

Abstract
Compared with simple ion receptors, which are able to bind either a cation or an anion, ion pair receptors bearing both a cation and an anion recognition site offer the promise of binding ion pairs or pairs of ions strongly as the result of direct or indirect cooperative interactions between co-bound ions. This critical review focuses on the recent progress in the design of ion pair receptors and summarizes the various binding modes that have been used to accommodate ion pairs (110 references).
1. Introduction
Over the past several decades, a large number of acyclic and macrocyclic compounds have been synthesized as cation receptors and evaluated for their ability to recognize cations.1–3 In recent years, as the importance of anions in biological and environmental systems has become increasingly recognized, attention has been directed towards the design and construction of anion receptors. As a consequence, both cation and anion recognition are now well-established branches of supramolecular chemistry.4–11 From this body of work it has become increasingly apparent that counterions can play a critical role in modulating the binding strength and selectivity of what appear to be the simplest of ion receptors. This appreciation has, in turn, led to the consideration that more elaborate systems, capable of forming simultaneously complexes with both a cation and an anion, might offer considerable advantages in terms of affinity or selectivity.12–14 Ion pair receptors might also permit a greater level of control over ion recognition, extraction and through-membrane transport than simple ion receptors. In fact, in many cases, ion pair receptors containing binding sites for both cations and anions display affinities for ion pairs or their constituent pairs of ions that are enhanced relative to simple ion receptors. Often this is the result of allosteric effects, such as those derived from favorable electrostatic interactions between the co-bound ions.12–14 However, in spite of their potential applications in various fields, such as salt solubilization, extraction, and membrane transport, the number of well-characterized ion pair receptors remains limited. This could reflect a combination of synthetic challenges (the systems reported to date have not been easy to prepare) and experimental complexities associated with tracking multiple ionic species as well as the high inherent lability of many ion pairs.12–14 Nevertheless, the potential importance of the field leads us to propose that the area could benefit from a critical review. Our goal is to provide such a review with an emphasis on the underlying binding phenomena.
To effect anion recognition, most ion pair receptors take advantage of hydrogen bonding donors (urea, amide, imidazolium, pyrrole, and hydroxyl group), Lewis acidic sites (boron, aluminium and uranyl), and positively charged polyammonium groups.4–11 In contrast, the majority of ion pair receptors rely on lone pair electron donors including crown ethers15 and π-electron donors, such as functionalized calixarenes, for cation recognition.16,17
One motivation for creating and studying ion pair receptors derives from the effects of ion pairing that have been noted in the case of simpler receptor systems. In the case of both anion and cation receptors, the reported binding constants have been found to vary significantly, especially in low dielectric constant media, depending on receptor concentration, the specifics of the ionic guest, their counterion concentrations, and nature of the other ions (if any) present in the medium.18–20 As a result, the interactions between the receptors and the guests are often complicated as the result of ion pairing effects. However, this complexity can be potentially overcome by decreasing the concentration of free counter ions as a result of complexing both cations and anions simultaneously, using, e.g., a ditopic receptor system, especially one that allows for the direct recognition of ion pairs. Here, we review recent efforts to prepare ion pair receptors.
In the limit, ion pair receptors can be classified as binding ion pairs in either a sequential or concurrent fashion. In the case of sequential binding, the receptor can bind one ion of the ion pair on its own. Once bound, this first ion enhances the affinity for the other ion of the ion pair through an allosteric effect or by providing an additional binding driving force, commonly a direct or solvent-mediated electrostatic interaction with its counter ion.12–14 By contrast, in the case of concurrent ion pair binding, the receptor literally forms a simultaneous complex with the anion and cation of the ion pair. Typically, this results in a complex where the two ions of the ion pair are in direct contact or spatially separated via one or more molecules of solvent or by the receptor skeleton.12–14
Another way of classifying ion pair receptors is by how they bind the cations and the anions of targeted ion pairs. Here, three different binding modes can be defined. These limiting modes are depicted in Fig. 1 and differ in how the ion pair is held within a host molecule. The first involves a contact ion pair, wherein the anion and the cation are in a direct contact (Fig. 1a); the second, termed a solvent-bridged ion pair, is where one or more solvent molecules bridges the gap between the anion and the co-bound cation (Fig. 1b), while the third consists of a host-separated ion pair, wherein the anion and the cation are bound relatively far from one another, usually by the receptor framework (Fig. 1c).12–14 Depending on the identities of co-bound ions, the separation distance between ion pairs, the nature of the constituent recognition sites, and the nature of the solvents, a given receptor can bind a given ion pair in one or more of these limiting modes.
