

Abstract
Background
Hedgehog (HH) signaling is critical for the expansion of granule neuron precursors (GNPs) within the external granular layer (EGL) during cerebellar development. Aberrant HH signaling within GNPs is thought to give rise to medulloblastoma (MB) - the most commonly-observed form of malignant pediatric brain tumor. Evidence in both invertebrates and vertebrates indicates that cyclic AMP-dependent protein kinase A (PKA) antagonizes HH signalling. Receptors specific for the neuropeptide pituitary adenylyl cyclase activating polypeptide (PACAP, gene name ADCYAP1) are expressed in GNPs. PACAP has been shown to protect GNPs from apoptosis in vitro, and to interact with HH signaling to regulate GNP proliferation. PACAP/ptch1 double mutant mice exhibit an increased incidence of MB compared to ptch1 mice, indicating that PACAP may regulate HH pathway-mediated MB pathogenesis.
Methods
Primary MB tumorsphere cultures were prepared from thirteen ptch1+/-/p53+/- double mutant mice and treated with the smoothened (SMO) agonist purmorphamine, the SMO antagonist SANT-1, the neuropeptide PACAP, the PKA activator forskolin, and the PKA inhibitor H89. Gene expression of gli1 and [3H]-thymidine incorporation were assessed to determine drug effects on HH pathway activity and proliferation, respectively. PKA activity was determined in cell extracts by Western blotting using a phospho-PKA substrate antibody.
Results
Primary tumor cells cultured for 1-week under serum-free conditions grew as tumorspheres and were found to express PAC1 receptor transcripts. Gli1 gene expression was significantly reduced by SANT-1, PACAP and forskolin, but was unaffected by purmorphamine. The attenuation of gli1 gene expression by PACAP was reversed by the PKA inhibitor H89, which also blocked PKA activation. Treatment of tumorsphere cultures with PACAP, forskolin, and SANT-1 for 24 or 48 hours reduced proliferation.
Conclusions
Primary tumorspheres derived from ptch1+/-/p53+/- mice exhibit constitutive HH pathway activity. PACAP antagonizes HH signalling in these cells in a manner blocked by the PKA antagonist H89. PACAP and pharmacological activation of PKA also inhibited proliferation. Our data suggests that regulation of HH signaling by PACAP/PKA signaling may provide an alternative to SMO inhibition for the treatment of MB.
Background
Medulloblastoma (MB) is a malignant pediatric brain tumor that is thought to arise in many cases from transformation of granule neuron precursors (GNPs) within the external granular layer (EGL) of the developing cerebellum reviewed in [1]. Medulloblastoma is classified into the following subtypes based on their phenotypic [2] and genetic characteristics [3]: classic, large cell, anaplastic, desmoplastic and MB with extensive nodularity. Analyses of human desmoplastic MB samples revealed altered activity of, and/or mutations in, molecules of the sonic hedgehog (HH) signaling pathway including patched-1 (ptch1) and glioma-associated oncogene homolog 1 (gli-1) [1,3-5]. HH signaling is pivotal in the regulation of CNS patterning and development [6-8] stem cell self-renewal [9] and MB pathogenesis [4,5,10], and has also been implicated in a variety of additional human cancers including lung, pancreatic, and prostate reviewed in [11]. Mutations in ptch1 (the gene that codes for the Patched-1 12 pass transmembrane protein, a negative regulator of the HH signaling cascade), are observed with Gorlin's (Basal Cell Nevus) Syndrome - an autosomal hereditary disease characterized by the development of MB, basal cell carcinomas, and rhabdomyosarcoma [12,13]. Ptch1-/- mice die in utero while ptch1+/- mice typically survive but develop cerebellar tumors closely resembling MB in about 15% of the cases [10].
Our present understanding of the HH signaling cascade suggests that binding of the hedgehog ligands to PTCH-1 de-represses smoothened (SMO), allowing for the cascade of events leading to the activation of cubitus interruptus (Ci) in flies and the Gli transcription factors in vertebrates reviewed in [14,17]. The Gli transcription factors bind to the promoters of several genes considered to be HH targets (including gli1 and ptch1) [16-19]. Presently, molecules that act directly on components of the canonical HH pathway, including several that target SMO, are in various stages of clinical trials [20]. On the other hand, the protein kinase A (PKA) signalling pathway is known to antagonize hedgehog (HH) signaling in both invertebrates [21] and vertebrates [22,23], suggesting an alternative approach to blocking overactive HH activity. Nonetheless, little is know about the significance of the PKA/HH interaction in the pathogenesis of MB and other tumors.
