

Abstract
The mammalian target of rapamycin (mTOR) assembles into two distinct complexes: mTOR complex 1 (mTORC1) is predominantly cytoplasmic and highly responsive to rapamycin, whereas mTOR complex 2 (mTORC2) is both cytoplasmic and nuclear, and relatively resistant to rapamycin. mTORC1 and mTORC2 phosphorylatively regulate their respective downstream effectors p70S6K/4EBP1, and Akt. The resulting activated mTOR pathways stimulate protein synthesis, cellular proliferation, and cell survival. Moreover, phospholipase D (PLD) and its product, phosphatidic acid (PA) have been implicated as one of the upstream activators of mTOR signaling. In this study, we investigated the activation status as well as the subcellular distribution of mTOR, and its upstream regulators and downstream effectors in endometrial carcinomas (ECa) and non-neoplastic endometrial control tissue. Our data show that the mTORC2 activity is selectively elevated in endometrial cancers as evidenced by a predominant nuclear localization of the activated form of mTOR (p-mTOR at Ser2448) in malignant epithelium, accompanied by overexpression of nuclear p-Akt (Ser473), as well as overexpression of vascular endothelial growth factor (VEGF)-A isoform, the latter a resultant of target gene activation by mTORC2 signaling via hypoxia-inducible factor (HIF)-2alpha. In addition, expression of PLD1, one of the two major isoforms of PLD in human, is increased in tumor epithelium. In summary, we demonstrate that the PLD1/PA-mTORC2 signal pathway is overactivated in endometrial carcinomas. This suggests that the rapamycin-insensitive mTORC2 pathway plays a major role in endometrial tumorigenesis and that therapies designed to target the phospholipase D pathway and components of the mTORC2 pathway should be efficacious against ECa.
Keywords: morphoproteomics, phospholipase D1, mTORC2, endometrial carcinoma
Introduction
Endometrial carcinoma (ECa) remains the most common gynecologic malignancy in the United States [1]. At presentation, over 85% of cases are stage I or II, and most patients with early-stage disease can be cured with surgical resection or radiotherapy or a combination of the two [2]. Hormonal therapy with progestins and/or chemotherapy utilizing cisplatin, doxorubicin, and paclitaxel thus far have been regimens with activity in advanced or recurrent ECa [3, 4]. However, the response rates (RR) to these systemic therapies have been consistently low. Progestins elicit a satisfactory response (usually in approximately 25 to 30%) in patients with high levels of hormonal receptor content. Treatment with cisplatin and doxorubicin produces a RR of 42% with a median survival of nine months, whereas the addition of paclitaxel slightly increases the RR to 57% with an overall survival of approximately 12 months [4]. The lack of effective treatment, together with the considerable toxicities, underscores the need for novel therapeutic strategies, particularly targeted therapy with or without conventional chemotherapy for advanced or recurrent ECa.
mTOR is a serine/threonine kinase that is an essential regulator of cell growth, cell cycle progression and angiogenesis [5]. Currently, synthetic mTOR inhibitors are being evaluated in clinical trials for treatment of patients with advanced or recurrent ECa [3]. In mammalian cells, mTOR assembles into two structurally and functionally distinct protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 contains mTOR, raptor, and mLST8. The assembly of mTORC1 predominantly occurs in the cytoplasm. The best characterized downstream effector and target of mTORC1 are p70S6K and 4EBP1 (eukaryotic initiation factor 4E binding protein 1), respectively [6, 7], both regulators of protein translation. In contrast, mTORC2, comprised of mTOR, rictor, mLST8 and mSin1, is abundant in both cytoplasmic and nuclear compartments. Upon growth factor stimulation, Akt is recruited to the plasma membrane and activated through phosphorylation at Thr308 by PDK1 and Ser473 by mTORC2 [5]. Activated Akt could subsequently phosphorylate and inactivate tuberin, causing increased mTORC1 activity [8]. VEGF expression is important for the neoangiogenesis, growth and metastasis of endometrial carcinoma, and it is regulated, at least in part, through the mTOR-dependent pathway [9, 10].
Recently, phospholipase D (PLD) and its metabolites phosphatidic acid (PA) have been implicated as one of the upstream regulators of the mTOR signaling. PLD in response to mitogenic signals catalyzes the hydrolysis of phos-phatidylcholine to choline and PA [11]. Binding of PA facilitates the association of mTOR with raptor to form mTORC1 and that of mTOR with rictor to form mTORC2. The effect of PA is competitive with rapamycin, a natural mTOR inhibitor, and as a consequence, elevated PLD activity may confer resistance to rapamycin [12]. Overexpression of PLD has been observed in a variety of human cancers, including breast cancer, gastric cancer, and renal cell carcinoma [13]. To date, only a few investigations have been performed to assess the expression of mTOR signaling cascade in invasive endometrial carcinomas [14, 15]. Furthermore, thesubcellular localization of the activated form of mTOR in these tumors has not been characterized. Determining the cytoplasmic and nuclear distribution of activated mTOR and thus understanding the relative extent of involvement of mTORC1 and mTORC2 in endometrial carcinoma is important, as mTORC1 is highly sensitive to rapamycin, whereas mTORC2 is relatively resistant to the effects of rapamycin [16]. Thus, the aim of the present study was to characterize the mTOR activation status via a morphoproteomic examination of the expression and subcellular distribution of mTOR and its upstream regulators and downstream effectors in endometrial carcinoma (ECa), as well as in proliferative-phase endometrium (PE), and secretory-phase endometrium (SE) [17,18].
