

Abstract
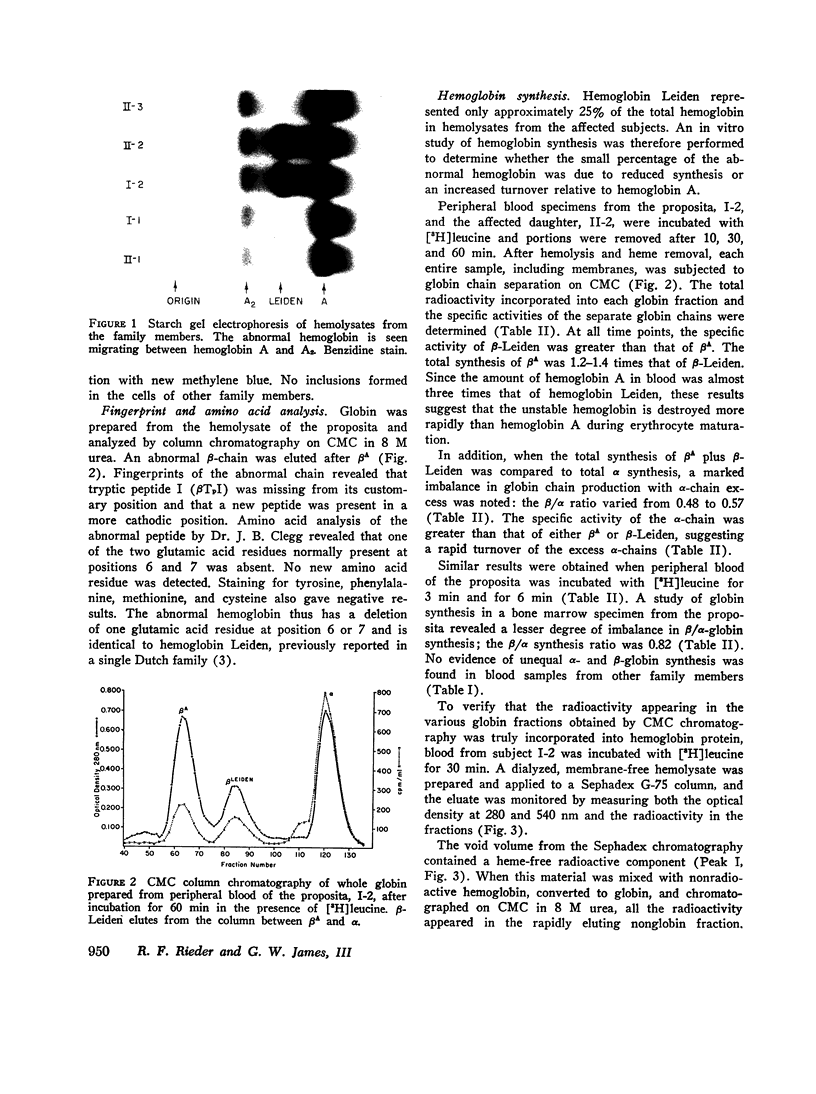
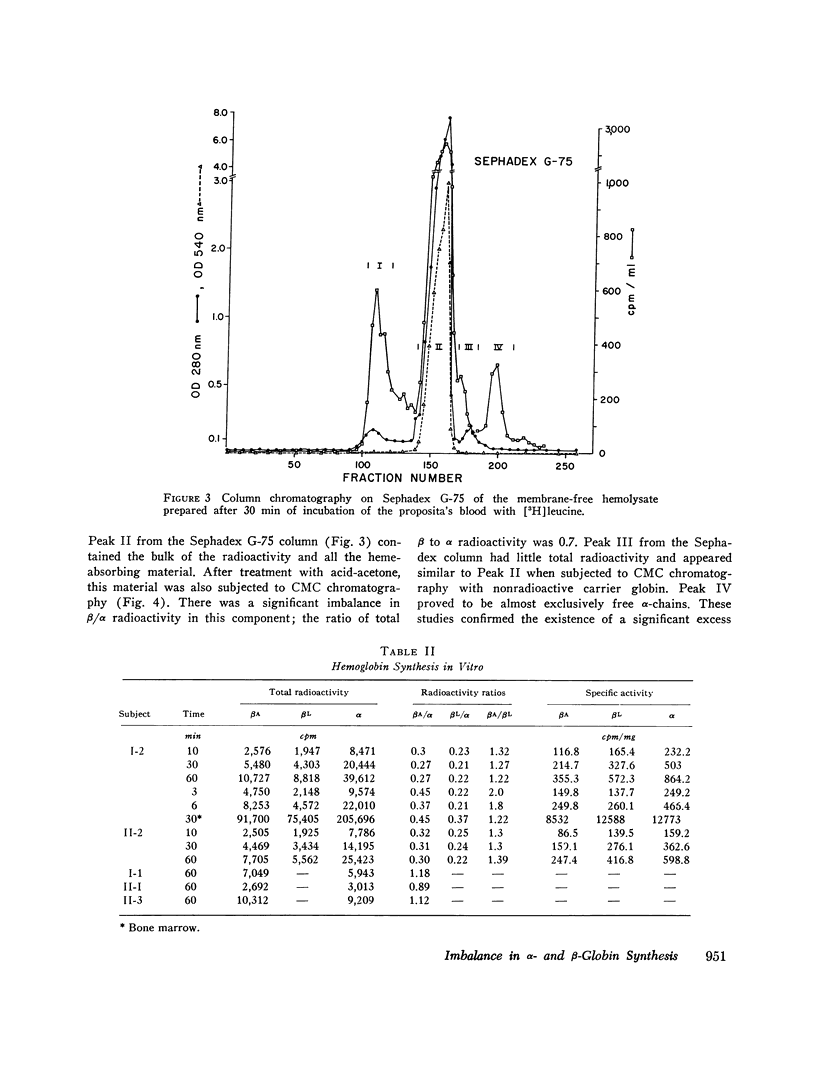
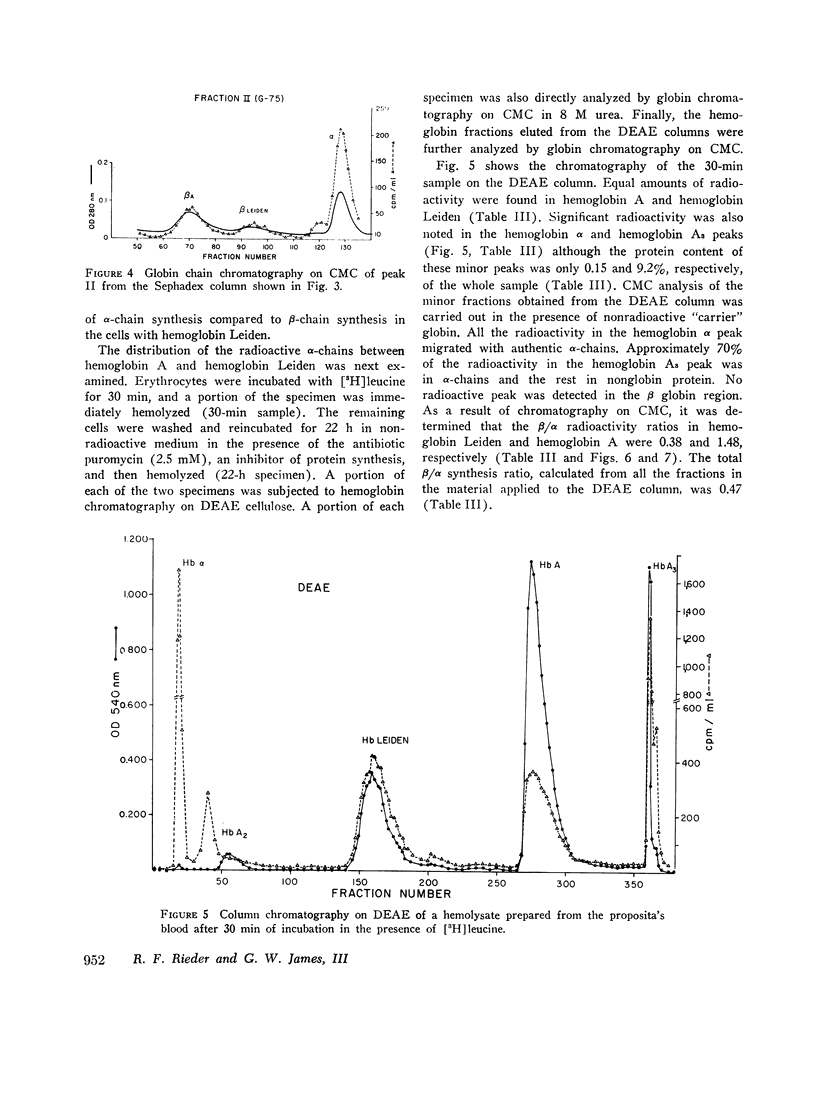
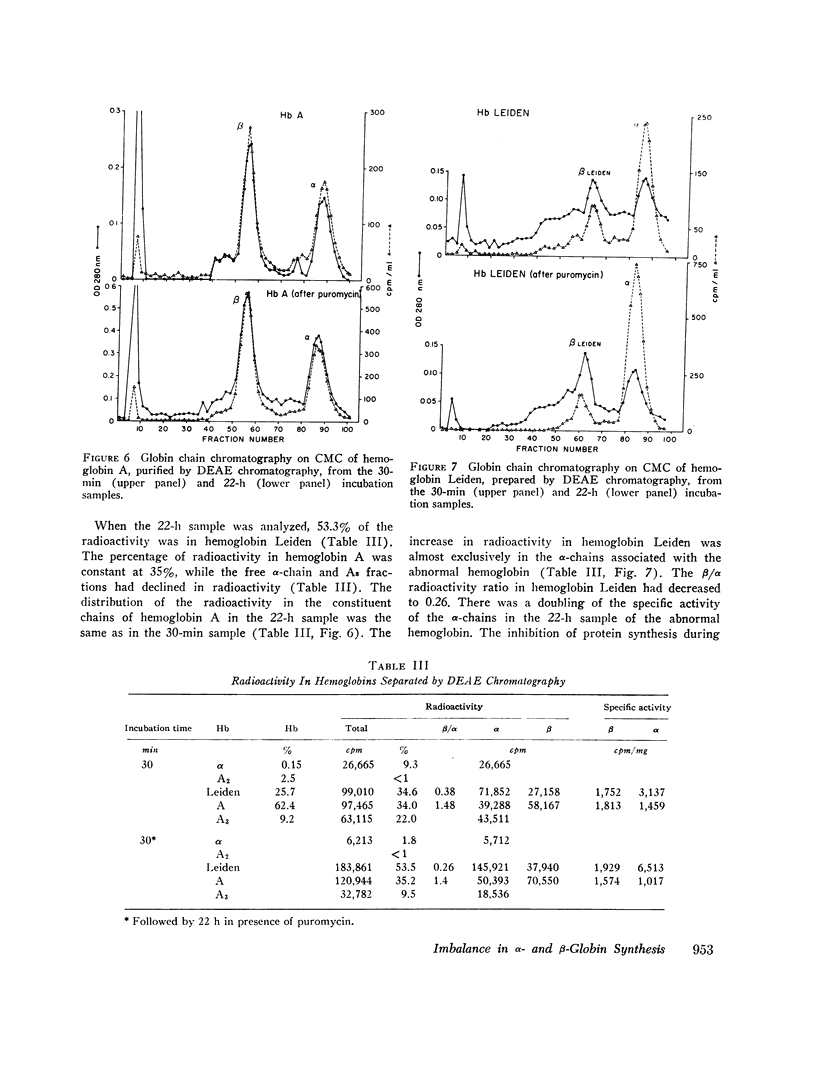
In contrast to findings in the thalasemia syndromes, studies of globin synthesis in subjects with structurally abnormal hemoglobins have generally revealed equal production of α and β polypeptide chains. However, in the present investigation of globin biosynthesis in vitro in blood and marrow from two subjects heterozygous for unstable hemoglobin Leiden, β6 or 7 Glu → O, a significant excess of α-chain production was revealed. A mother and daughter of northern European ancestry with mild compensated hemolytic anemia were found to have 25% hemoglobin Leiden. Increased hemolysis occurred after the ingestion of a sulfonamide and during infections. Normal levels of hemoglobin A2, 3.0 and 2.7%, and hemoglobin F, 0.8 and 0.6%, were found in the two subjects. Similar percentages of the minor hemoglobins were demonstrated in other family members without hemoglobin Leiden. After incubation of peripheral blood with [3H]-leucine, the βA/βLeiden synthesis ratio was 1.3, and the specific activity of βLeiden was 1.3-2 times βA. These results indicate preferential destruction of the unstable hemoglobin Leiden. However, in contrast to previous studies of other unstable hemoglobins, there was excess synthesis of α-chains. The total β/α synthesis ratio was 0.47-0.63 in peripheral blood and 0.82 in marrow. A pool