PACAP is a 38-amino-acid peptide originally identified on the basis of its ability to induce production of cAMP in pituitary cells [24]. PACAP regulates a variety of biological functions including neuroblast proliferation [25,26], and neuroblast survival [27]. In the developing brain PACAP binds to, and signals through, the G-protein coupled receptor PAC1, increasing cAMP production and PKA activity [28]. PAC1 is also coupled to other signaling cascades in some cells, including phospholipase C (PLC) [29], phosphatidylinositol 3-Kinase (PI3-K) [30], src [31], and the MAP kinase pathways [32-34].
Gene transcripts for the HH targets genes ptch1 and gli1 are expressed within the EGL in the developing rat and mouse cerebella [26,35], indicating that the HH pathway is active in these cells. PAC1 gene transcripts are also present in the EGL [26], suggesting that the PACAP and HH pathways might interact within the proliferating cells in this layer. Moreover, the robust proliferative action of SHH on cultured GNPs was completely blocked by either treatment with PACAP or pharmacological induction of cAMP/PKA [26]. The potential significance of a PACAP/HH interaction in MB was recently demonstrated. First, PAC1 gene transcripts were found to be abundantly expressed in MB preneoplastic lesions in ptch1 mutant mice [36]. Second, deletion of a single copy of PACAP in ptch1+/- mutant mice was found to increase the incidence of MB approximately 2.5 fold [36]. Taken together, these data suggest that PACAP may regulate HH signaling in MB pathogenesis. In this work we investigate the interaction between HH and PACAP/PKA signaling in murine primary tumorspheres derived from MB tumors arising in ptch1+/-/p53+/- double mutant mice. Our data suggests that regulation of HH signaling by PACAP/PKA may provide a novel alternative to SMO inhibition for the treatment of medulloblastoma (MB).
Methods
Animal Generation, Care, and Assessment of Medulloblastoma
These studies utilized mice with single copy mutations in both the ptch1 and the p53 genes. The creation of these mice has been reported previously [10,37]. Generation of ptch1+/-/p53+/- mice were achieved by crossing p53-/- and ptch1+/- mice as previously described by Wetmore, Eberhart and Curran, 2001 [38]. Mice were housed according to UCLA Institutional Guidelines, including standard light-dark cycles with food and water ad libitum. All mice were monitored daily for evidence of tumor presence or illness.
Drugs and Stock Concentrations
PACAP-38, SANT-1 (a HH pathway antagonist), purmorphamine (a HH pathway agonist), and H-89 (a PKA antagonist) were obtained from Calbiochem. DMSO (vehicle) and forskolin (a cAMP/PKA inducer) were obtained from Sigma. Treatment concentrations for the drugs, unless otherwise stated, were as follows: 10 nM PACAP, 5 μM forskolin, 3 μM purmorphamine, 1 μM SANT-1 and 30 μM H89. When DMSO was used as drug solvent its concentration was adjusted to 0.25% (v/v) for all samples, except for experiments where H89 was used, in which case the final DMSO concentration was adjusted to 0.55%. In all experiments using H89, cells were pre-treated with this agent 30 minutes prior to addition of other drugs.
Isolation of Primary Tumorspheres, Maintenance, and Treatment
GNP-like tumor cells were isolated in a manner similar to that of Zhao, H et al., 2008 [39]. A total of thirteen individual tumor isolations and preparations were performed. Briefly, tumors were minced in 1 mL of trypsin/DNAse, placed into 1.5 mL eppendorf tubes and centrifuged at 750 × g for 5 min at 4°C. Pellets were resuspended in 800 μL of cold DNAse and immediately triturated with a 1000 μL filtered pipet, followed by trituration with a 200 μL pipet, and lastly with a fire polished Pasteur pipet. After another centrifugation as above, 500 μL of DNAse was added to the pellet with a trituration. This sample was resuspended in 4.5 ml of DMEM-F12 and applied to a 40 μm strainer (BD Falcon). The cell suspension was carefully layered on top of a 32%/60% percoll gradient and centrifuged at 2985 × g for 20 min at 4°C. Cells collected from the layer between the 32% and 60% percoll were washed once with cold DMEM-F12 by centrifuging at 2985 × g for 10 min. The supernatant was aspirated and the resulting pellet was resuspended in serum free medium comprising of DMEM/F12 medium (Invitrogen), Heparin [2 μg/mL](Sigma), Penicillin/Streptomycin/Glutamine [1%] (Invitrogen), bFGF [0.2 μg/mL] (Peprotech), EGF [0.02 μg/mL](Sigma), and B27 supplement [1:50] (Invitrogen). The cells were then counted and plated into 6-well plates containing 2 mL of medium such that the final cell concentrations were either 1 × 106 cells/mL or 0.5 × 106 cells/mL. Cells were then cultured for 6 or 7 days. During this period the cells were supplemented with additional bFGF (0.05 μg) and EGF (0.04 μg) on days 3 and 5. On day 6 or 7, serum-free media was added when necessary to equalize all volumes at 2 mL, and then drug treatments were added to the cells in the same wells as indicated in the figure legends.