Materials and methods
Specimens
A total of 83 cases including 33 cases of ECa, 25 cases of PE, and 25 cases of SE were retrieved from the surgical pathology files at the Department of Pathology, the Lyndon B Johnson General Hospital and the Memorial Hermann Hospital at Texas Medical Center. Among the 33 cases of ECa, 11 (33%) are International Federation of Gynecology and Obstetrics (FIGO) grade 1, 19 (58%) are FIGO grade 2, 3 (9%) are FIGO grade 3, and one patient (3%) had been treated with medroxyprogesterone prior to hysterectomy.
The tissues were made anonymous and dissociated from any clinical information. The hematoxylin and eosin (H&E) stained slides were reviewed and the diagnoses were confirmed by two pathologists. Corresponding formalin-fixed, paraffin-embedded tissue blocks were obtained and tissue microarrays were constructed with a tissue arrayer (Beecher Instruments, Sun Prairie, WI) by selecting a 1-mm tissue core from each case. H&E staining of the microarray sections demonstrated histologically representative areas of the tumors.
Immunohistochemical staining
Antibodies utilized in this study included: phosphorylated (p)-mTOR at Serine 2448, p-Akt at Serine 473 (Cell Signaling Technology, Inc, Beverly, MA); vascular endothelial growth factor (VEGF)-A isoform (DakoCytomation, Carpentaria, CA); and phospholipase D 1 (PLD1), and phospholipase D 2 (PLD2) (Santa Cruz Biotechnology, Inc, Santa Cruz, CA). After deparaffinization in xylene and rehydration in ethanol, heat-induced epitope retrieval was performed on tissue microarray sections. The tissue was treated with 3% H2O2 and washed with trisbuffered saline (TBS)/Tween-20. A few drops of diluted blocking serum were added on the tissue and incubated at room temperature. The sections were then incubated with the primary antibodies. (Those incubated with anti-phosphospecific probes were incubated overnight at 4 degrees centigrade, per the vendors recommended procedure). The rest of the staining procedure was performed on a DAKO autostainer (Carpinteria, CA, United States) programmed to incubate each slide with diluted biotinylated secondary antibodies, rinse slides, and incubate with DAB solution (3, 3′-diaminobenzidine chromogen solution). The slides were then counterstained with hematoxylin, dehydrated, and cover-slipped. Positive and negative controls were run in parallel with the samples.
Tissue microarray scoring
Chromogenic signals were evaluated by bright-field microscopy and semi-quantified with regard to percentage of cells stained (0-100%) and the staining intensity (0: non-staining, 1+: weak staining, 2 +: moderate staining, and 3 +: strong staining). In addition, the cellular compartmentalization of each signal was indicated as plasmalemmal (membranous), cytoplasmic, or nuclear. Cases in which no core was available were excluded from analysis.
Statistical analysis
Pearson's chi-square test was used to determine statistical significance of differences in frequency of strong positive (staining intensity 2-3+) expression of analytes between ECa and benign endometrial tissues (PE and SE). Med-Calc (Mariakerke, Belgium) software was used for statistical analysis and P values less than 0.05 were considered statistically significant.
Results
Expression and subcellular localization of p-mTOR (Ser2448)
Analysis of different subcellular fractions of the endometrial epithelial cells revealed that plasmalemmal/cytoplasmic p-mTOR (Ser2448) immunopositivity was the main staining pattern in the non-neoplastic cells (Figure 1 and 2). 19 of 24 PE (79%) and 11 of 23 SE (46%) showed moderate to strong (2-3+) plasmalemmal / cytoplasmic expression of p-mTOR (Ser2448), whereas only one third of PE and SE had weak (1+) nuclear p-mTOR (Ser2448) staining. In contrast, nuclear staining of p-mTOR was the predominant pattern in ECa and was detected in all 32 cases examined. Moderate to strong (2-3+) nuclear p-mTOR (Ser2448) staining was detected in 9 of 32 (30%) of ECa cases, whereas none of the 47 non-cancerous cases (24 PE and 23 SE) showed moderate to strong (2-3+) nuclear p-mTOR expression (Figure 1 and 2, Table 1). Statistical analysis revealed that moderate to strong (2-3+) nuclear p-mTOR (Ser2448) expression was more frequently observed in ECa compared with benign endometrial tissues (PE and SE) (p < .001, see Table 1). These results suggest a selective overactivation of mTORC2 in endometrial carcinomas.
Figure 1.