Fig. 1.
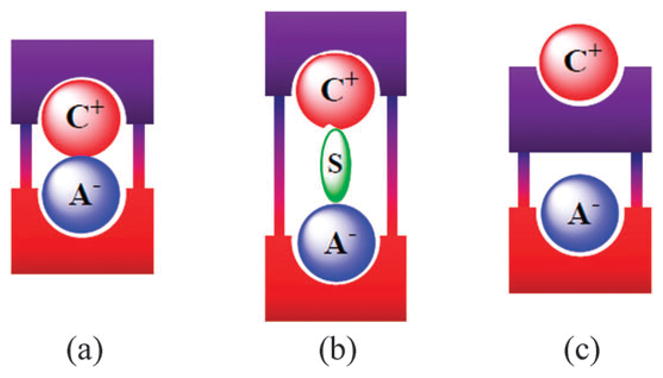
Limiting ion-pair interactions relevant to receptor-mediated ion-pair recognition: (a) contact, (b) solvent-bridged, and (c) host-separated. In this schematic, the anion is shown as “A−”, the cation as “C+”, and the solvent is represented as “S”.
2. Ion pair receptors based on Lewis acidic groups for anion recognition
One of the earliest examples of an ion pair receptor was reported by Reetz and coworkers in 1991. In this system, 1, a crown ether moiety introduced for cation recognition was coupled to a Lewis-acidic boron center for anion recognition to provide compound 1 (Fig. 2).21 It was demonstrated by 11B and 13C NMR spectroscopy that, in contrast to simple ion receptors, such as a crown ether analogue without the boronic center and an arylboroic acid ester without the crown ether, receptor 1 forms a 1 : 1 complex with KF where the K+ cation is bound within the crown ether and the F− anion is covalently bound to the boronic center. One consequence is that this receptor allows the otherwise insoluble salt, KF, to dissolve in dichloromethane within 4 h. The crystal structure of the KF complex of receptor 1 was solved via standard X-ray diffraction means. It reveals that the K+ ion is bound to the crown ether but interacts with an F− anion bound to a neighboring ion pair receptor. As the result of this close intermolecular ion pair contact, a supramolecular polymer is stabilized in the solid state. This same research group also reported in 1994 that ion pair receptor 2 (Fig. 2), consisting of an aluminium phenolate moiety surrounded by a crown ether, is able to bind LiCl as an ion pair simultaneously via interactions with the Lewis acidic aluminium moiety and the crown ether ring. Ion pair binding was seen both in solution and in the solid state.22
Fig. 2.

Structures of 1, 1·KF and 2.