of free α-chains was demonstrated by starch gel electrophoresis and DEAE column chromatography. The synthesis of globin chains was balanced in family members without hemoglobin Leiden. This degree of predominance of α-chain synthesis in subjects with hemoglobin Leiden resembles the findings in heterozygous β-thalassemia. However, the relatively normal hemoglobin content of the cells with this abnormal hemoglobin suggests the possibility of an absolute excess α-chain production in the hemoglobin Leiden syndrome.
Full text
PDF





Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Adams J. G., 3rd, Winter W. P., Rucknagel D. L., Spencer H. H. Biosynthesis of hemoglobin Ann Arbor: evidence for catabolic and feedback regulation. Science. 1972 Jun 30;176(4042):1427–1429. doi: 10.1126/science.176.4042.1427. [DOI] [PubMed] [Google Scholar]
- Bank A., O'Donnell J. V., Braverman A. S. Globin chain synthesis in heterozygotes for beta chain mutations. J Lab Clin Med. 1970 Oct;76(4):616–621. [PubMed] [Google Scholar]
- Benz E. J., Jr, Forget B. G. Defect in messenger RNA for human hemoglobin synthesis in beta thalassemia. J Clin Invest. 1971 Dec;50(12):2755–2760. doi: 10.1172/JCI106778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg J. B., Naughton M. A., Weatherball D. J. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol. 1966 Aug;19(1):91–108. doi: 10.1016/s0022-2836(66)80052-9. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Weatherall D. J., Milner P. F. Haemoglobin Constant Spring--a chain termination mutant? Nature. 1971 Dec 10;234(5328):337–340. doi: 10.1038/234337a0. [DOI] [PubMed] [Google Scholar]