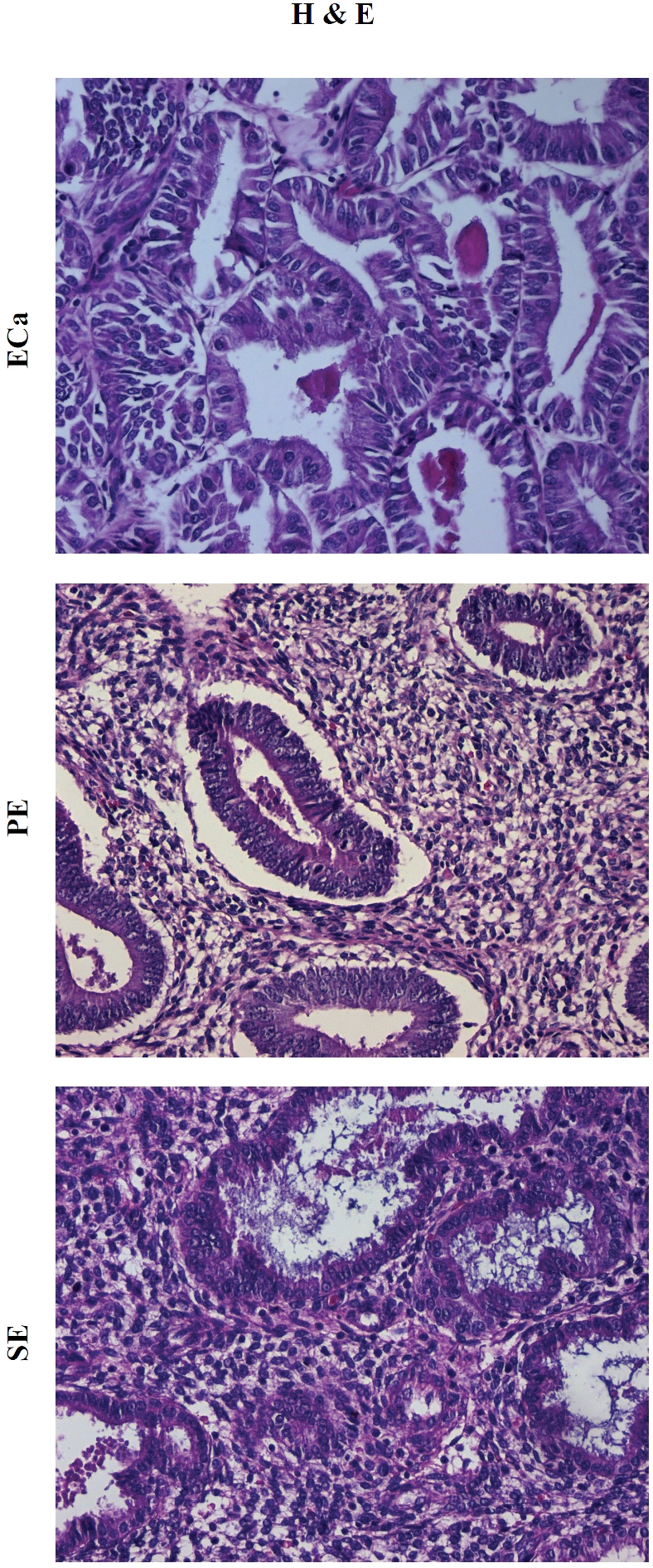
Representative hematoxylin-eosin (H&E) depictions of endometrial carcinoma (ECa), prolifera-tive-phase endometrium (PE), and secretory-phase endometrium (SE), respectively. Original magnification × 400.
Figure 2.
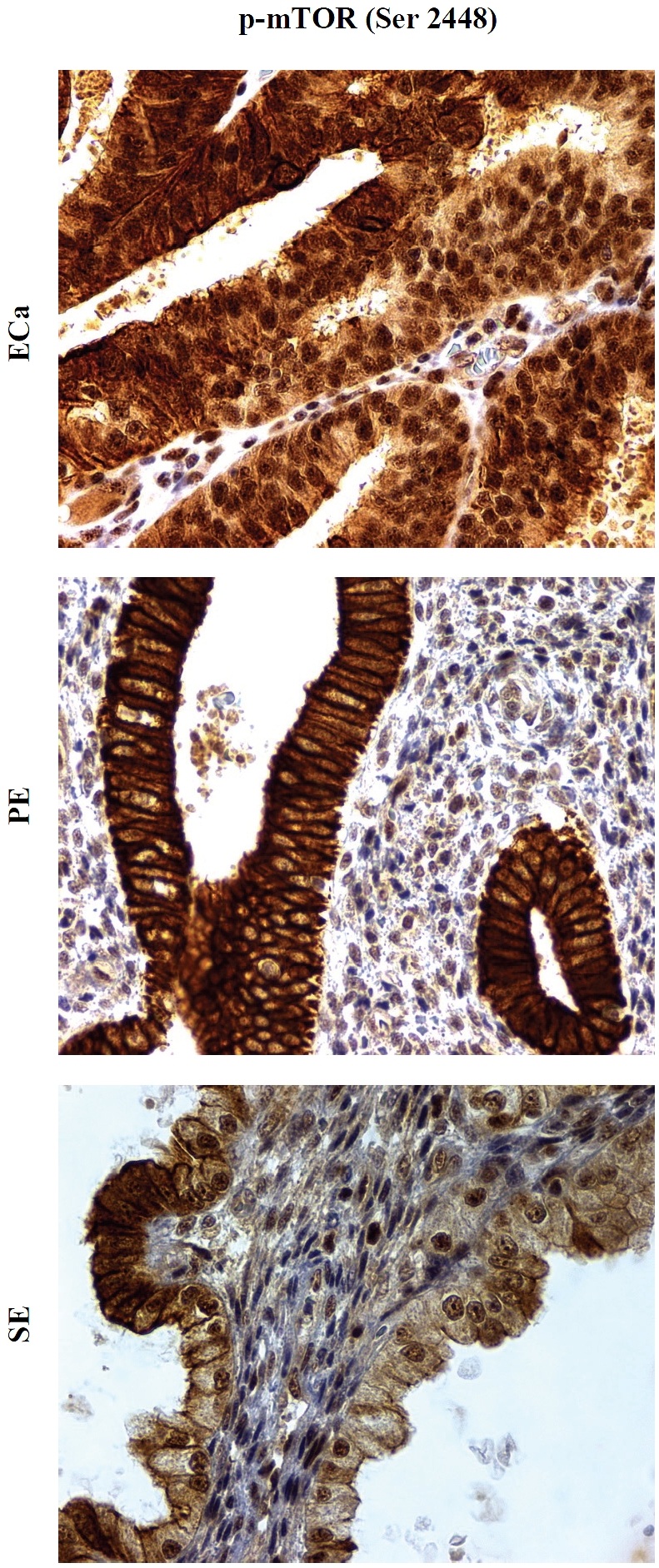
Expression of p-mTOR (Ser2448) in endometrial carcinoma (ECa), proliferative-phase endometrium (PE), and secretory-phase endometrium (SE). Phosphospecific antibodies against p-mTOR at serine 2448 show brown chromogenic signals in the plasmalemmal/cytoplasmic compartments of proliferative and secretory endometrium, and the signals are largely confined to the nucleus of tumor cells, the latter most consistent with mTORC2. Original magnification × 400.
Table 1.
Morphoproteomic Detection and Subcellular Compartmentalization of p-mTOR (Ser 2448), p-Akt (Ser 473), VEGF-A and PLD Analytes in Endometrial Carcinoma versus Proliferative and Secretory Endometrial Glands
| Protein analytes | Staining intensity | ECa | PE | SE | Frequency of 2-3+ expression in ECa vs. PE & SE: p value# |