Reinhoudt and coworkers reported the synthesis and the ion pair binding property of receptor 3 (Fig. 3) in 1994.23 This system contains a Lewis-acidic uranyl (UO2)2+ center covalently linked to two benzo[15]crown-5 units. Cyclic voltammetry and FAB-mass spectrometric analysis revealed that receptor 3 can complex K+ and H2PO4− concurrently.23 In the resulting complex, the K+ cation is sandwich-complexed by the two crown ether rings, while the phosphate anion interacts with the Lewis-acidic uranyl group. This team also reported ion pair receptor 4 in 1994. This latter system employs a calix[4]arene tetraethylester subunit as a cation recognition site, a subunit that is known to bind the Na+ cation selectively and strongly. In accord with design expectations, this system was reported to capture Na+ and H2PO4− concurrently and to do so with a 1 : 1 (4 : NaH2PO4) stoichiometry.24 Reinhoudt and coworkers also replaced the benzo[15]crown-5 of receptor 3 with a calix[4]crown-6, a moiety known to complex the Cs+ cation well. This gave receptor 5.25 Once in hand, the ability of 5 to transport CsCl and CsNO3 through a supported liquid membrane (SLM) was assessed. This SLM contained a porous polymeric support (Accurel) impregnated with o-nitrophenyl n-octyl ether (NPOE) as the hydrophobic barrier. Although the Cl− anion is more hydrophilic than NO3−, a higher rate of flux was observed through the hydrophobic membrane for CsCl (1.20 × 10−7 mol m−2 s−1) than for CsNO3 (0.89 × 10−7 mol m−2 s−1) in the presence of 5.25 On the other hand, when mono-functional analogues of 5, control systems lacking the uranyl– salophen complex subunit, were used, selectivity for CsNO3 over CsCl was seen. This observation was considered consistent with the proposal that both binding sites are necessary to achieve efficient complexation and transport of CsCl.25
Fig. 3.

Ion pair receptors 3–5.
In 2003, Rissanen et al. described that the uranyl–salophen complexes 7 and 8 (Fig. 4), receptors bearing appended aromatic groups, behave as ion pair receptors for tetraalkylammonium halide salts.26 Compared with the control receptor 9 lacking aromatic side arms, receptors 7 and 8 were found to bind tetramethylammonium chloride and tetrabutylammonium chloride with improved efficiency. Evidence for binding came from 1H NMR spectroscopic studies, as well as from an X-ray crystal structure. These analyses provided support for a stabilizing cation–π interaction between the tetramethylammonium cations and the appended aromatic rings. Evidence of Lewis acid– anion (chloride) interactions were also seen.26
Fig. 4.

Uranyl–salophen based ion pair receptors.
Interestingly, unlike 9, ion pair receptor 7 was found to form complexes with alkaline halide salts. These complexes consist of contact ion pairs wherein the halide anions are bound to the uranyl center and directly interact with the alkaline cations that in turn are bound to the oxygen atoms and the π-electron surfaces of the benzylic groups. Solid state structural analyses revealed that KCl, RbCl, CsF, and CsCl form capsule-like dimeric (2 : 2) complexes with 7, with the presence of a stabilizing π–metal interaction between the metal ions and the benzylic aromatic rings being specifically inferred.27
Receptor 10, having one benzylic side arm, was also found to bind the CsCl ion pair but with a different binding stoichiometry. Specifically, X-ray diffraction analysis revealed that 10 forms a 4 : 2 (10 : CsCl) complex where four receptor molecules are assembled to provide a capsule-like arrangement.
As a complement to the above studies, 1H NMR spectroscopic titrations of 7–9 were carried out with various quaternary ammonium and iminium salts, such as acetylcholine chloride, N-methylpyridinium iodide, and N-methylisoquinolinium iodide. On the basis of these studies, it was concluded that receptors 7 and 8, bearing pendant aromatic rings, bind quaternary salts much more effectively than does the control compound 9, which lacks a cation recognition site. Such findings provide additional support for the conclusion that the enhanced binding ability demonstrated by 7 and 8 reflects in part the stabilization provided by π–cation interactions.28
In 1995, Shinkai et al. synthesized the Lewis acidic Zn– porphyrin 11 strapped with calix[4]arene having a cation recognition site (Fig. 5).29 The Na+ and K+ cations were found to bind to the upper amide groups thereby creating a cavity between these positively charged ions and the Zn center of the porphyrin subunit. The I− anion was then taken up into this cavity, being bound there by a combination of hydrogen bonding interactions involving the lower amide groups and direct coordination to the Lewis acidic Zn–porphyrin center. Presumably, as the result of this latter binding interaction, a change in the color of the system is induced. Proton NMR spectroscopic analyses revealed that the K+ complex of 11 binds the I− anion more efficiently than its Na+ counterpart, a finding that is ascribable to the different electron accepting character of the Na+ cation and the larger distance between the Na+ cation and the Zn(II) center.29
Fig. 5.
