,2). Solar ultraviolet (UV) radiation, particularly its UVB (290–320 nm) component, is the foremost cause of nonmelanoma skin cancers in humans, particularly in Caucasian individuals (2,3). UVB radiation is a complete carcinogen and causes excessive generation of reactive oxygen species (ROS) thus resulting in an oxidative stress in the skin (4,5). Studies have shown that UVB radiation produces a variety of adverse effects that includes DNA damage (6,7), mutations in key regulatory genes (7,8), inflammation (9,10), immunosuppression (11,12), photoaging and skin cancer (2,13). UVB is directly absorbed by cellular DNA leading to the formation of DNA lesions primarily cyclobutane primidine dimers (CPDs) and pyrimidyne-(6-4)-pyrimidone photoproducts (14,15). In contrast, 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG) is induced by ROS and has been proposed as a key biomarker of oxidative DNA damage relevant to carcinogenesis (16,17).
Mechanisms that contribute to UVB-induced mutagenesis and carcinogenesis comprise inactivation of tumor suppressor genes and/or activation of oncogenes (18,19). Cellular responses to DNA-damage by UVB radiation are usually multifaceted and often regulated by more than one transcription factors, for instance by both p53 and nuclear factor kappa B (NF-κB). Studies have shown that DNA damage induced by UVB radiation triggers a rapid increase in p53, enhancing p21 synthesis and shutting off cell replication and DNA synthesis, allowing extended time for either DNA repair or apoptosis induction in the cells carrying UVB-damaged DNA (20,21). In both cases, the risk of UVB induced skin cancer is reduced. Studies have shown that UVB-mediated activation of NF-κB plays an imperative role in inflammation, cell proliferation and skin carcinogenesis (22,23).
Since skin cancer is a significant problem associated with mortality and morbidity intensive efforts are required to develop novel strategies for the prevention of UV responses. There has been substantial interest in the identification of botanical agents capable of affording protection to skin from the adverse effects of solar UVB radiation. These botanical antioxidants for human use should have the ability to ameliorate the adverse biological effects of UV radiation. Pomegranate (Punica granatum L.) fruit widely consumed fresh and in beverage as juice or wine has been extensively used in traditional medicine in various parts of the world. Pomegranate fruit extract (PFE) derived from the tree Punica granatum contains several polyphenols (such as catechins, gallic and ellagic acids) and anthocyanidins (such as delphinidin, cyanidin, and pelargonidin) (24), and its antioxidant activity is superior than that of red wine and green tea (25). Studies have shown that PFE possesses strong anti-inflammatory (24), anti-proliferative (26,27), and anti-tumorigenic properties (24,26). We recently demonstrated that PFE treatment of normal human epidermal keratinocytes resulted in inhibition of UVB-mediated activation of NF-κB and mitogen activated protein kinases (MAPK) pathways (28). The objective of this study was to investigate the effect of oral feeding of PFE on early biomarkers of photocarcinogenesis employing SKH-1 hairless mice.
Materials and Methods
Materials
The mono- and poly-clonal antibodies (PCNA, ODC, p53 and IKKα) were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). CPD antibody was procured from Kamiya Biomedical Company (Seattle, WA). The monoclonal and polyclonal antibodies for COX-2 and p21 were obtained from Cell Signaling Technology (Beverly, MA). The 8-oxodG antibody was purchased from Chemicon International Inc. (Temecula, CA). IκBα and IκBα (Phospo) antibodies were obtained from New England Biolabs (Beverly, MA). NF-κB/p65 antibody was procured from Geneka Biotechnology (Montreal, Canada). Anti-mouse secondary horseradish peroxidase conjugate was obtained from GE Healthcare (Piscataway, NJ). The DC BioRad Protein assay kit was purchased from BioRad Laboratories (Herculus, CA). Novex pre-cast Tris-Glycine gels were obtained from Invitrogen (Carlsbad, CA).
Preparation of pomegranate fruit extract
Fresh fruit of pomegranate was peeled, and its edible portion (seed coat and juice) was squeezed in 70% acetone-30% distilled water (1:20, by w/v). The red extract was filtered through filter paper (Whatman No. 1). The filtrate was condensed, freeze-dried and stored at 4°C (24).
Animals and treatment
Female SKH-1 hairless mice (6-weeks-old) obtained from Charles River Laboratories (Wilmington, MA) were used in this study. After their arrival in the animal facility, the animals were allowed to acclimatize for one week before the start of the experiments. The animals were fed Purina Chow diet and water ad libitum. The mice were maintained at standard conditions: temperature of 24 ± 2°C, relative humidity of 50% ± 10%, and 12 h room light/12 h dark cycle. For UVB irradiation, the mice were housed in specially designed cages where they were held in dividers separated by Plexiglas. Sixty-four female SKH-1 hairless mice, maintained as described, were divided into four groups of twenty four animals each (except in control and PFE alone groups, where 8 animals were used). The mice in the first group received drinking water and served as a control, and those in the second group received PFE (0.2%, wt/vol) dissolved in drinking water. The mice in the third group received a single UVB (180 mJ/cm2) exposure with a custom designed Research Irradiation Unit (Daavlin, Bryan, OH) that consists of a fixture mounted on fixed legs. Mounted within the exposure unit are four UVB lamps and the exposure system is controlled using Daavlin Flex Control Integrating Dosimeters. The UV lamps emit UVB (280–320 nm; ~80% of total energy) and UVA (320–375 nm; ~20% of total energy), with UVC emission being insignificant. In this system dose units can be entered in MilliJoules/cm2 for UVB; variations in energy output are automatically compensated for the delivery of the desired dose. Using this system, the mice were exposed to accurate dosimetery of UVB radiation. The mice in the fourth group received PFE (0.2%, wt/vol) for 14 days before a single UVB (180 mJ/cm2) irradiation and continued till the termination of the experiment. The mice were then sacrificed at 1, 8 and 24 h post UVB exposure and skin biopsies were harvested for biochemical analysis.
Edema and Hyperplasia
To assess the inhibitory effect of oral feeding of PFE on UVB-induced skin edema the mice were exposed to UVB (180 mJ/cm2) and 24 h post UVB exposure bifold-skin thickness was measured. Atleast four determinations were made at different dorsal skin sites per mouse in each group. For the hyperplasia study, skin was removed, fixed in 10% formalin, and embedded in paraffin. Vertical sections (5 μm) were cut, mounted on a glass slide, and stained with hematoxylin and eosin.