|---|---|---|---|---|---|
| Nuclear | 1+ | 23/32(70%) | 8/24(33%) | 7/23(32%) | |
| p-mTOR (Ser2448) | 2-3+ | 9/32 (30%) | 0/24 (0%) | 0/23 (0%) | < .001 |
| Cytoplasmic p-AKT (Ser473) | 1+ | 27/33(82%) | 25/27 (93%) | 22/22(100%) | |
| 2-3+ | 6/33 (18%) | 2/27 (7%) | 0/22 (0%) | .084 | |
| Nuclear p-AKT (Ser473) | 1+ | 13/33 (39%) | 20/27(74%) | 22/22(100%) | |
| 2-3+ | 20/33(61%) | 7/27(26%) | 0/22 (0%) | < .001 | |
| VEGF-A+ | 1+ | 13/32(41%) | 19/23(83%) | 14/22(64%) | |
| 2-3+ | 18/32(55%) | 4/23 (17%) | 4/22(19%) | .001 | |
| Cytoplasmic PLD1 | 1+ | 13/33(39%) | 7/24 (29%) | 6/21 (29%) | |
| 2+ | 12/33(37%) | 0/24 (0%) | 0/21 (0%) | < .001 | |
| Cytoplasmic PLD2 | 1+ | 0/33 (0%) | 0/24 (0%) | 0/21 (0%) | |
| 2-3+ | 33/33(100%) | 24/24 (100%) | 21/21(100%) | .21 |
1+: weak; 2+: moderate; 3+: strong.
Both plasmalemmal and cytoplasmic staining.
Significance of differences in frequency of positive strong (2-3+) expression in ECa and benign endometrial tissues (PE and SE) is determined by Pearson's chi-square test (degrees of freedom: 1).