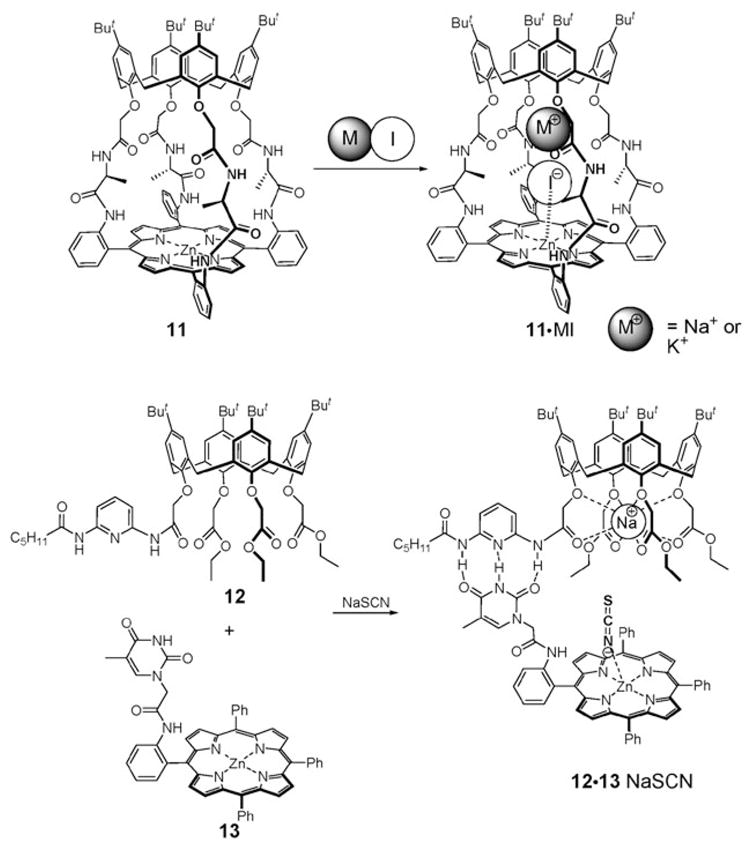
Ion pair receptors based on calix[4]arenes and Zn–porphyrins and their proposed mode of interaction with ion pairs.
In 1995, the self-assembled ditopic receptor 12·13 (Fig. 5) was synthesized by Reinhoudt and coworkers.30 This self-assembled system and its constituents were then evaluated for their ability to capture ion pairs. In the presence of the Na+ cation, receptor 12 was found to interact with the anion receptor 13 to form the self-assembled system 12·13. This complex is stabilized via hydrogen bonding interactions involving the diaminopyridine component and the thymine moiety. In the absence of an appropriate cation bound in the ethyl ester cavity, 12 does not interact with 13. Such a finding is ascribed to the fact that under these latter conditions, the diaminopyridine subunit is intramolecularly hydrogen bonded to the ester group and thus less prone to interact with the thymine group of 13. Complexation of the Na+ cation by the ester groups of 12 serves to break the intramolecular hydrogen bonds and thus permits intermolecular self-assembly between receptors 12 and 13. The self-assembled bifunctional receptor 12·13 shows a higher affinity for the SCN− anion (Ka ≈ 25 000 M−1) as compared to the isolated anion receptor 13 (Ka ≈ 10 M−1).30
In 2002, Kim and Hong synthesized the ditopic receptors 14a,b (Fig. 6) and examined their interactions with various sodium salts.31 These systems consist of a Lewis acidic Zn–porphyrin covalently linked to a benzocrown-5 subunit. Changes in the UV/Vis spectra were used to monitor the resulting binding events. On this basis, it was concluded that both of these receptors bind NaCN in chloroform with high selectivity over other sodium salts. This binding of NaCN was accompanied by a color change from red to green as a result of the cyanide anion binding to the Lewis acidic Zn center. Upfield shifts were also observed for the proton signals corresponding to the crown ether moiety. These shifts were taken as evidence that the sodium cation is bound to the crown ether ring, implying that the NaCN is bound to receptors 14a,b in a ditopic fashion. More specific evidence for the strong nature of the complexation process came from two-phase extraction experiments. In notable contrast to what is seen for normal Zn–porphyrins without an appended crown ether moiety, receptors 14a,b were found capable of extracting NaCN from an aqueous phase into CH2Cl2, and forming kinetically stable complexes on the NMR time scale.