Immunohistochemical detection of CPDs and 8-oxodG
The skin biopsies were frozen in optimal cutting temperature (OCT) compound under liquid nitrogen immediately after removal and were stored at −80 °C for further use. Briefly, 5-μm-thick frozen skin sections were thawed and fixed in cold acetone for 10 min and washed in PBS (pH: 7.4). The sections were then kept in 70 mmol/L NaOH in 70% ethanol for 2 min to denature nuclear DNA followed by neutralization for 1 min in 100 mmol/L Tris-HCl (pH 7.5) in 70% ethanol. Non-specific binding sites were blocked by 30 min incubation at room temperature in PBS containing10% goat serum. Sections were then incubated with mouse monoclonal antibodies to 8-oxodG and CPD. For negative control the sections were incubated without primary antibody. After overnight incubation at 4°C and three washes in PBS, the sections were incubated for 30 min with biotinylated rabbit anti-mouse link antibody and then for 30 min with streptavidin conjugated to horse radish peroxidase. After washing, sections were incubated with diaminobenzidine plus peroxidase substrate and counterstained with methyl green.
Preparation of epidermal skin lysate and nuclear lysate
Epidermis from the whole skin was separated as described earlier (24) and was homogenized in ice-cold lysis buffer [50mM Tris-HCl, 150mM NaCl, 1mM EGTA, 1mM EDTA, 20mM NaF, 100mM Na3VO4, 0.5% NP-40, 1% Triton X-100, 1mM PMSF (pH 7.4)] with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III; Calbiochem, La Jolla, CA). The homgenate was then centrifuged at 14,000 x g for 25 min at 4° C and the supernatant (total cell lysate) was collected, aliquoted and stored at −80° C. For the preperation of nuclear lysate, 0.2 g of the epidermis was homogenized into 1.0 ml of ice-cold phosphate-buffered saline (pH 7.6) and centrifuged at 12,000 x g for 5 min at 4°C. The pellet was resuspended in 1 ml of cold buffer containing 10 mM HEPES (pH 7.9), 2 mM MgCl2, 10 mM KCI, 1 mM dithiothreitol, 0.1 mM EDTA and 0.1 mM PMSF. After homogenization in a tight-fitting Dounce homogenizer, the homogenates were left on ice for 10 min then were centrifuged at 25,000 x g for 10 min. The nuclear pellet was resuspended in 0.1 ml of the buffer containing 10 mM HEPES (pH 7.9), 300 mM NaCI, 50 mM KCI, 0.1 mM EDTA, 1 mM dithiothreitol, 0.1 mM PMSF, and 10% glycerol with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III, Calbiochem, La Jolla, CA). The suspension was gently shaken for 20 min at 4°C. After centrifugation at 25000 x g for 10 min, the nuclear extracts (supernatants) were collected and quickly frozen at −80°C. The protein content in the lysates was measured by DC BioRad assay (BioRad Laboratories, Hercules, CA) as per the manufacturer’s protocol.
Western blot analysis
For western analysis, 25–50 μg of the protein was resolved over 8–12% Tris-glycine gels and transferred to a nitrocellulose membrane. The blot containing the transferred protein was blocked in blocking buffer (5% nonfat dry milk, 1% Tween 20; in 20 mM TBS, pH 7.6) for 1h at room temperature followed by incubation with appropriate monoclonal primary antibody in blocking buffer for 2 h to overnight at 4 °C. This was followed by incubation with anti-mouse or anti-rabbit secondary antibody horse-radish peroxidase for 1 h and then washed several times and detected by chemiluminescence and autoradiography using XAR-5 film obtained from Eastman Kodak Co.
Ornithine decarboxylase (ODC) enzyme activity
The epidermis from the dissected skin was separated as described earlier (24) and homogenized at 4°C in a glass-to-glass homogenizer in 10 volumes of ODC buffer [50 mM Tris-HCl buffer (pH 7.5) containing 0.1 mM EDTA, 0.1 mM dithiothreitol, 0.1 mM pyridoxal-5-phosphate, 1mM 2-mercaptoethanol and 0.1% Tween-80]. The homogenate was centrifuged at 100,000 g at 4 °C and the supernatant was used for enzyme determination. ODC enzyme activity was determined in epidermal cytosolic fraction by measuring the release of 14CO2 from the D,L-[14C] ornithine by the method described earlier (24). Briefly, 400 μl of the supernatant was added to 0.95 ml of the assay mixture [35 mM sodium phosphate (pH 7.2), 0.2 mM pyridoxal phosphate, 4mM dithiothreitol, 1 mM EDTA, 0.4 mM L-ornithine containing 0.5 μCi of DL-[1-14C] ornithine hydrochloride] in 15 ml corex centrifuge tube equipped with rubber stoppers and central well assemblies containing 0.2 ml ethanolamine and methoxyethanol in 2:1(v/v) ratio. After incubation at 37 °C for 60 min, the reaction was terminated by the addition of 1.0 ml of 2 M citric acid, using a 21G needle/syringe. The incubation was continued for 1 h. Finally, the central well containing the ethanolamine: methoxyethanol mixture to which 14CO2 has been trapped was transferred to a vial containing 10 ml of toluene-based scintillation fluid and 2 ml of ethanol. The radioactivity was measured in a Beckman LS 6000 SC liquid scintillation counter. Enzyme activity was expressed as picomoles CO2 released/h/mg protein.
Immunostaining of hydrogen peroxide (H2O2)
Immunohistochemical detection of H2O2 in normal as well as UV-irradiated skin was performed following the procedure as described earlier (29). Briefly, 6 μm thick frozen skin sections were incubated with 0.1 M Tris–HCl buffer, pH 7.5, containing 1 mg/mlglucose and 1 mg/ml DAB for 5–6 h at 37°C. Sections were then washed in distilled water and counterstained with methyl green (2% for 60 min). The DAB–peroxidase reaction gave a brown reaction product and methyl green a blue nuclear counterstain.