Abbreviations: ECa (endometrial carcinoma); PE (proliferative endometrial glands); SE (secretory endometrial glands); mTOR (mammalian target of rapamycin); VEGF (vascular endothelial growth factor)-A; PLD (phospholipase D)
Expression and subcellular localization of p-Akt (Ser473), a readout of mTORC2 activity
The expression of p-Akt at Ser473 is seen in both cytoplasmic and nuclear compartments in all groups. 20 of 33 ECa (61%) showed moderate to strong (2-3+) nuclear p-Akt (Ser473) immunopositivity; however, only 7 of 27 PE (26%) and none of the 22 SE cases (0%) had moderate to strong (2-3+) nuclear p-Akt (Ser473) staining (Figure 3). Moderate to strong nuclear p-Akt (Ser473) expression was more frequently observed in ECa compared with benign endometrial tissues (PE and SE) (p < .001, see Table 1). The cytoplasmic expression of p-Akt (Ser473) does not show statistically significant difference between the cancerous (ECa) and non-cancerous groups (PE and SE) (p = 0.08, see Table 1). These results correlate with the predominant nuclear staining of p-mTOR (Ser 2448) in ECa vis-a-vis benign endometrial epithelium.
Figure 3.

Expression of downstream effectors of mTORC2 pathway in endometrial carcinoma (ECa), proliferative-phase endometrium (PE), and secretory-phase endometrium (SE). Phosphospecific antibodies against p-Akt at serine 473 show chromogenic signals in the cytoplasm and nucleus of epithelial cells in all three groups (left column) with relatively overexpression of nuclear p-Akt (Ser473) in the ECa. Antibodies recognizing VEGF-A show chromogenic signals in the plasmalemmal/cytoplasmic fractions of both non-neoplastic and neoplastic epithelial cells (right column) with relative overexpression in the ECa. Original magnification × 400.
Expression of VEGF-A, the product of a target gene of mTORC2 signaling via hypoxia-inducible factor-2alpha [19, 20]
The plasmalemmal and cytoplasmic expression of VEGF-A was weak (1+) in most cases of PE and SE, with less than 20% showing moderate to strong (2-3+) VEGF-A staining in both groups, whereas moderate and strong VEGF-A expression was detected in a substantially higher proportion of ECa cases (55%, 18/32) (Figure 3 and Table 1). Moderate to strong (2-3+) VEGF-A expression was more frequently observed in ECa than in benign endometrial tissues (PE and SE) (p = 0.001, see Table 1).
Expression of PLD1 and PLD2, upstream facilitators of mTORC1/ mTORC2 signaling
The expression of PLD1 is seen in both cytoplasm and nucleoli in all groups. Moderate (2+) cytoplasmic PLD1 expression is observed in 37% ECa but none in PE or SE; in addition, 71% PE and 71% SE show a complete lack of PLD1 expression (Figure 4). Moderate to strong (2-3+) PLD1 expression was more frequently observed in ECa compared with benign endometrial tissues (PE and SE) (p < 0.001, see Table 1). PLD2 shows a moderate to strong (2-3+) cytoplasmic signal in all groups (figure not shown). No marked differences in the PLD2 expression level are noted among the three groups (p = 0.21, see Table 1).
Figure 4.
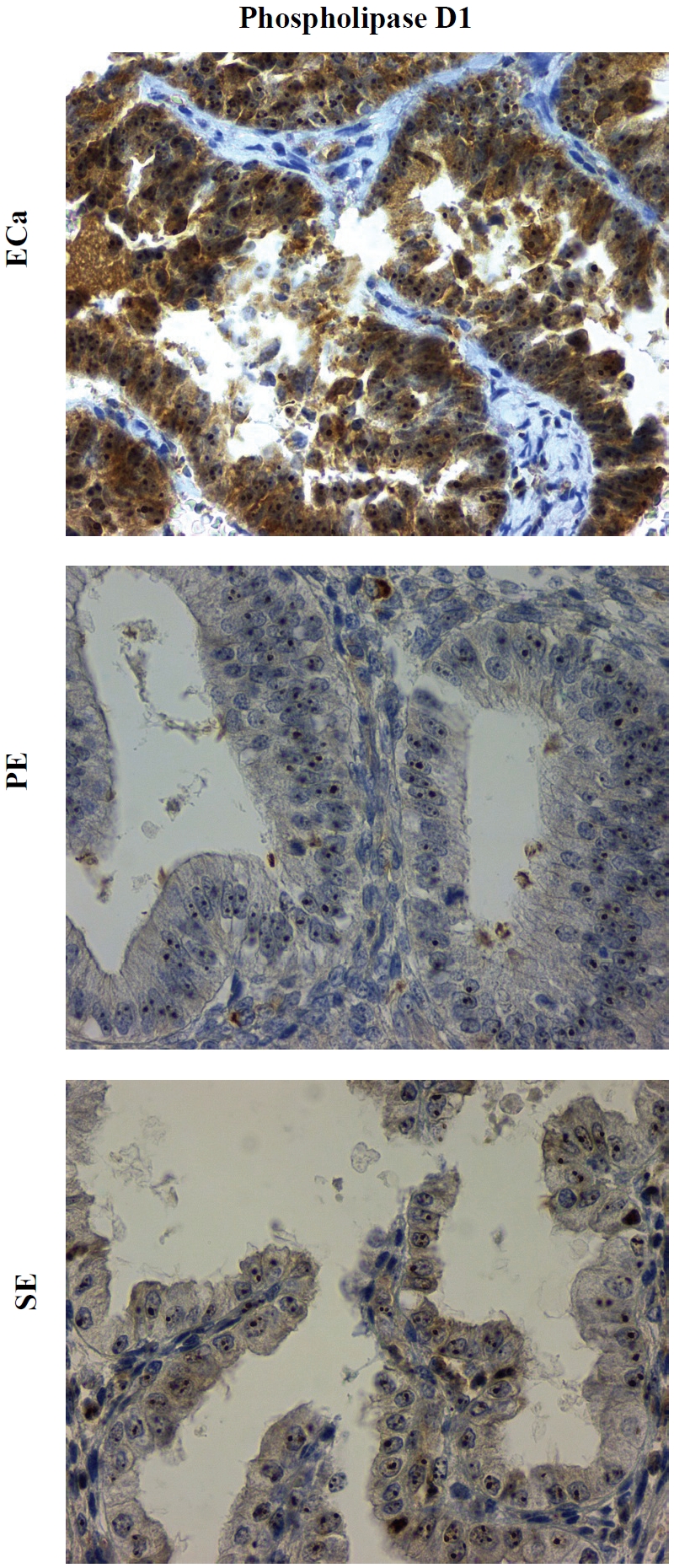
Expression of PLD1 in endometrial carcinoma (ECa), proliferative-phase endometrium (PE), and secretory-phase endometrium (SE). Isoform-specific antibodies against PLD1 show chromogenic signals in both cytoplasm and nucleoli in both non-neoplastic and neoplastic epithelial cells but with relative cytoplasmic overexpression in the ECa. Original magnification × 400.