RNA Extraction and RT-PCR
Tumorspheres were pelleted in 1.5 mL eppendorf tube by microcentrifugation (750 × g for 5 minutes at 4°C). RNA was extracted from the samples using the TRIzol reagent (Invitrogen) according to manufacturer's instructions. cDNA was generated using the iScript cDNA synthesis kit (BioRad). Semi-quantitative RT-PCR for pac1 and SYBR-Green quantitative real-time RT-PCR for gapdh and gli1 were performed as described previously [26,36]. Individual sample differences were calculated based on the ratio of standard quantities of gli1 and gapdh transcripts determined by the standard curve method.
Western Blotting
Whole-cell lysates, prepared in NP-40 lysis buffer along with 1 mM PMSF, 1 μg/ml each of aprotinin, leupeptin, and pepstatin, 1 mM Na3VO4, 1 mM NaF (Sigma), were analyzed by immunoblotting. Equal amounts of proteins from whole-cell extracts were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. The membranes were subjected to Western blot analysis with rabbit anti-phospho-PKA substrate antibody (1:2000; Cell Signaling Technology) or mouse anti-β-actin antibody (1:5000; Cell Signaling Technology). The signals from the primary antibody were amplified by horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse immunoglobulin G (Santa Cruz Biotechnology) and detected with enhanced chemiluminescence (GE Healthcare).
Proliferation Studies
Thymidine incorporation in tumorsphere cultures was performed using the trichloroacetic acid (TCA) precipitation method. Dissociated cells isolated from tumors by percoll gradient as described above, were seeded in 12-well tissue culture plates at the initial density of 1×106 cells/well and cultured in serum free media as indicated above. After 24 hours, the tumorspheres were treated for either 24 or 48 hours with indicated drugs. [3H]-thymidine (NEN/PerkinElmer), 2 μCi/well, was added to the wells during the last 18 hours of treatment. Tumorspheres were harvested and centrifuged at 800 × g for 10 min at 4°C. After removal of the supernatant, the pellets were washed twice with 1 mL of PBS then lysed in 500 μl of NaOH (0.5 M) for 10 minutes at room temperature. Cell extracts were transferred into fresh tubes and DNA was precipitated with 500 μl of cold 20% TCA on ice for 20 min. The samples were centrifuged at 20,000 × g for 20 min at 4°C. Supernatants were aspirated and the DNA-containing pellets washed with 500 μl of 10% TCA. After a 10 min centrifugation, pellets were finally reconstituted in 500 μl of 0.1 M NaOH and transferred into scintillation vials. Incorporated radioactivity was counted on a scintillation counter (Beckman) using Aquasafe™ scintillation cocktail (Wallac).
Statistics
Statistical analyses were performed using the GraphPad Prism 4.0 software. All P values were calculated using Student's t-test for comparison between two samples only, and ANOVA followed by Bonferroni's post hoc test for multiple comparisons. A value of P < 0.05 was considered significant. All data were normalized to vehicle controls.
Results
Primary Mouse Medulloblastoma Cells Grow as Tumorspheres and Exhibit Constitutive Hedgehog Pathway Activity
Cell lines derived from human or murine MB tumors have been used to understand the biology of MB, but recent evidence indicate that propagated MB cell lines lose much or all of their responsiveness to HH pathway antagonism [40]. We therefore harvested fresh primary MB tumor cells for each experiment. We utilized ptch1+/-/p53+/- double mutant mice for our studies because mutations in the p53 tumor suppressor gene in the context of ptch1 haploinsufficiency results in a higher incidence of MB than that in mice with the ptch1 mutation alone [38]. Tumor cells were harvested and purified by percoll gradient, and single cell suspensions were plated in serum-free media for 1 week using a modification of methods reported by others to preserve HH pathway-dependent proliferation [39]. After several days of culture the cells formed aggregates of increasing size, referred to as "tumorspheres" (Figure 1A). To determine if the HH pathway was active in these cells, we treated these cultures at the end of the 1-week period with the HH pathway antagonist SANT-1 (a known inhibitor of smoothened (SMO), 1 μM [41]) and measured the expression of the HH target gene gli1 by real time RT-PCR. SANT-1 significantly decreased gli1 expression (Figure 1B) relative to vehicle. These data implied that the HH pathway is constitutively active in tumorsphere cultures generated using our experimental paradigm.