Lipid peroxidation (LPO) assay
After treatment, skin samples were harvested and washed with PBS, and microsomal fraction was prepared as described earlier (29). Briefly, cells were homogenized with a Polytron homogenizer in PBS buffer containing potassium chloride (1.19%, w/v) and centrifuged at 18,000g for 15 min at 4°C to prepare microsomal fraction. The LPO assay was performed in microsomal fraction obtained from the different treatment groups. The generation of malondialdehyde (MDA) was employed as a marker of LPO and estimated by the method of Wright et al. (30). Briefly, microsomal fraction (2.0 mg protein) was incubated for 1 h at 37 °C in the presence of ferric ions (1 mM) and ADP (5mM) in Ca2+-free phosphate buffer (0.1 M; pH: 7.4) containing MgCl2 (0.1mM). The reaction was terminated by addition of 0.6 ml of 10% (w/v) trichloroacetic acid followed by 1.2 ml of 0.5% (w/v) 2-thiobarbituric acid. The mixture was heated for 20 min at 90 °C in a water bath. After cooling, the MDA levels were measured in the clear supernatant by recording absorbance at 532 nm. The final concentration of MDA generated during the reaction was calculated using a molar extinction coefficient of 1.56X105/M/cm.
Statistical analysis
The results are expressed as the mean ± SE. Statistical analysis of all the data between groups receiving UVB exposure alone and that with PFE treatment plus UVB exposure was performed by Student’s t-test. The P value <0.05 was considered statistically significant.
Results
Oral feeding of PFE inhibits UVB-induced cutaneous edema, hyperplasia and infiltration of leukocytes
Acute exposure of skin to UVB irradiation leads to cutaneous edema, hyperplasia, erythema, infiltration of leukocytes, dilation of dermal blood vessels and vascular hyperpermeability (22,31,32). In the present study, we assessed the effect of oral feeding of PFE on skin edema in UVB exposed SKH-1 hairless mouse. Bifold-skin thickness as an indicator of vascular permeability and edema was measured 24 h post UVB radiation. Exposure of mouse skin to UVB radiation resulted in a significant increase in bifold-skin thickness compared to control and PFE alone treated animals (Figure 1a). Oral feeding of PFE resulted in a significant inhibition against UVB-induced bifold-skin thickness when compared to UVB alone group (Figure 1a). We next evaluated the effect of oral feeding of PFE on UVB-mediated induction of epidermal hyperplasia and infiltration of leukocytes, 24 h post UVB radiation. Hematoxylin-eosin staining of mouse dorsal skin sections revealed that UVB irradiation to the mouse skin resulted in hyperplasia, and mixed cell infiltration in the dermis, which comprised mostly neutrophils with some mononuclear cells admixed when compared to control treated animals (Figure 1b). However, these effects of UVB-induced infiltration of leukocytes and epidermal hyperplasia were inhibited by oral feeding of PFE (Figure 1b).
Figure 1. Inhibitory effect of oral feeding of PFE on UVB-induced cutaneous edema, hyperplasia and infiltration of leukocytes in SKH-1 hairless mice.
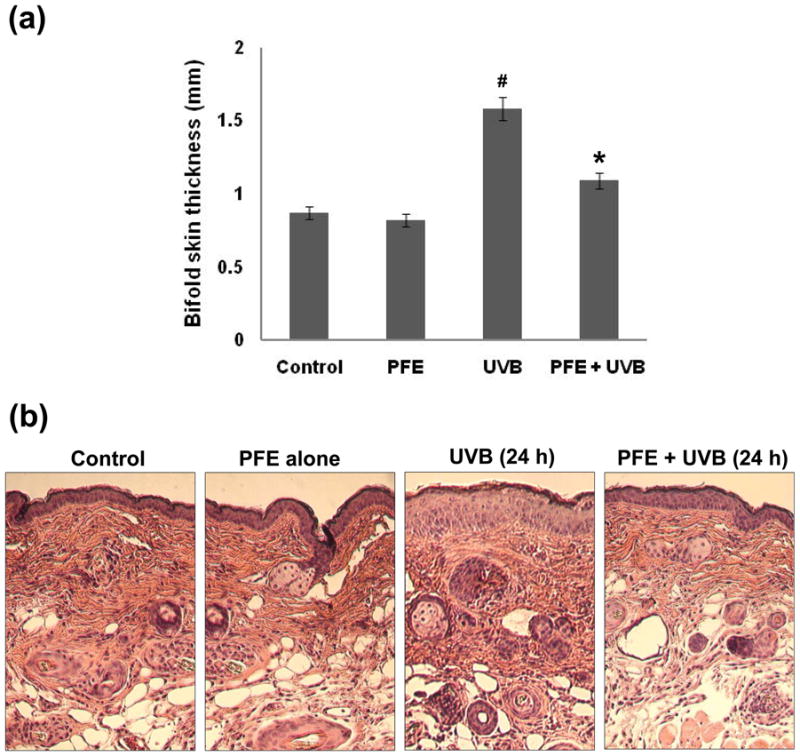
The groups of mice were either unexposed (control), received PFE in drinking water (0.2%, wt/vol), exposed to UVB radiation (180 mJ/cm2), received PFE in drinking water (0.2%, wt/vol) for 14 days before a single UVB (180 mJ/cm2) irradiation and continued till the termination of the experiment. [a] Twenty-four hours after UVB irradiation, the skin edema was determined by measuring the bifold-skin thickness. At least four determinations were made at different dorsal skin sites per mouse in each group. The data represents the mean ± SE of 8 mice (#p < 0.001 vs control; *p < 0.001 vs UVB). [b] At 24 hr post UVB irradiation, the animals were sacrificed and skin biopsies were processed for hyperplasia study by hematoxylin and eosin staining. Representative pictures are shown.
Oral feeding of PFE inhibits UVB-induced lipid peroxidation (LPO) and generation of hydrogen peroxide (H2O2) in SKH-1 hairless mice
One of the hallmarks of UVB-induced oxidative stress is the formation of oxidized macromolecules, including LPO (29,33). Therefore, we evaluated the effect of oral feeding of PFE on UVB-mediated increase in LPO, which is a well-accepted marker of photo-oxidative damage. UVB irradiation to mouse skin resulted in a significant induction of LPO in the epidermal microsomal fraction 24 h after UVB irradiation. Oral feeding of PFE significantly inhibited the UVB-mediated enhancement of LPO (Figure 2a). We next assessed the effect of oral feeding of PFE on UVB-mediated generation of H2O2 in mouse skin. Immunohistochemical analysis data revealed that UVB irradiation of mouse skin resulted in increased production of H2O2 in the skin, 24 h post UVB irradiation compared to control and PFE alone groups (Figure 2b). In skin samples obtained 24 h after UVB exposure the numbers of H2O2 positive cells were significantly higher when compared to control group (Figure 2c). Oral feeding of PFE abrogated the numbers of H2O2 producing cells compared to the UVB irradiated animals (Figure 2b,c).