Discussion
Morphoproteomics [18] attempts to define the biology of tumors by: utilizing phosphospecific immunohistochemical probes directed against putative sites of activation of signal transduction molecules and characterizing their subcellular compartmentalization (nuclear, cytoplasmic and plasmalemmal); assessing the correlative expression of effectors downstream of an activated pathway and of protein analytes associated with its enhancement; and comparing the signal intensity of such analytes in tumor cells versus those in their non-neoplastic counterpart as an indication of overexpression. Applied to this study, we have shown the overexpression of the following protein analytes in endometrial carcinoma: nuclear p-mTOR (Ser 2448) and nuclear p-Akt (Ser 473), indicative of mTORC2 pathway expression [21, 5]; VEGF-A, consistent with downstream signaling by the mTORC2 pathway via hypoxia-induciblefactor-2alpha [19,20,22]; and phospholipase D1, whose product, phosphatidic acid facilitates mTOR complex formation and which appears to be involved in the activation of mTORC2 [12, 23]. A computer-assisted search of the National Library of Medicine's MEDLINE database provides clinical correlates for our observations and conclusion that the mTORC2 pathway predominates and is facilitated by the phospholipase D1 pathway in endometrial carcinoma. Moreover, such correlates provide insight into new therapeutic strategies that target the phospholipase D-mTORC2 pathway in the treatment of patients with endometrial carcinoma.
We believe that our findings of the preferential mTORC2 pathway activation in endometrial carcinoma in contrast to the preponderant mTORC1 pathway activation in proliferative and secretory endometrium are in accord with the observation of McCampbell and co-workers, who demonstrated loss of inhibitory insulin receptor substrate (IRS)-1 phosphorylation in the majority of endometrial carcinoma as compared with normal proliferative endometrium [24, 25]. Such concordance is strengthened and explained, at least in part by the report of Tzatsos who observed that binding of IRS with raptor of mTORC1 is necessary to effect inhibitory phosphorylation of IRS-1 atserine 636/639 [26].
Additionally, the expression of IRS-1 and/or IRS-2 in the majority of the endometrial carcinomas reported by McCampbell et al [24] is also consistent with our findings of mTORC2 in ECa, as well as the established literature. This is based on several findings. First, signaling by insulin-like growth factor (IGF)-1 receptor leads to translocation of IRS-1 and IRS-2 to the nucleus [27], the latter, IRS-2 has been shown to form complexes with rictor/mTORC2 [28]. Second, IGF stimulates downstream effectors in the mTORC2 pathway, namely HIF-2alpha and VEGF-A [20, 29], the latter is shown to be overexpressed in our study. Moreover, the overexpression of phospholipase D1 in our series of endometrial carcinomas could explain the seemingly paradoxical finding reported by McCampbell et al., of the frequent expression of phospho-S6 ribosomal protein (S235), a downstream effector of p70S6K signaling in the absence of the expected p70S6K-induced phosphorylative inactivation of IRS-1 in endometrial carcinomas. Phosphatidic acid, the product of phospholipase D activity can directly phosphorylate p70S6K on threonine 389 [30] and thereby, account for activation of S6 ribosomal protein in the absence of mTORC1 signaling. Finally, a phase 2 clinical trial using everolimus, a rapalog with both mTORC1 and mTORC2 inhibitory properties produced a clinical benefit response in 21% (6 out of 28 patients) of patients with recurrent endometrial carcinoma at 20 weeks of therapy [31].