Figure 1.

Primary Medulloblastoma Cells Form Tumorspheres and Exhibit Constitutive Hedgehog Pathway Activity. (A)Primary medulloblastoma cells from ptch1+/-/p53+/- mice cultured in a serum-free environment formed tumorsphere-like aggregates within 24 hours, which became progressively larger at three and five days.(B and C) Quantitative real time RT-PCR analysis of gli1 gene expression in 6-7 day old tumorsphere cultures treated for 6 h with either the smoothened antagonist SANT-1 (1 μM) (B) or the smoothened agonist purmorphamine (3 μM) (C) Gene expression experiments were performed at least three times and with at least n = 3 samples for each treatment. (**P < 0.01).
To determine if the HH pathway could be activated beyond basal levels in these tumorsphere cultures, we treated them with the SMO agonist purmorphamine at a concentration (3 μM) shown to induce maximum HH signalling without cellular toxicity [42-44]. Purmorphamine failed to induce gli1 expression above control levels, suggesting that the HH pathway was already maximally activated in the tumorspheres under these experimental conditions (Figure 1C).
Pituitary adenylyl cyclase activating polypeptide (PACAP) and Forskolin Inhibit Hedgehog Pathway Activity in Primary Tumorspheres
Because the HH pathway exhibited a significant level of constitutive activity in primary tumorsphere cultures, we investigated if this activity could be inhibited by either PACAP (10 nM) or forskolin (5 μM), an activator of adenylyl cyclase. We first performed RT-PCR analysis for the PACAP receptor PAC1 and confirmed that its gene expression is preserved in tumorspheres cultures (Figure 2B). Treatment with 10 nM PACAP and 5 μM forskolin resulted in significant down-regulation of gli1 in these cultures compared to vehicle treated controls (Figure 2A).
Figure 2.
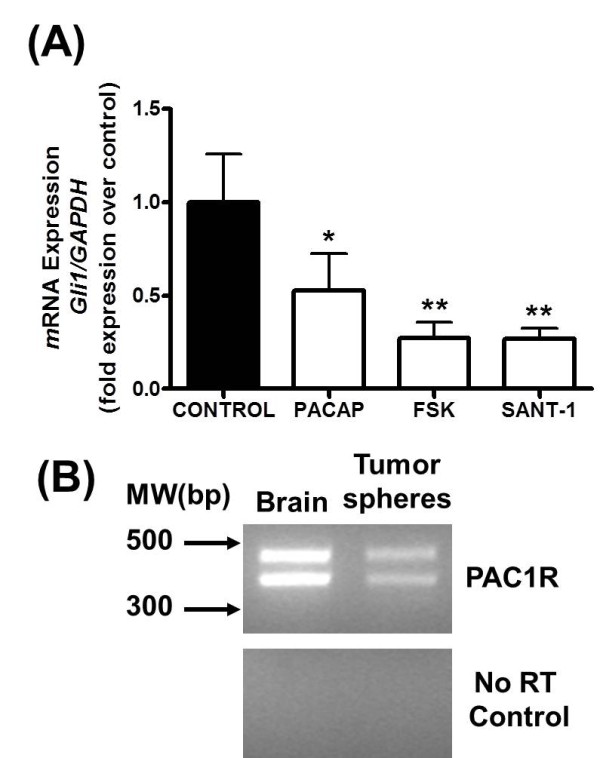
PACAP and Forskolin Inhibit HH pathway activity in Primary Tumorspheres. (A) Expression of the HH target gene gli1 in 6-7 day old primary tumorspheres treated for 6 hours with either vehicle, 10 nM PACAP, 5 μM forskolin, or 1 μM SANT-1, determined by real time RT-PCR. Experiments were performed three times with at least n = 3 for each treatment (*P < 0.05, **P < 0.01). (B) RT-PCR analysis demonstrating gene expression of the PAC1 PACAP receptor in primary tumorspheres. Total brain (left lane) was used as a positive control. Negative controls (lower panel) utilized the same samples, but were not subjected to reverse transcriptase (RT).