Figure 2. Inhibitory effect of oral feeding of PFE on UVB-induced generation of hydrogen peroxide and lipid peroxidation in SKH-1 hairless mice.

Animals in each group were treated as described in Figure 1. [a] Twenty-four hours after UVB irradiation, the animals were sacrificed, and epidermal microsomal fraction was prepared. Generation of malondialdehyde was used as a marker of LPO. The data represents the mean ± SE of 8 mice (#p < 0.001 vs control; *p <0.001 vs UVB). [b] Twenty-four hours after UVB irradiation, the animals were sacrificed, and immunohistochemical detection of H2O2 was performed using the DAB–peroxidase reaction. Representative pictures are shown. [c] The numbers of H2O2 positive cells were counted in five different areas of the sections under a microscope. The numbers of H2O2 positive cells are represented as percent of H2O2 positive cells. The data represents the mean ± SE of 8 mice (#p < 0.001 vs control; *p <0.001 vs. UVB).
Oral feeding of PFE inhibits UVB-induced epidermal ODC activity and protein expression
ODC the rate-limiting enzyme in polyamine biosynthesis is upregulated in skin tumors compared to normal skin (34). Studies have shown that increase in ODC activity plays a critical role in skin tumor promotion in animal models (35). Therefore, we studied the effect of oral feeding of PFE on UVB-mediated induction of epidermal ODC activity in SKH-1 hairless mice. UVB irradiation to SKH-1 hairless mouse skin resulted in a significant increase in epidermal ODC activity, at 24 h post UVB treatment compared to control and PFE-alone groups. Oral feeding of PFE significantly inhibited UVB-induced epidermal ODC activity when compared to UVB alone group (Figure 3a). We next examined the effect of oral feeding of PFE on UVB-induced epidermal ODC protein expression. UVB exposure to SKH-1 hairless mice resulted in a marked increase in epidermal ODC protein expression in time-dependent manner (i.e. at 1, 8 and 24 h post UVB) compared to control group as determined by western blot analysis (Figure 3b). Oral feeding of PFE resulted in inhibition in ODC protein expression when compared to UVB alone groups at all time points (Figure 3b).
Figure 3. Inhibitory effect of oral feeding of PFE on UVB-induced epidermal ODC activity and protein expression in SKH-1 hairless mice.

Animals in each group were treated as described in Figure 1. [a] At 24 h post UVB irradiation, the animals were sacrificed, epidermal cytosolic fraction was prepared and ODC activity was determined. The data represents the mean ± SE of 8 mice (#p < 0.001 vs control; *p <0.001 vs. UVB). [b] At different time points post UVB irradiation, animals were sacrificed. The epidermis was separated, epidermal protein lysates were prepared and western blot analysis was performed to determine the protein expression of ODC. Equal loading was confirmed by stripping the western blot and reprobing it for β-actin. The representative blots are shown from three independent experiments.
Oral feeding of PFE inhibits UVB-induced epidermal cyclooxygenase (COX-2) and proliferating cell nuclear antigen (PCNA) protein expression
Studies have demonstrated that the expression of proinflammatory enzyme COX-2 is induced by UVB exposure (29,36). Therefore, we determined the effect of oral feeding of PFE on UVB-induced epidermal COX-2 protein expression. Western blot analysis revealed that UVB exposure to SKH-1 hairless mice resulted in a marked increase in epidermal COX-2 protein expression in a time-dependent manner compared to control group (Figure 4). Oral feeding of PFE resulted in inhibition COX-2 protein expression. (Figure 4). It is well known that exposure of skin to UVB radiation induces proliferative potential of epidermal cells (37). We, therefore next investigated the effect of oral feeding of PFE on UVB-induced expression of PCNA (marker of proliferation). We found that oral feeding of PFE resulted in inhibition of PCNA protein expression when compared to UVB alone group. (Figure 4).
Figure 4. Inhibitory effect of PFE on UVB-induced epidermal COX-2 and PCNA protein expression in SKH-1 hairless mice.

At different time points post UVB irradiation, animals were sacrificed. The epidermis was separated, epidermal protein lysates were prepared and western blot analysis was performed to determine the protein expression of COX-2 and PCNA. Equal loading was confirmed by stripping the western blot and reprobing it for β-actin. The representative blots are shown from three independent experiments.
Oral feeding of PFE inhibits UVB-induced formation of CPDs and 8-oxodG in SKH-1 hairless mice
It is well documented that both CPDs and 8-oxodG are formed after UVB irradiation and considered as important biomarkers of DNA damage (6,16). Employing immunohistochemical analysis, we assessed the effect of oral feeding of PFE on UVB-mediated DNA damage in mouse epidermis. UVB irradiation to SKH-1 hairless mouse skin resulted in increased formation of both CPDs (Figure 5a) and 8-oxodG (Figure 5c) when compared to their respective control groups. In skin samples obtained 24 h after UVB exposure the numbers of CPD positive cells (Figure 5b) and 8-oxodG positive cells (Figure 5d) were significantly higher when compared to their respective control groups. Oral feeding of PFE to SKH-1 hairless mice resulted in marked inhibition of UVB-induced formation of CPDs (Figure 5a,b) and 8-oxodG (Figure 5c,d) when compared to their respective UVB alone groups. These observations suggest that endogenous defense mechanism may play a role in the repair of UVB-induced DNA damage in mice that received PFE.
Figure 5. Inhibitory effect of PFE on UVB-induced formation of cyclobutane primidine dimers and 8-oxo-7,8-dihydro-2′-deoxyguanosine in SKH-1 hairless mice.

Twenty four hours post-UVB irradiation, the animals were sacrificed, skin biopsies were taken and frozen in OCT. Skin sections, 6 μm thick, were cut and processed for immunostaining. Immunohistochemical staining for CPDs [a], and 8-oxodG [c] was performed using appropriate antibodies. Representative pictures are shown. The number of CPD positive cells (b) and 8-oxodG positive cells (d) after immunostaining were counted in five different areas of the sections under a microscope. The numbers of CPD and 8-oxodG positive cells are represented as percent of CPD and 8-oxodG positive cells respectively. The data represents the mean ± SE of 8 mice (#p < 0.001 vs control; *p <0.001 vs. UVB).