Most importantly, the constellation of findings from our morphoproteomic analysis of this series of patients with endometrial carcinoma, when viewed in the context of the scientific literature raises new therapeutic options. Metformin is one such consideration for the following reasons: (1) metformin is an AMP dependent kinase (AMPK) agonist and has been shown to phosphorylatively inactivate IRS-1 at serine 789, downregulate Akt phosphorylated at serine 473, mTOR phosphorylated at serine 2448, IGF-1R and IRS-1 levels [32]; (2) metformin reduces IGF-1 levels [33] and IGF-1-induced HIF-1alpha expression[34]; (3) metformin directly inhibits the phosphorylation of p70S6K, independent of the AMPK pathway [35]; and (4) metformin has been shown to downregulate the levels of lysophosphatidylcho-line and phosphatidylcholine the substrates for phospholipase D [36]. In a preclinical study on endometrial cancer cell lines, metformin has been shown to inhibit cell proliferation, mediated at least in part through the inhibition of the mTOR pathway [37].
In summary, morphoproteomic analysis reveals the overexpression of nuclear p-mTOR (Ser 2448) and p-Akt (Ser 473) and of VEGF-A isoform and phospholipase D1 in endometrial carcinoma vis-a-vis benign proliferative and secretory endometrium. This constellation of findings supports the presence of the mTORC2 pathway in the biology of endometrial cancer and suggests new possible therapeutic approaches to target the phospholipase D1-mTORC2 pathway.
Acknowledgments
The authors would like to thank Richard A. Breckenridge (ASCP) and Pamela K. Johnston (ASCP) for their technical assistance and Bheravi Patel for secretarial and graphic support.
References
- 1.Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005;366:491–505. doi: 10.1016/S0140-6736(05)67063-8. [DOI] [PubMed] [Google Scholar]
- 2.Sorosky JI. Endometrial cancer. Obstet Gynecol. 2008;111:436–447. doi: 10.1097/AOG.0b013e318162f690. [DOI] [PubMed] [Google Scholar]
- 3.Delmonte A, Sessa C. Molecule-targeted agents in endometrial cancer. Curr Opin Oncol. 2008;20(5):554–549. doi: 10.1097/CCO.0b013e32830b0deb. [DOI] [PubMed] [Google Scholar]
- 4.Pectasides D, Pectasides E, Economopoulos T. Systemic therapy in metastatic or recurrent endometrial cancer. Cancer Treat Rev. 2007;33(2):177–190. doi: 10.1016/j.ctrv.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12(4):487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation pre-initiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 7.Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167(3):399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 9.Kaku T, Kamura T, Kinukawa N, Kobayashi H, Sakai K, Tsuruchi N, Saito T, Kawauchi S, Tsuneyoshi M, Nakano H. Angiogenesis in endometrial carcinoma. Cancer. 1997;80(4):741–747. doi: 10.1002/(sici)1097-0142(19970815)80:4<741::aid-cncr13>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 10.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–158. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 11.Exton JH. Regulation of phospholipase D. Biochim Biophys Acta. 1999;1439(2):121–133. doi: 10.1016/s1388-1981(99)00089-x. [DOI] [PubMed] [Google Scholar]
- 12.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29(6):1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, Deguchi T, Ohishi N, Yagi K, Nozawa Y. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem Biophys Res Commun. 2000;278(1):140–143. doi: 10.1006/bbrc.2000.3719. [DOI] [PubMed] [Google Scholar]
- 14.Darb-Esfahani S, Faggad A, Noske A, Weichert W, Buckendahl AC, Muller B, Budczies J, Roske A, Dietel M, Denkert C. Phospho-mTOR and phospho-4EBP1 in endometrial adenocarcinoma: association with stage and grade in vivo and link with response to rapamycin treatment in vitro. J Cancer Res Clin Oncol. 2009;135(7):933–941. doi: 10.1007/s00432-008-0529-5. [DOI] [PubMed] [Google Scholar]
- 15.Lu KH, Wu W, Dave B, Slomovitz BM, Burke TW, Munsell MF, Broaddus RR, Walker CL. Loss of tuberous sclerosis complex-2 function and activation of mammalian target of rapamycin signaling in endometrial carcinoma. Clin Cancer Res. 2008;14(9):2543–2550. doi: 10.1158/1078-0432.CCR-07-0321. [DOI] [PubMed] [Google Scholar]
- 16.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 17.Lin F, Zhang PL, Yang XJ, Prichard JW, Lun M, Brown RE. Morphoproteomic and molecular concomitants of an overexpressed and activated mTOR pathway in renal cell carcinomas. Annals of clinical and laboratory science. 2006;36(3):283–293. [PubMed] [Google Scholar]
- 18.Brown RE. Morphogenomics and morphoproteomics: a role for anatomic pathology in personalized medicine. Archives of pathology & laboratory medicine. 2009;133(4):568–579. doi: 10.5858/133.4.568. [DOI] [PubMed] [Google Scholar]