Attenuation of Gli1 Expression in Primary Tumorspheres by PACAP was Associated with PKA Activation and was Reversed by the PKA Inhibitor H89
As both PACAP and forskolin typically exert their effects in a PKA dependent manner, we investigated if their effects could be reversed by the PKA inhibitor H89. To confirm their effects on PKA activity, we performed Western blot analyses using an antibody that detects proteins containing a phospho-serine/threonine residue with arginine at the -3 and -2 positions, which are typically found on proteins phosphorylated by PKA. We observed that treatment of primary tumorspheres with either 10 nM PACAP or 5 μM forskolin induced similar increases in PKA activity (Figure 3A) compared to vehicle treated controls. Treatment with H89 (30 μM) reversed the inductions of PKA activity by these treatments (Figure 3A). Moreover, treatment with H89 reversed the reduction in gli1 expression by 10 nM PACAP (Figure 3B). These data indicate that PACAP attenuates HH signaling in primary tumorspheres, most likely via a PKA dependent mechanism.
Figure 3.
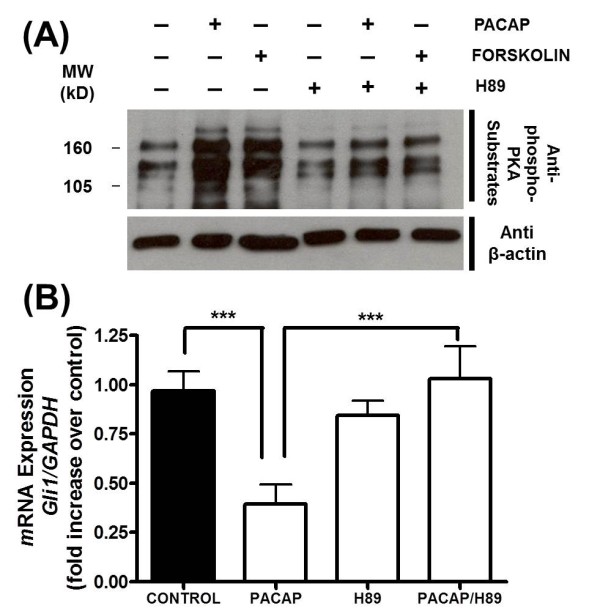
Attenuation of Gli1 Expression in Primary Tumorspheres by PACAP was Associated with PKA Activation and was Reversed by the PKA Inhibitor H89. (A) Western blot analysis of PKA activity in tumorsphere cultures treated with vehicle, 10 nM PACAP, 5 μM forskolin, 30 μM H89, 10 nM PACAP plus 30 μM H89 or 5 μM forskolin plus 30 μM H89. Blot was incubated with a rabbit phospho-PKA-substrate antibody (top) or a mouse β-actin antibody (bottom). The blot shown is representative of 3 experiments. (B) Gli1 gene expression in primary tumorspheres treated with either vehicle, 10 nM PACAP, 30 μM H89, or the combination of PACAP and H89. Experiments were performed at least 3 times and with at least n = 3 for each treatment (***P < 0.001).
PACAP and Forskolin Inhibit Cellular Proliferation in Primary Tumorspheres
The above studies suggest that PACAP treatment or stimulation of PKA activity by other methods might provide strategies to inhibit the growth of MB tumors. We thus investigated the effects of PACAP (10 nM) and forskolin (5 μM) on cellular proliferation using [3H]-thymidine to measure DNA synthesis. As predicted, proliferation was reduced by 24 or 48 hour treatment with PACAP in a dose dependent manner. Forskolin (5 μM) and SANT-1 (1 μM), also decreased proliferation at these time periods (Figures. 4A and 4B). These data suggest that PACAP and PKA activation might provide therapeutic alternatives to SMO antagonism in the treatment of MB.
Figure 4.

PACAP and Forskolin Inhibit Proliferation in Primary Tumorsphere Cultures. [3H]-thymidine incorporation assays were performed in cultures treated with vehicle, increasing concentrations of PACAP, 5 μM forskolin, or 1 μM SANT-1 for 24 hours (A) or 48 hours (B). In each case, [3H]-thymidine was added for the last 18 hr of the experiment. Experiment was performed three times, with 3-4 samples for each treatment (** P < 0.01, *** P < 0.001).