Fig. 6.

Ditopic receptors used for the recognition of sodium or potassium cyanide ion pairs and schematic representations of their proposed binding modes.
A similar approach to ditopic ion pair receptor construction was reported by Chen et al. in 2005.32 This research group prepared and then studied the diazacrown ether-capped Zn–porphyrins 15a,b (Fig. 6), focusing on KCN and NaCN recognition and sensing. In this case, changes in the UV/Vis and 1H NMR spectra led to the suggestion that, depending on the size of the crown ether ring, these receptors would selectively bind NaCN and KCN in a ditopic binding fashion in methanol. Specifically, it was found that receptor 15a binds NaCN about 56 times more strongly than KCN, whereas receptor 15b, a system having a bigger crown ether, is 13 times more selective for KCN than for NaCN.
In 2003, Peacock and coworkers reported the water-soluble ion pair receptor 16 (Fig. 7), which consists of a dibenzo-24-crown-8 subunit linked to a Zn(H2O)-complexed cyclen via a glycine bridge.33 Binding experiments, carried out using 1H NMR spectroscopy, UV/Vis spectroscopy, and ITC (isothermal titration calorimetry), revealed that receptor 16 binds H2PO4 2− 3–6 times more strongly than the cyclen· Zn(H2O) control compound 17. This finding provides support for the suggestion that an enhancement in the inherent affinity is obtained as the result of the ditopic binding mode permitted in the case of 16 as the result of the crown ether ring. While both sodium and potassium phosphates were bound by receptor 17 and with essentially identical affinities, potassium phosphate was bound by receptor 16 approximately twice as well as the corresponding sodium salt (cf. Table 1). This finding is rationalized in term of the size of the crown ether present in 16, which was expected to favor the K+ cation over the Na+ cation. The difference in selectivity seen for 16 and 17 was taken as evidence that the crown ring and the Zn(H2O)-complexed cyclen moieties bind the potassium cation and the phosphate anion, respectively. It was thus concluded that compound 16 works as an ion pair receptor. Analogous cation-dependant results were observed for receptor 18 (Fig. 7) having a smaller crown ether.34 In this case, it was found that the affinities for LiH2PO4, NaH2PO4, and KH2PO4 displayed by this two-component receptor were at least twice as high as those of the Zn(H2O)-complexed cyclen 17; again, this was interpreted in terms of the crown ether ring playing a critical role in recognizing these particular ion pairs.34
Fig. 7.

Zn(H2O)-complexed cyclen-based water soluble ion pair receptors used for the recognition of cyanide alkali metal salts.
Table 1.