- De Jong W. W., Went L. N., Bernini L. F. Haemoglobin Leiden: deletion of beta-6 or 7 glutamic acid. Nature. 1968 Nov 23;220(5169):788–790. doi: 10.1038/220788a0. [DOI] [PubMed] [Google Scholar]
- Esan G. J., Morgan F. J., O'Donnell J. V., Ford S., Bank A. Diminished synthesis of an alpha chain mutant, hemoglobin I (alpha-16 lys leads to glu). J Clin Invest. 1970 Dec;49(12):2218–2221. doi: 10.1172/JCI106440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill F., Atwater J., Schwartz E. Hemoglobin Lepore trait: globin synthesis in bone marrow and peripheral blood. Science. 1972 Nov 10;178(4061):623–625. doi: 10.1126/science.178.4061.623. [DOI] [PubMed] [Google Scholar]
- Housman D., Forget B. G., Skoultchi A., Benz E. J., Jr Quantitative deficiency of chain-specific globin messenger ribonucleic acids in the thalassemia syndromes. Proc Natl Acad Sci U S A. 1973 Jun;70(6):1809–1813. doi: 10.1073/pnas.70.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby M. A., Mason R. G., Honig G. R. Unbalanced globin chain synthesis in congenital dyserythropoietic anemia. Blood. 1973 Dec;42(6):843–850. [PubMed] [Google Scholar]
- Huehns E. R. The unstable haemoglobins. Bull Soc Chim Biol (Paris) 1970;52(11):1131–1146. [PubMed] [Google Scholar]