Discussion
A large and diverse array of cancer types show evidence of overactive HH signalling [11]. This has triggered an intensive search for agents that selectively inhibit this pathway [20]. As a result, several molecules have been discovered or designed that directly interact and inhibit the activity of SMO, some of which are in clinical trials [20]. Despite the promise of this approach, two potential problems are that 1) some tumors acquire mutations in SMO that render them relatively resistant to SMO antagonists [45], and 2) SMO is critically involved in the growth of bones and other tissues [11,46], and appears to regulate stem cell biology [9] and [47]. Thus, there is a need for molecules that act downstream of SMO in the HH signalling pathway and that inhibit HH signalling specifically in tumor cells. PKA is thought to act in the HH pathway downstream of SMO, phosphorylating and triggering proteosome-mediated degradation of Gli2 and Gli3 [48,50]. Thus PKA activators might be expected to block HH signalling even in the context of drug-resistant SMO mutations. However, given that PKA is used in so many biological processes, it is unlikely that general activation of PKA will be useful as a treatment strategy. An alternative strategy would be to activate PKA specifically in tumor cells. This could potentially be achieved by targeting appropriate heterotrimeric G protein-coupled receptors expressed on the cells. The data reported here suggest that a potentially-effective strategy to treat MB tumors exhibiting overactive HH signaling would be to activate PACAP receptors. We investigated this possibility by using genetically engineered mice that spontaneously develop MB, and a tissue culture system that preserves a significant level of constitutive HH activity in tumor cells. We found that PACAP inhibited proliferation and the expression of the HH target gene gli1 in these cells. Moreover, the reduction of gli1 gene expression by PACAP was reversed by PKA inhibition, and was mimicked by stimulation of the cAMP/PKA pathway by forskolin. Overall, the data suggest that PACAP, via activation of PKA, inhibits HH signalling and HH-driven proliferation in MB cells. However, our findings do not rule out alternative mechanisms of PACAP/PKA action in these assays. For example, despite its action on gli1 in MB tumorspheres, the ability of PACAP to inhibit proliferation might be unrelated to its effects in HH pathway activity. In this respect, PACAP (and PKA activation in general) is known on inhibit the proliferation of many types of cells [51,53], some of which might not exhibit HH pathway activity. In any case, the findings reported here suggest that PACAP or PAC1 agonists could be useful as alternatives to, or in conjunction with, SMO antagonists, for the treatment of MB and other HH dependent tumors.
Conclusions
Primary murine medulloblastoma derived tumorspheres from ptch1+/-/p53+/- mice, cultured in a serum-free environment, express the PACAP receptor PAC1, and exhibit constitutive HH pathway activity as evidenced by sensitivity to HH pathway antagonist SANT-1. Treatment of tumorspheres with PACAP or the cAMP/PKA activator forskolin significantly reduces gli1 expression in a manner that is reversed by the PKA inhibitor H89. PACAP and forskolin also block tumorsphere proliferation. These finding suggest that PACAP or PKA activation may be useful for therapeutic intervention in the treatment of MB.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JRC: designed and performed the experiments, analyzed and interpreted the data, and wrote the manuscript; DZR: helped in performing experiments; PN: helped with experimental design, technical expertise and data interpretation; HD: performed all animal care, husbandry, and genotyping; LML: helped with experimental design and interpretation of the data; JAW: coordinated the design and execution of experiments, supervised and participated in data analysis, and interpretation, and editing of the manuscript. All authors have read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Joseph R Cohen, Email: joseph.r.cohen@ucla.edu.
Daniel Z Resnick, Email: danielresnick@berkeley.edu.
Pawel Niewiadomski, Email: niewiap@stanford.edu.
Hongmei Dong, Email: hmdong@ucla.edu.
Linda M Liau, Email: lliau@mednet.ucla.edu.
James A Waschek, Email: jwaschek@mednet.ucla.edu.
Acknowledgements
We would like to thank the members of Dr. Waschek's laboratory for their valuable insight and support throughout this work. We would also like to thank Drs. Matthew P. Scott and Owen N. Witte for generously providing the ptch1 and p53 mutant mice, respectively. This present work was supported by NIH grant CA110384 to JW. JRC was supported by the Molecular and Cellular Neurobiology and Intellectual and Developmental Disabilities Research Center training grants (5T32DH007032-30 and 5T32MH19384-13, respectively).