Association constants corresponding to the interaction of receptor 16 and the control cyclen·Zn(H2O) system 17 with NaH2PO4 and KH2PO4 in HEPES buffer at pH 7.4 at RT
| Ligand | Substrate | K/M−1 |
|---|---|---|
| 16 | NaH2PO4 | (4.93 ± 0.72) × 104 |
| 16 | KH2PO4 | (9.32 ± 1.60) × 104 |
| 17 | NaH2PO4 | (1.60 ± 0.09) × 104 |
| 17 | KH2PO4 | (1.52 ± 0.19) × 104 |
In 2001, Willem and coworkers reported the synthesis of receptors 19a,b (Fig. 8) comprised of a triphenyltin species linked to various sized benzocrown ethers via an ester bridge. These researchers demonstrated that these receptors were capable of complexing thiocyanate anion salts. The resulting complexes were characterized by their unusual zwitterionic nature (i.e., a degree of intramolecular charge separation), which is reflected in a relatively large distance between the Na+ cation and the formally negatively charged tin center.35 A combination of 1H NNR and 117Sn NMR spectroscopy, as well as X-ray crystal structural analyses, established that for NaSCN and KSCN, respectively, the Na+ and K+ cations are complexed by the crown ether component, whereas the NCS− anion interacts with the Sn atom (Fig. 8).
Fig. 8.
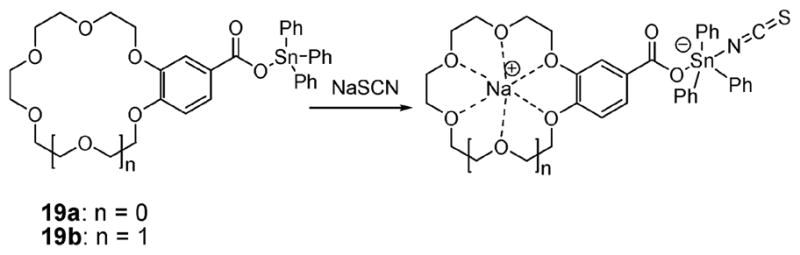
Ion pair receptors based on a combination of a Lewis acidic tin center and a benzocrown ether. Also shown are their proposed binding modes.
In 2007, Jurkschat et al. reported the bis(organostannyl)-methane derivative 20 bearing a crown ether.36,37 On the basis of 1H, 13C, 19F, and 119Sn NMR spectroscopy, as well as ESI mass spectrometry, it was proposed that receptor 20 binds NaF as an ion pair in acetonitrile, wherein the constituent ions are spatially separated by the host molecule (Fig. 9). Presumably, as a consequence of being able to effect concurrent cation and anion binding, receptor 20 was found capable of dissolving solid NaF in acetonitrile, a solvent in which it is otherwise insoluble. On the other hand, an X-crystal structural analysis revealed that, in the presence of methanol, the Na+ and F− ions bound to receptor 20 are bridged by a methanol molecule, thus forming a solvent-separated ion pair.38
Fig. 9.

Structure of the ion pair receptor 20. Also shown are the proposed ditopic binding modes that are thought to pertain in acetonitrile and methanol, respectively.
3. Ion pair receptors based on urea groups for anion recognition
The synthesis of the first urea-based ditopic ion pair receptors and their interactions with alkali halide salts were reported by Reinhoudt et al. in 1996.39 These researchers attached either two or four urea moieties to the upper rim of a calix[4]arene core that possessed the cation-recognizing tetra(ethyl ester) groups on the lower rim. This gave the ion pair receptors 21 and 22a,b (Fig. 10). In CDCl3, receptor 21 was found to adopt a pinched cone conformation, presumably as the result of intramolecular hydrogen bonding between the trans-like urea groups (Fig. 11). These researchers proposed that these internal hydrogen bonds preclude anion binding to the urea moieties in the absence of Na+. However, in the presence of the Na+, cation–ester interactions at the lower rim of the calix[4]arene serve to alter the conformation of calix[4]arene, as shown in Fig. 11. This proposed molecular motion serves to break the hydrogen bonds between the urea groups. As a result, halide ions, such as Cl− and Br−, are bound to the urea groups on the upper rim of the calixarene, as evidenced by downfield shift of the urea protons observed in the 1H NMR spectra. Interestingly, no evidence of Cs+ cation binding within the cavity was seen. Likely, this reflects the fact that the Cs+ ion is too big to fit in the cavity. It was also shown via 1H NMR spectroscopic analysis that receptors 22a,b, as well as 21, are able to extract certain solid sodium halide salts into chloroform solution with several potassium halide salts being partially solubilized (Table 2). Such findings are in agreement with the higher affinity displayed by the calix[4]arene tetra(ethyl ester) derivatives towards the Na+ cation than the K+ cation.36 The binding mechanism proposed in the case of NaCl and NaBr is conceptually similar to the allosteric action of enzymes and biological receptors, wherein the binding of one ion induces a major conformational change in the receptor such that the affinity to the counter-ion is significantly improved.