Oral feeding of PFE enhances UVB-mediated increase in p53 and p21 protein expression in SKH-1 hairless mice
Studies have shown that the expression levels of p53 as well as its downstream target p21 were increased in response to UVB radiation (21). Therefore, we evaluated the effect of oral feeding of PFE on UVB-mediated increase in the expression of p53 and p21 proteins. The western blot data demonstrated that UVB irradiation resulted in an increase in p53 and p21 proteins in time-dependent manner compared to their respective control groups (Figure 6). Oral feeding of PFE resulted in a further enhancement in UVB-exposure-mediated increase in these proteins in SKH-1 hairless mouse epidermis (Figure 6).
Figure 6. Oral feeding of PFE enhances UVB-mediated increase in p53 and p21 protein expression in SKH-1 hairless mice.

At different time points post UVB irradiation, animals were sacrificed. The epidermis was separated, epidermal protein lysates were prepared and western blot analysis was performed to determine the protein expression of p21and p53. Equal loading was confirmed by stripping the western blot and reprobing it for β-actin. The representative blots are shown from three independent experiments.
Oral feeding of PFE inhibits UVB-induced activation of IKKα and NFκB, and phosphorylation and degradation of IκBα in SKH-1 hairless mice
NF-κB is present in the cytosol as a heterodimer usually consisting of its p50 and p65 subunits bound to its inhibitory proteins IκB. One of the critical events in NF-κB activation is its dissociation from inhibitory protein IκB (14). We determined whether oral feeding of PFE to SKH-1 hairless mice inhibits UVB-mediated activation of NF-κB. UVB irradiation resulted in phosphorylation and degradation of IκBα protein in time-dependent manner in the epidermal cytosol. Our data clearly demonstrated that oral feeding of PFE markedly inhibited UVB-mediated phosphorylation and degradation of IκBα (Figure 7). Because IKKα activity is necessary for IκBα protein phosphorylation/degradation, we also measured IKKα protein level. UVB radiation resulted in the activation of IKKα protein that in turn phosphorylate and degrade IκBα protein. Oral feeding of PFE inhibited UVB-induced activation of IKKα (Figure 7). We then investigated whether oral feeding of PFE inhibits UVB-induced activation and nuclear translocation of NF-κB/p65 in skin of SKH-1 hairless mice. Employing western blot analysis, we found that UVB exposure resulted in the activation and nuclear translocation of NF-κB/p65, which was markedly inhibited by oral feeding of PFE (Figure 7). Lamin was used as a loading control for NFκB.
Figure 7. Inhibitory effect of oral feeding of PFE on UVB-induced activation of IKKα and NF-κB, and phosphorylation and degradation of IκBα in SKH-1 hairless mice.

At different time points post UVB irradiation, animals were sacrificed. The epidermis was separated and epidermal cytosolic and nuclear protein lysates were prepared and western blot analysis was performed to determine the protein expression. Equal loading was confirmed by stripping the western blot and reprobing it for β-actin or lamin. The representative blots are shown from three independent experiments.
Discussion
The UVB component of solar UV radiation is believed to be the major cause of the variety of cutaneous disorders including skin cancers (14,38,39). Botanical antioxidants consumed by the human population have gained considerable attention as photoprotective and photochemopreventive agents against skin cancers (2,14,38). Previously, we have shown that treatment of NHEK with PFE inhibited UVB-mediated activation of NF-κB and MAPK pathways (28). This study was designed to assess the photochemopreventive effect of PFE after single UVB irradiations to the skin of SKH-1 hairless mice, a well-accepted model of photodamage studies. Our data clearly demonstrate that oral feeding of PFE affords significant inhibition against UVB-induced skin edema and hyperplasia.
UVB radiation to mammalian skin is known to alter cellular function via oxidation of macromolecules, DNA damage, generation of ROS, and alterations in signaling pathways (2,5,14). The incidence of LPO in the biological membrane is a free radical-mediated event that is regulated by the availability of substrates in the form of polyunsaturated fatty acids, pro-oxidants which promote peroxidation (40,41). LPO is highly detrimental to cell membrane structure and function, and its elevated level has been linked to damaging effects such as loss of fluidity, inactivation of membrane enzymes, increases cell membrane permeability which may ultimately lead to disruption of cell membrane potential (42,43). As we observed that UVB-induced leukocyte infiltration in the skin, and inflammatory leukocytes are the major source of H2O2 production that plays an important role in inflammatory skin diseases and skin cancer. Our results clearly demonstrate that oral feeding of PFE would result in reduction of the risk factors associated with UVB radiation by inhibiting UVB-mediated LPO and production of H2O2.
ODC, a rate-limiting enzyme that catalyzes the biosynthesis of polyamines, plays an important role in the regulation of cell transformation and development of cancer (44,45). Aberrations in ODC regulation, and subsequent polyamine accumulation, are closely associated with neoplastic transformation (46). In addition, high levels of ODC gene products are consistently detected in virtually all-animal tumors, and in certain tissues predisposed to tumorigenesis (44,46). Agents that have the ability to block induction of ODC can prevent tumor formation. Therefore, ODC inhibition was shown to be a promising tool for screening inhibitors of skin tumorigenesis. In the present study, oral feeding of PFE resulted in a significant inhibition of UVB-mediated induction of epidermal ODC activity and protein expression. It is logical to consider that oral feeding of PFE inhibited the action of the tumor promoter and/or the enzymatic pathway(s) that regulates the ODC induction.
COX-2 has been implicated in UVB-induced skin inflammation and photocarcinogenesis, and its overexpression has been shown to enhance cell proliferation, induce angiogenesis, regulate antiapoptotic cellular defenses and augment immunological response through production of PGE2 (47,48). Considerable body of evidence suggest that inhibition of COX-2 expression or activity is important for not only alleviating inflammation, but also for prevention of cancer. Our results demonstrate the inhibitory effect of oral feeding of PFE against UVB-mediated induction of epidermal COX-2 protein expression. These results indicate that, in addition to ODC, PFE also targets COX-2 expression in exerting its photoprotective effects.