- Kacian D. L., Gambino R., Dow L. W., Grossbard E., Natta C., Ramirez F., Spiegelman S., Marks P. A., Bank A. Decreased globin messenger RNA in thalassemia detected by molecular hybridization. Proc Natl Acad Sci U S A. 1973 Jun;70(6):1886–1890. doi: 10.1073/pnas.70.6.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie-Injo L. E., Ganesan J., Clegg J. B., Weatherall D. J. Homozygous state for Hb Constant Spring (slow-moving Hb X components). Blood. 1974 Feb;43(2):251–259. [PubMed] [Google Scholar]
- Nienhuis A. W., Anderson W. F. Isolation and translation of hemoglobin messenger RNA from thalassemia, sickle cell anemia, and normal human reticulocytes. J Clin Invest. 1971 Nov;50(11):2458–2460. doi: 10.1172/JCI106745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder R. F. Synthesis of hemoglobin Gun Hill: increased synthesis of the heme-free beta-GH globin chain and subunit exchange with a free alpha-chain pool. J Clin Invest. 1971 Feb;50(2):388–400. doi: 10.1172/JCI106506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solal M. C., Labie D. A new case of hemoglobin Genova 2 2 28(B10) Leu leads to Pro. Further studies on the mechanism of instability and defective synthesis. Biochim Biophys Acta. 1973 Jan 25;295(1):67–76. doi: 10.1016/0005-2795(73)90074-3. [DOI] [PubMed] [Google Scholar]
- Steadman J. H., Yates A., Huehns E. R. Idiopathic Heinz body anaemia: Hb-Bristol (beta67 (E11) Val to Asp). Br J Haematol. 1970 Apr;18(4):435–446. doi: 10.1111/j.1365-2141.1970.tb01457.x. [DOI] [PubMed] [Google Scholar]