Fig. 10.

Structures of ion pair receptors 21 and 22a,b.
Fig. 11.

Proposed ion-induced molecular motions and associated binding modes for receptor 21 thought to pertain in the presence of NaCl.
Table 2.
Percentage of MX complex formed with 21 and 22a,b after liquid–solid extraction using chloroform as the organic phase
| 21 |
22a |
22b |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Na+ | K+ | Cs+ | Na+ | K+ | Cs+ | Na+ | K+ | Cs+ | |
| Cl− | 100 | — | — | 100 | 29 | — | 100 | 30 | — |
| Br− | 100 | 16 | — | 100 | 62 | — | 100 | 75 | — |
| I− | 100 | 100 | — | 100 | 100 | — | 100 | 100 | — |
Reinhoudt and coworkers also investigated the ability of (i) cation receptors 23a,b, (ii) a mixture of 23 with 24, and (iii) ditopic receptors 25a,b to transport salts through SLMs (Fig. 12).40 On this basis, they concluded that the mixture of 23a and 24 transports CsCl more efficiently than the corresponding mono-functional calix[4]crown-6 analogue 24 does on its alone. Moreover, ditopic receptors 25a and 25b proved capable of transporting KCl and CsCl, respectively. Presumably, this reflects the fact that these receptors can form complexes with both the constituent cations and anions concurrently. It was also noted that receptor 25b transports CsCl much more efficiently than the cation carrier 23b or the anion carrier 24. By contrast, at higher concentrations of KCl, 25a is less effective for transporting KCl than is the mono-functional cation carrier 23a. This finding was attributed to the very low rate of diffusion of the bifunctional carrier complex.40
Fig. 12.

Cation, anion and ditopic carriers for cation and anion transport.
In 2002, a similar synthetic strategy was applied by Kang and Nam and used to synthesize the ion pair receptor 26 (Fig. 13).41 This system is based on a 1,3-alternate calix[4]-arene skeleton strapped by a crown-5 subunit, a moiety that is known to form very strong and very selective complexes with the K+ cation. To permit anion recognition, two urea moieties were introduced onto the calix[4]arene framework. In the presence of 1.0 molar equiv. of KClO4, the association constants (Ka) of 26 for Cl− (5420 M−1), Br− (1550 M−1), and I− (808 M−1) were recorded to be 3.9–5.8 times larger than those measured in its absence. A year later, it was reported that receptor 27, which possesses a strong and selective binding site for the Na+ cation, binds the Cl− and the Br− anions 7.1 and 20 times more effectively, respectively, in the presence of the Na+ cation than it does in the absence of a crown-bound cation.42
Fig. 13.

Calix[4]arene and urea based ion pair receptors 26–28.
In 2003, Webber and Beer reported the calix[4]arene-based ion pair receptor 28, a system that consists of a calix[4]semitube for cation binding and two urea groups for anion recognition.43 On the basis of 1H NMR spectroscopic studies, receptor 28 was found to bind a range of sodium and potassium halide and acetate salts in a cooperative fashion in CDCl3/CD3CN (2/1, v/v). Anion binding enhancements of over thirty-fold in the case of Br− were seen (Table 3). Extraction experiments carried out by these researchers also revealed that the ion pair receptor 28 can solubilize certain sodium and potassium salts in chloroform (cf. Table 4).