- 19.Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA. HIF alpha expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene. 2008;27(19):2746–2753. doi: 10.1038/sj.onc.1210927. [DOI] [PubMed] [Google Scholar]
- 20.Akeno N, Robins J, Zhang M, Czyzyk-Krzeska MF, Clemens TL. Induction of vascular endothelial growth factor by IGF-I in osteoblast-like cells is mediated by the PI3K signaling pathway through the hypoxia-inducible factor-2alpha. Endocrinology. 2002;143(2):420–425. doi: 10.1210/endo.143.2.8639. [DOI] [PubMed] [Google Scholar]
- 21.Rosner M, Hengstschlager M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet. 2008;17(19):2934–2948. doi: 10.1093/hmg/ddn192. [DOI] [PubMed] [Google Scholar]
- 22.Akeno N, Czyzyk-Krzeska MF, Gross TS, Clemens TL. Hypoxia induces vascular endothelial growth factor gene transcription in human osteoblast-like cells through the hypoxia-inducible factor-2alpha. Endocrinology. 2001;142(2):959–962. doi: 10.1210/endo.142.2.8112. [DOI] [PubMed] [Google Scholar]
- 23.FangY, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294(5548):1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 24.McCampbell AS, Harris HA, Crabtree JS, Winneker RC, Walker CL, Broaddus RR. Loss of inhibitory insulin receptor substrate-1 phosphorylation is an early event in mammalian target of rapamycin-dependent endometrial hyperplasia and carcinoma. Cancer Prev Res (Phila) 2010;3(3):290–300. doi: 10.1158/1940-6207.CAPR-09-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCampbell AS, Broaddus RR, Walker CL. Loss of inhibitory insulin receptor substrate-1 phos-phorylation: An early event in endometrial hyperplasia and progression to carcinoma. Cell Cycle. 2010;9(14):2698–2699. doi: 10.4161/cc.9.14.12618. [DOI] [PubMed] [Google Scholar]
- 26.Tzatsos A. Raptor binds the SAIN (Shc and IRS-1 NPXY binding) domain of insulin receptor substrate-1 (IRS-1) and regulates the phosphoryla-tion of IRS-1 at Ser-636/639 by mTOR. J Biol Chem. 2009;284(34):22525–22534. doi: 10.1074/jbc.M109.027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun H, Tu X, Prisco M, Wu A, Casiburi I, Baserga R. Insulin-like growth factor I receptor signaling and nuclear translocation of insulin receptor substrates 1 and 2. Mol Endocrinol. 2003;17(3):472–486. doi: 10.1210/me.2002-0276. [DOI] [PubMed] [Google Scholar]
- 28.Singh BK, Singh A, Mascarenhas DD. A nuclear complex of rictor and insulin receptor substrate- 2 is associated with albuminuria in diabetic mice. Metab Syndr Relat Disord. 2010;8(4):355–363. doi: 10.1089/met.2010.0011. [DOI] [PubMed] [Google Scholar]
- 29.Carroll VA, Ashcroft M. Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 2006;66(12):6264–6270. doi: 10.1158/0008-5472.CAN-05-2519. [DOI] [PubMed] [Google Scholar]
- 30.Lehman N, Ledford B, Di Fulvio M, Frondorf K, McPhail LC, Gomez-Cambronero J. Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. Faseb J. 2007;21(4):1075–1087. doi: 10.1096/fj.06-6652com. [DOI] [PubMed] [Google Scholar]
- 31.Slomovitz BM, Lu KH, Johnston T, Coleman RL, Munsell M, Broaddus RR, Walker C, Ramondetta LM, Burke TW, Gershenson DM, Wolf J. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer. 2010 doi: 10.1002/cncr.25515. 000:000-000 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123(1):271–279. doi: 10.1007/s10549-010-0763-9. [DOI] [PubMed] [Google Scholar]
- 33.Berker B, Emral R, Demirel C, Corapcioglu D, Unlu C, Kose K. Increased insulin-like growth factor-I levels in women with polycystic ovary syndrome, and beneficial effects of metformin therapy. Gynecol Endocrinol. 2004;19(3):125–133. doi: 10.1080/09513590400007309. [DOI] [PubMed] [Google Scholar]
- 34.Treins C, Murdaca J, Van Obberghen E, Giorgetti-Peraldi S. AMPK activation inhibits the expression of HIF-1alpha induced by insulin and IGF-1. Biochem Biophys Res Commun. 2006;342(4):1197–1202. doi: 10.1016/j.bbrc.2006.02.088. [DOI] [PubMed] [Google Scholar]
- 35.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8(1):88–96. doi: 10.4161/cc.8.1.7499. [DOI] [PubMed] [Google Scholar]
- 36.Wanninger J, Neumeier M, Weigert J, Liebisch G, Weiss TS, Schaffler A, Aslanidis C, Schmitz G, Scholmerich J, Buechler C. Metformin reduces cellular lysophosphatidylcholine and thereby may lower apolipoprotein B secretion in primary human hepatocytes. Biochim Biophys Acta. 2008;1781(6-7):321–325. doi: 10.1016/j.bbalip.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation-implications for a novel treatment strategy. Gynecol Oncol. 2010;116(1):92–98. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]