References
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- Ellison D. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol. 2002;28(4):257–282. doi: 10.1046/j.1365-2990.2002.00419.x. [DOI] [PubMed] [Google Scholar]
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2003;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–845. [PubMed] [Google Scholar]
- Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sörensen N, Berthold F, Henk B, Schmandt N, Wolf HK, von Deimling A, Wainwright B, Chenevix-Trench G, Wiestler OD, Wicking C. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085–2088. [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14(3):357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Palma V, Lim DA, Dahmane N, Sánchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Alvarez-Buylla A, Ruiz i Altaba A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132(2):335–344. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Barakat MT, Humke EW, Scott MP. : Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med. 2010;16(8):337–348. doi: 10.1016/j.molmed.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Christiansen J, Wicking C, Zaphiropoulos PG, Chidambaram A, Gerrard B, Vorechovsky I, Bale AE, Toftgard R, Dean M, Wainwright B. A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J Biol Chem. 1996;271(21):12125–12128. doi: 10.1074/jbc.271.21.12125. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272(5268):1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Palma V, Dahmane N. Hedgehog-Gli signaling and the growth of the brain. Nat Rev Neurosci. 2002;3(1):24–33. doi: 10.1038/nrn704. [DOI] [PubMed] [Google Scholar]
- Dellovade T, Romer JT, Curran T, Rubin LL. The hedgehog pathway and neurological disorders. Annu Rev Neurosci. 2006;29:539–563. doi: 10.1146/annurev.neuro.29.051605.112858. [DOI] [PubMed] [Google Scholar]
- Cohen MM Jr. The hedgehog signaling network. Am J Med Genet A. 2003;123A(1):5–28. doi: 10.1002/ajmg.a.20495. [DOI] [PubMed] [Google Scholar]
- Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990;10(2):634–642. doi: 10.1128/mcb.10.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agren M, Kogerman P, Kleman MI, Wessling M, Toftgård R. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli-binding site. Gene. 2004;330:101–114. doi: 10.1016/j.gene.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Mas C, Ruiz i Altaba A. Small molecule modulation of HH-GLI signaling: current leads, trials and tribulations. Biochem Pharmacol. 2010;80(5):712–723. doi: 10.1016/j.bcp.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Li W, Ohlmeyer JT, Lane ME, Kalderon D. Function of protein kinase A in HH signal transduction and Drosophila imaginal disc development. Cell. 1995;80:553–562. doi: 10.1016/0092-8674(95)90509-X. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Bitgood MJ, McMahon A. Protein kinase A is as common negative regulator of SHH signaling in the vertebrate embryo. Genes Dev. 1996;10:647–648. doi: 10.1101/gad.10.6.647. [DOI] [PubMed] [Google Scholar]
- Epstein DJ, Marti E, Scott MP, McMahon AP. Antagonizing cAMP-dependent protein kinase A in the dorsal CNS activates a conserved SHH signaling pathway. Development. 1996;122:2885–2894. doi: 10.1242/dev.122.9.2885. [DOI] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164:567–574. doi: 10.1016/0006-291X(89)91757-9. [DOI] [PubMed] [Google Scholar]
- Nicot A, DiCicco-Bloom E. Regulation of neuroblast mitosis is determined by PACAP receptor isoform expression. Proc Natl Acad Sci USA. 2001;98(8):4758–4763. doi: 10.1073/pnas.071465398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot A, Lelievre V, Tam J, Waschek JA, DiCicco-Bloom E. Pituitary adenylate cyclase-activating polypeptide and sonic hedgehog interact to control cerebellar granule precursor cell proliferation. J Neurosci. 2002;22:9244–9254. doi: 10.1523/JNEUROSCI.22-21-09244.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Gonzalez BJ, Basille M, Fournier A, Vaudry H. Neurotrophic activity of pituitary adenylate cyclase-activating polypeptide on rat cerebellar cortex during development. Proc Natl Acad Sci USA. 1999;96:9415–9420. doi: 10.1073/pnas.96.16.9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Gonzalez BJ, Basille M, Yon L, Fournier A, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to functions. Pharmacol Rev. 2000;52(2):269–324. [PubMed] [Google Scholar]
- Pantaloni C, Brabet P, Bilanges B, Dumuis A, Houssami S, Spengler D, Bockaert J, Journot L. Alternative Splicing in the N-Terminal Extracellular Domain of the Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Receptor Modulates Receptor Selectivity and Relative Potencies of PACAP-27 and PACAP-38 in Phospholipase C Activation. J Biol Chem. 1996;271:22146–22151. doi: 10.1074/jbc.271.36.22146. [DOI] [PubMed] [Google Scholar]