- Weatherall D. J., Clegg J. B., Knox-Macaulay H. H., Bunch C., Hopkins C. R., Temperley I. J. A genetically determined disorder with features both of thalassaemia and congenital dyserythropoietic anaemia. Br J Haematol. 1973 Jun;24(6):681–702. doi: 10.1111/j.1365-2141.1973.tb01696.x. [DOI] [PubMed] [Google Scholar]
- Weissman S. M., Jeffries I., Karon M. The synthesis of alpha, beta, and delta peptide chains by reticulocytes from subjects with thalassemia or hemoglobin Lepore. J Lab Clin Med. 1967 Feb;69(2):183–193. [PubMed] [Google Scholar]
- White J. M., Brain M. C., Ali M. A. Globin synthesis in sideroblastic anaemia. I. Alpha and beta peptide chain synthesis. Br J Haematol. 1971 Mar;20(3):263–275. doi: 10.1111/j.1365-2141.1971.tb07037.x. [DOI] [PubMed] [Google Scholar]
- White J. M., Brain M. C. Defective synthesis of an unstable haemoglobin: haemoglobin Koln (beta 98 Val-Met). Br J Haematol. 1970 Feb;18(2):195–209. doi: 10.1111/j.1365-2141.1970.tb01434.x. [DOI] [PubMed] [Google Scholar]
- White J. M., Dacie J. V. In vitro synthesis of Hb Hammersmith (CDI phe-ser). Nature. 1970 Feb 28;225(5235):860–861. doi: 10.1038/225860a0. [DOI] [PubMed] [Google Scholar]
- White J. M., Lang A., Lorkin P. A., Lehmann H., Reeve J. Synthesis of haemoglobin Lepore. Nat New Biol. 1972 Feb 16;235(59):208–210. doi: 10.1038/newbio235208a0. [DOI] [PubMed] [Google Scholar]
- White J. M. The synthesis of abnormal haemoglobins. Biochimie. 1972;54(5):657–663. doi: 10.1016/s0300-9084(72)80159-7. [DOI] [PubMed] [Google Scholar]