PCNA is an auxiliary protein of DNA polymerase-δ and its high levels of expression correlate cell proliferation, suggesting that PCNA is an excellent marker of cellular proliferation, which also serves as an effective predictive indicator of initiated cancer cells (49). PCNA has been shown to play a role in the regulation of p21 activity by modulating the rate at which p21 is degraded (50). Studies have shown that phosphorylation of specific residues within the PCNA-binding motif can modulate the p21-PCNA interactions (51). Therefore, PCNA is regarded as an important target for p21 as well as a reliable biomarker for cell proliferation. We found that UVB irradiation enhanced epidermal PCNA protein expression compared to control and PFE alone fed groups. Oral feeding of PFE to mice inhibited UVB-induced PCNA protein expression. This suggests that inhibiting cell proliferation could be one of the mechanisms by which PFE protects epidermal damaged cells from entering the cell cycle, thus affording these cells additional time for DNA damage repair to avoid replication error that otherwise often results in carcinogenic mutation.
The p53 tumor suppressor gene plays a decisive role in protecting cells from DNA-damage as a consequence of UVB exposure (52). In the keratinocytes, UV-DNA damage induces an elevation of p53 protein followed by the induction of its direct transcriptional target p21, a cyclin dependent kinase inhibitor (21). It has been shown that p53 can play direct and indirect role in UVB-induced, transcription-coupled DNA repair (53). DNA damage elicited by UVB triggers p53 accumulation and transcriptional activation, leading to cell cycle arrest allowing more time for the repair of damaged DNA or elimination of damaged cells by apoptosis (54). Studies have shown that early responses in the skin of SKH-1 mice after an acute UVBexposure include increased number of cells expressing both p21 and p53 (55). Our data demonstrated that UVB irradiation resulted in an increase in p53 and p21 proteins, and oral feeding of PFE resulted in a further enhancement in UVB-exposure-mediated increase in these proteins in SKH-1 hairless mouse epidermis. UVB is a potent carcinogen known to damage DNA directly or through the generation of ROS. Both CPDs and 8-oxodG are formed in epidermal DNA after UVB irradiation and are considered as important biomarkers of DNA damage. Our data clearly demonstrated that oral feeding of PFE to SKH-1 hairless mice resulted in marked reduction in the number of CPDs and 8-oxodG positive cells and these may be due to enhanced DNA repair. Our data also suggest that the repair of 8-oxodG at the surface is better than at the base which is in agreement to the earlier published work on human skin (56).
Activated NF-κB is a crucial factor for the immunoinflammatory responses and is also implicated in tumorigenesis (57). Therefore, NF-κB has emerged as one of the most promising molecular targets in the prevention of cancer. NF-κB is sequestered in the cytoplasm as a heterotrimer consisting of p50, p65 and IκBα subunits. Upon phosphorylation and subsequent proteolytic degradation of IκBα, NF-κB activates and translocates to the nucleus where it binds to DNA and activates the target genes by binding to the DNA regulatory element (58). In the present study, we have demonstrated that oral feeding of PFE inhibited UVB-induced NF-κB and IKKα activation, and phosphorylation and degradation of IκBα protein. Because PFE inhibited IκBα phosphorylation and degradation, this study suggests that the effect of PFE on NF-κB/p65 is through inhibition of phosphorylation and subsequent proteolytic degradation of IκBα.
Our data suggest that oral feeding of PFE to mice affords substantial protection from the adverse effects of UVB radiation via modulation in early biomarkers of photocarcinogenesis and provide suggestion for its photochemopreventive potential.
Acknowledgments
This work was supported by a grant from R21 AT002429-02 awarded to FA.
References
- 1.Afaq F, V, Adhami M, Ahmad N, Mukhtar H. Botanical antioxidants for chemoprevention of photocarcinogenesis. Front Biosci. 2002;7:d784–d792. doi: 10.2741/afaq. [DOI] [PubMed] [Google Scholar]
- 2.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Jul 7; doi: 10.3322/caac.20073. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Afaq F, V, Adhami M, Mukhtar H. Photochemoprevention of ultraviolet B signaling and photocarcinogenesis. Mutat Res. 2005;571:153–173. doi: 10.1016/j.mrfmmm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 5.Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. 2006;126:2565–2575. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- 6.Afaq F, Syed DN, Malik A, Hadi N, Sarfaraz S, Kweon MH, Khan N, Zaid MA, Mukhtar H. Delphinidin, an anthocyanidin in pigmented fruits and vegetables, protects human HaCaT keratinocytes and mouse skin against UVB-mediated oxidative stress and apoptosis. J Invest Dermatol. 2007;127:222–232. doi: 10.1038/sj.jid.5700510. [DOI] [PubMed] [Google Scholar]
- 7.de Gruijl FR, Rebel H. Early events in UV carcinogenesis--DNA damage, target cells and mutant p53 foci. Photochem Photobiol. 2008;84:382–387. doi: 10.1111/j.1751-1097.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 8.Melnikova VO, Pacifico A, Chimenti S, Peris K, Ananthaswamy HN. Fate of UVB-induced p53 mutations in SKH-hr1 mouse skin after discontinuation of irradiation: relationship to skin cancer development. Oncogene. 2005;24:7055–7063. doi: 10.1038/sj.onc.1208863. [DOI] [PubMed] [Google Scholar]
- 9.Halliday GM, Lyons JG. Inflammatory doses of UV may not be necessary for skin carcinogenesis. Photochem Photobiol. 2008;84:272–283. doi: 10.1111/j.1751-1097.2007.00247.x. [DOI] [PubMed] [Google Scholar]