- Straub SG, Sharp GW. A Wortmannin-Sensitive Signal Transduction Pathyway Involved in the Stimulation of Insulin Release by Vasoactive Intestinal Polypeptide and Pituitary Adenylate Cyclase-Activating Polypeptide. J Biol Chem. 1996;271:1660–1668. doi: 10.1074/jbc.271.3.1660. [DOI] [PubMed] [Google Scholar]
- SW Koh. Signal Transduction Through the Vasoactive Intestinal Peptide Receptor Stimulates Phosphorylation of the Kinase pp60c-Src. Biochem Biophys Res Commun. 1991;174:452–458. doi: 10.1016/0006-291X(91)91437-H. [DOI] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP-38) Protects Cerebellar Granule Neurons from Apoptosis by Activating the Mitogen-Activated Protein Kinase (MAP Kinase) Pathway. J Neurosci. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrie AF, Clohessy AM, Buensuceso CS, Rogers MV, Allen JM. Pituitary Adenylyl Cyclase-Activating Peptide Stimulates Extracellular Signal-Regulated Kinase 1 or 2 (ERK1/2) Activity in In Ras-Independent, Mitogen-Activated Protein Kinase/ERK Kinase1 or 2-Dependent Manner in PC12 Cells. J Biol Chem. 1997;272:19666–19671. doi: 10.1074/jbc.272.32.19666. [DOI] [PubMed] [Google Scholar]
- Lelievre V, Pineau N, Du J, C-H Wen, T.B Nguyen, Janet J, J-M Muller, JA Waschek. Differential Effects of Peptide Histidine Isoleucine (PHI) and Related Peptides on Stimulation and Suppression on Neuroblastoma Cell Proliferation:A Novel VIP-Independent Action of PHI via MAP Kinase. J Biological Chemistry. 1998;273:19685–19690. doi: 10.1074/jbc.273.31.19685. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Charytoniuk D, Watroba L, Faure H, Sales N, Ruat M. Discrete localizations of hedgehog signalling components in the developing and adult rat nervous system. Eur J Neurosci. 1999;11(9):3199–3214. doi: 10.1046/j.1460-9568.1999.00777.x. [DOI] [PubMed] [Google Scholar]
- Lelievre V, Seksenyan A, Nobuta H, Yong WH, Chhith S, Niewiadomski P, Cohen JR, Dong H, Flores A, Liau LM, Kornblum HI, Scott MP, Waschek JA. Disruption Of The PACAP Gene Promotes Medulloblastoma In PTC1 Mutant Mice. Develop Biol. 2008;313(1):359–370. doi: 10.1016/j.ydbio.2007.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61(2):513–516. [PubMed] [Google Scholar]
- Zhao H, Ayrault O, Zindy F, Kim JH, Roussel MF. Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev. 2008;22(6):722–727. doi: 10.1101/gad.1636408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, Curran T. Shh Pathway Activity Is Down-Regulated in Cultured Medulloblastoma Cells. Implications for Preclinical Studies Cancer Res. 2006;66(8):4215–4222. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA. 2002;99(22):14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Walker J, Zhang J, Ding S, Schultz PG. Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem Biol. 2004;11(9):1229–1238. doi: 10.1016/j.chembiol.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat Chem Biol. 2006;2(1):29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- Fontaine C, Cousin W, Plaisant M, Dani C, Paraldi P. Hedgehog signaling alters adipocyte maturation of human mesenchymal stem cells. Stem Cells. 2008;26(4):1037–1046. doi: 10.1634/stemcells.2007-0974. [DOI] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13(3):249–260. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- Zhou JX, Jia LW, Liu WM, Miao CL, Liu S, Cao YJ, Duan EK. Role of sonic hedgehog in maintaining a pool of proliferating stem cells in the human fetal epidermis. Hum Reprod. 2006;21(7):1698–1704. doi: 10.1093/humrep/del086. [DOI] [PubMed] [Google Scholar]
- Pan Y, Wang C, Wang B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev Biol. 2009;326(1):177–189. doi: 10.1016/j.ydbio.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. doi: 10.1016/S0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Tempé D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26(11):4316–4326. doi: 10.1128/MCB.02183-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschek JA, Dicicco-Bloom E, Nicot A, Lelievre V. Hedgehog signaling: new targets for GPCRs coupled to cAMP and protein kinase A. Ann N Y Acad Sci. 2006;1070:120–128. doi: 10.1196/annals.1317.089. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after thediscovery. Pharmacol Rev. 2009;61(3):283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- Fila T, Trazzi S, Crochemore C, Bartesaghi R, Ciani E. Lot1 is a key element of the pituitary adenylate cyclase-activating polypeptide (PACAP)/cyclic AMP pathway that negatively regulates neuronal precursor proliferation. J Biol Chem. 2009;284(22):15325–15338. doi: 10.1074/jbc.M109.002329. [DOI] [PMC free article] [PubMed] [Google Scholar]