- 10.Nichols JA, Katiyar SK. Skin photoprotection by natural polyphenols: anti-inflammatory, antioxidant and DNA repair mechanisms. Arch Dermatol Res. 2010;302:71–83. doi: 10.1007/s00403-009-1001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz T. 25 years of UV-induced immunosuppression mediated by T cells- from disregarded T suppressor cells to highly respected regulatory T cells. Photochem Photobiol. 2008;84:10–18. doi: 10.1111/j.1751-1097.2007.00223.x. [DOI] [PubMed] [Google Scholar]
- 12.Halliday GM, Rana S. Waveband and dose dependency of sunlight-induced immunomodulation and cellular changes. Photochem Photobiol. 2008;84:35–46. doi: 10.1111/j.1751-1097.2007.00212.x. [DOI] [PubMed] [Google Scholar]
- 13.Afaq F, Mukhtar H. Botanical antioxidants in the prevention of photocarcinogenesis and photoaging. Exp Dermatol. 2006;15:678–684. doi: 10.1111/j.1600-0625.2006.00466.x. [DOI] [PubMed] [Google Scholar]
- 14.Afaq F, V, Adhami M, Mukhtar H. Photochemoprevention of ultraviolet B signaling and photocarcinogenesis. Mutat Res. 2005;571:153–173. doi: 10.1016/j.mrfmmm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 15.Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 16.Arad S, Zattra E, Hebert J, Epstein EH, Jr, Goukassian DA, Gilchrest BA. Topical thymidine dinucleotide treatment reduces development of ultraviolet-induced basal cell carcinoma in Ptch-1+/− mice. Am J Pathol. 2008;172:1248–1255. doi: 10.2353/ajpath.2008.071117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooke MS, Loft S, Olinski R, Evans MD, Bialkowski K, Wagner JR, Dedon PC, Møller P, Greenberg MM, Cadet J. Recommendations for standardized description of and nomenclature concerning oxidatively damaged nucleobases in DNA. Chem Res Toxicol. 2010;23:705–707. doi: 10.1021/tx1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ming M, Han W, Maddox J, Soltani K, Shea CR, Freeman DM, He YY. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene. 2010;29:492–502. doi: 10.1038/onc.2009.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lotti LV, Rotolo S, Francescangeli F, Frati L, Torrisi MR, Marchese C. AKT and MAPK signaling in KGF-treated and UVB-exposed human epidermal cells. J Cell Physiol. 2007;212:633–642. doi: 10.1002/jcp.21056. [DOI] [PubMed] [Google Scholar]
- 20.van Schanke A, van Venrooij GM, Jongsma MJ, Banus HA, Mullenders LH, van Kranen HJ, de Gruijl FR. Induction of nevi and skin tumors in Ink4a/Arf Xpa knockout mice by neonatal, intermittent, or chronic UVB exposures. Cancer Res. 2006;66:2608–2615. doi: 10.1158/0008-5472.CAN-05-2476. [DOI] [PubMed] [Google Scholar]
- 21.Abd Elmageed ZY, Gaur RL, Williams M, Abdraboh ME, Rao PN, Raj MH, Ismail FM, Ouhtit A. Characterization of coordinated immediate responses by p16INK4A and p53 pathways in UVB-irradiated human skin cells. J Invest Dermatol. 2009;129:175–183. doi: 10.1038/jid.2008.208. [DOI] [PubMed] [Google Scholar]
- 22.Afaq F, Ahmad N, Mukhtar H. Suppression of UVB-induced phosphorylation of mitogen-activated protein kinases and nuclear factor kappa B by green tea polyphenol in SKH-1 hairless mice. Oncogene. 2003;22:9254–9264. doi: 10.1038/sj.onc.1207035. [DOI] [PubMed] [Google Scholar]
- 23.Afaq F, V, Adhami M, Ahmad N, Mukhtar H. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea Constituent (-)-epigallocatechin-3-gallate. Oncogene. 2003;22:1035–1044. doi: 10.1038/sj.onc.1206206. [DOI] [PubMed] [Google Scholar]
- 24.Afaq F, Saleem M, Krueger CG, Reed JD, Mukhtar H. Anthocyanin- and hydrolyzable tannin-rich pomegranate fruit extract modulates MAPK and NF-kappaB pathways and inhibits skin tumorigenesis in CD-1 mice. Int J Cancer. 2005;113:423–433. doi: 10.1002/ijc.20587. [DOI] [PubMed] [Google Scholar]
- 25.Gil MI, Tomas-Barberan FA, Hess-Pierce B, Holcroft DM, Kedar AA. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J Agric Food Chem. 2000;10:4581–4589. doi: 10.1021/jf000404a. [DOI] [PubMed] [Google Scholar]
- 26.Khan N, Hadi N, Afaq F, Syed DN, Kweon MH, Mukhtar H. Pomegranate fruit extract inhibits prosurvival pathways in human A549 lung carcinoma cells and tumor growth in athymic nude mice. Carcinogenesis. 2007;28:163–173. doi: 10.1093/carcin/bgl145. [DOI] [PubMed] [Google Scholar]
- 27.Malik A, Afaq F, Sarfaraz S, Adhami VM, Syed DN, Mukhtar H. Pomegranate fruit juice for chemoprevention and chemotherapy of prostate cancer. Proc Natl Acad Sci U S A. 2005;102:14813–14818. doi: 10.1073/pnas.0505870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Afaq F, Malik A, Syed D, Maes D, Matsui MS, Mukhtar H. Pomegranate fruit extract modulates UV-B-mediated phosphorylation of mitogen-activated protein kinases and activation of nuclear factor kappa B in normal human epidermal keratinocytes. Photochem Photobiol. 2005;81:38–45. doi: 10.1562/2004-08-06-RA-264. [DOI] [PubMed] [Google Scholar]
- 29.Afaq F, V, Adhami M, Ahmad N. Prevention of short-term ultraviolet B radiation-mediated damages by resveratrol in SKH-1 hairless mice. Toxicol Appl Pharmacol. 2003;186:28–37. doi: 10.1016/s0041-008x(02)00014-5. [DOI] [PubMed] [Google Scholar]
- 30.Wright JR, Colby HD, Miles PR. Cytosolic factors which affect microsomal lipid peroxidation in lung and liver. Arch Biochem Biophys. 1981;206:296–304. doi: 10.1016/0003-9861(81)90095-3. [DOI] [PubMed] [Google Scholar]
- 31.Yano K, Kajiya K, Ishiwata M, Hong YK, Miyakawa T, Detmar M. Ultraviolet B-induced skin angiogenesis is associated with a switch in the balance of vascular endothelial growth factor and thrombospondin-1 expression. J Invest Dermatol. 2004;122:201–208. doi: 10.1046/j.0022-202X.2003.22101.x. [DOI] [PubMed] [Google Scholar]
- 32.Pearse AD, Gaskell SA, Marks R. Epidermal changes in human skin following irradiation with either UVB or UVA. J Invest Dermatol. 1987;88:83–87. doi: 10.1111/1523-1747.ep12465094. [DOI] [PubMed] [Google Scholar]
- 33.Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat Res. 2005;571:107–120. doi: 10.1016/j.mrfmmm.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Gilmour SK. Polyamines and nonmelanoma skin cancer. Toxicol Appl Pharmacol. 2007;224:249–256. doi: 10.1016/j.taap.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feith DJ, Bol DK, Carboni JM, Lynch MJ, Sass-Kuhn S, Shoop PL, Shantz LM. Induction of ornithine decarboxylase activity is a necessary step for mitogen-activated protein kinase kinase-induced skin tumorigenesis. Cancer Res. 2005;65:572–578. [PubMed] [Google Scholar]
- 36.Sharma SD, Katiyar SK. Dietary grape seed proanthocyanidins inhibit UVB-induced cyclooxygenase-2 expression and other inflammatory mediators in UVB-exposed skin and skin tumors of SKH-1 hairless mice. Pharm Res. 2010 Feb 9; doi: 10.1007/s11095-010-0050-9. [DOI] [PubMed] [Google Scholar]
- 37.Gu M, Dhanalakshmi S, Singh RP, Agarwal R. Dietary feeding of silibinin prevents early biomarkers of UVB radiation-induced carcinogenesis in SKH-1 hairless mouse epidermis. Cancer Epidemiol Biomarkers Prev. 2005;14:1344–1349. doi: 10.1158/1055-9965.EPI-04-0664. [DOI] [PubMed] [Google Scholar]
- 38.Adhami VM, Syed DN, Khan N, Afaq F. Phytochemicals for prevention of solar ultraviolet radiation-induced damages. Photochem Photobiol. 2008;84:489–500. doi: 10.1111/j.1751-1097.2007.00293.x. [DOI] [PubMed] [Google Scholar]
- 39.Erb P, Ji J, Kump E, Mielgo A, Wernli M. Apoptosis and pathogenesis of melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:283–295. doi: 10.1007/978-0-387-77574-6_22. [DOI] [PubMed] [Google Scholar]
- 40.Katiyar SK, Meeran SM. Obesity increases the risk of UV radiation-induced oxidative stress and activation of MAPK and NF-kappaB signaling. Free Radic Biol Med. 2007;42:299–310. doi: 10.1016/j.freeradbiomed.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider LA, Bloch W, Kopp K, Hainzl A, Rettberg P, Wlaschek M, Horneck G, Scharffetter-Kochanek K. 8-Isoprostane is a dose-related biomarker for photo-oxidative ultraviolet (UV) B damage in vivo: a pilot study with personal UV dosimetry. Br J Dermatol. 2006;154:1147–1154. doi: 10.1111/j.1365-2133.2006.07192.x. [DOI] [PubMed] [Google Scholar]
- 42.Bresgen N, Jaksch H, Lacher H, Ohlenschläger I, Uchida K, Eckl PM. Iron-mediated oxidative stress plays an essential role in ferritin-induced cell death. Free Radic Biol Med. 2010;48:1347–1357. doi: 10.1016/j.freeradbiomed.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53:135–159. [PubMed] [Google Scholar]
- 44.Auvinen M, Paasinen A, Andersson LC, Hölttä E. Ornithine decarboxylase activity is critical for cell transformation. Nature. 1992;360:355–358. doi: 10.1038/360355a0. [DOI] [PubMed] [Google Scholar]
- 45.Thomas T, Thomas TJ. Polyamine metabolism and cancer. J Cell Mol Med. 2003;7:113–126. doi: 10.1111/j.1582-4934.2003.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auvinen M, Laine A, Paasinen-Sohns A, Kangas A, Kangas L, Saksela O, Andersson LC, Hölttä E. Human ornithine decarboxylase-overproducing NIH3T3 cells induce rapidly growing, highly vascularized tumors in nude mice. Cancer Res. 1997;57:3016–3025. [PubMed] [Google Scholar]
- 47.Athar M, An KP, Morel KD, Kim AL, Aszterbaum M, Longley J, Epstein EH, Jr, Bickers DR. Ultraviolet B(UVB)-induced cox-2 expression in murine skin: an immunohistochemical study. Biochem Biophys Res Commun. 2001;280:1042–1047. doi: 10.1006/bbrc.2000.4201. [DOI] [PubMed] [Google Scholar]
- 48.Herschman HR. Regulation of prostaglandin synthase-1 and prostaglandin synthase-2. Cancer Metastasis Rev. 1994;13:241–256. doi: 10.1007/BF00666095. [DOI] [PubMed] [Google Scholar]
- 49.Lee KM, Lee KW, Byun S, Jung SK, Seo SK, Heo YS, Bode AM, Lee HJ, Dong Z. 5-deoxykaempferol plays a potential therapeutic role by targeting multiple signaling pathways in skin cancer. Cancer Prev Res. 2010;3:454–465. doi: 10.1158/1940-6207.CAPR-09-0137. [DOI] [PubMed] [Google Scholar]
- 50.Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene. 1998;17:2437–2444. doi: 10.1038/sj.onc.1202189. [DOI] [PubMed] [Google Scholar]
- 51.Scott MT, Morrice N, Ball KL. Reversible phosphorylation at the C-terminal regulatory domain of p21(waf1/cip1) modulates proliferating cell nuclear antigen binding. J Biol Chem. 2000;275:11529–11537. doi: 10.1074/jbc.275.15.11529. [DOI] [PubMed] [Google Scholar]
- 52.Smith ML, Fornace AJ., Jr p53-mediated protective responses to UV irradiation. Proc Natl Acad Sci USA. 1997;94:12255–12257. doi: 10.1073/pnas.94.23.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKay BC, Francis MA, Rainbow AJ. Wild type p53 is required for heat shock and ultraviolet light enhanced repair of a UV-damaged reporter gene. Carcinogenesis. 1997;18:245–249. doi: 10.1093/carcin/18.2.245. [DOI] [PubMed] [Google Scholar]
- 54.Berg RJ, van Kranen HJ, Rebel HG, de Vries A, van Vloten WA, Van Kreijl CF, van der Leun JC, de Gruijl FR. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc Natl Acad Sci USA. 1996;93:274–278. doi: 10.1073/pnas.93.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Javeri A, Huang XX, Bernerd F, Mason RS, Halliday GM. Human 8-oxoguanine-DNA glycosylase 1 protein and gene are expressed more abundantly in the superficial than basal layer of human epidermis. DNA Repair (Amst) 2008;7:1542–1550. doi: 10.1016/j.dnarep.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 57.Chiang YM, Lo CP, Chen YP, Wang SY, Yang NS, Kuo YH, Shyur LF. Ethyl caffeate suppresses NF-kappaB activation and its downstream inflammatory mediators, iNOS, COX-2, and PGE2 in vitro or in mouse skin. Br J Pharmacol. 2005;146:352–363. doi: 10.1038/sj.bjp.0706343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]