Table 3.
Stability data for the anion complexes of 28, [28·Na+] and [28·K+] as determined in CDCl3/CD3CN (2/1, v/v)
|
Ka/M−1 |
|||
|---|---|---|---|
| Free 28 | [28·Na+] | [28·K+] | |
| Cl− | 60 | N/A | >730 |
| Br− | 20 | 620 | 550 |
| I− | 15 | 280 | 310 |
| OAc− | 110 | N/A | 710 |
Table 4.
Solid–liquid extraction data for Group 1 halide and acetate salts recorded using 28 as the receptor and CDCl3 as the solvent. The results are given as the percentage of complex formation
| Complex (%) [lattice energy/kJ mol−1] |
||
|---|---|---|
| Na+ | K+ | |
| Cl− | 0 [786] | 0 [715] |
| Br− | 3 [747] | 13 [682] |
| I− | 27 [704] | 95 [649] |
| OAc− | 0 [763] | 13 [682] |
In 2003, Kilburn and coworkers reported ditopic receptor 29 (Fig. 14), a system that coordinates alkali metal cations via the oxygen atoms, while binding the anion in a hydrogen bonding donor pocket formed by the two thiourea groups.44 In the absence of the Na+ cation, receptor 29 binds the acetate anion selectively over the diphenyl phosphate anion. However, in the presence of the Na+ cation, the selectivity is reversed. These observations are rationalized as follows: in the absence of the Na+ cation, electrostatic repulsion between the phenoxy groups of the diphenyl phosphate anion and the ether oxygen atoms of the receptor prevents the receptor from binding the diphenyl phosphate anion. However, in the presence of the Na+ cation, the phenoxy groups interact favorably with the bound Na+ cation, leading to the observed strong binding of the ion pair.
Fig. 14.
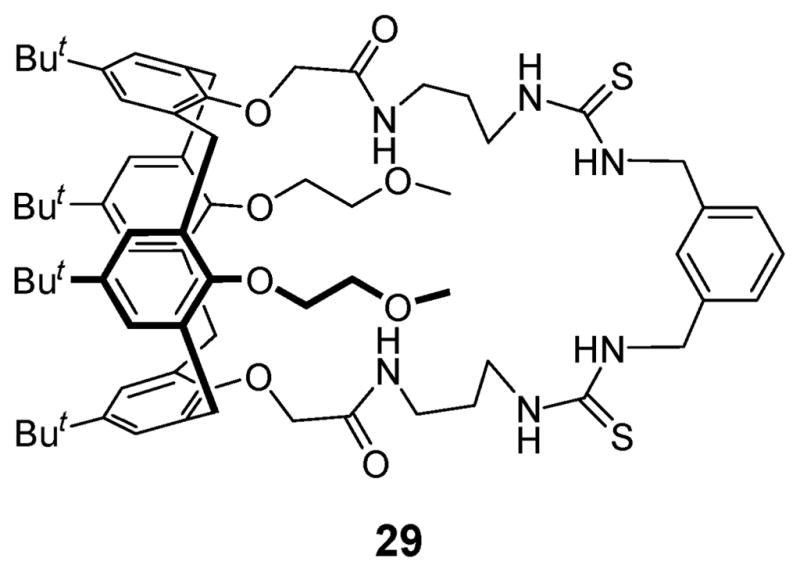
Structure of ditopic receptor 29.
Nabeshima et al. synthesized the more sophisticated tritopic ion receptor 30 (Fig. 15) and examined the effects of cations on its anion binding efficiency.45 This receptor, reported in 2005, is composed of an oxygen-abundant cone calix[4]arene cavity and two bipyridine groups for hard and soft cation recognition, respectively; it also contains two appended urea moieties for anion recognition. It was demonstrated by 1H NMR spectroscopy that either Na+ cation binding to the calix[4]arene cavity or Ag+ cation binding to the bipyridine moieties enhances the affinity of 30 for the NO3− and CF3SO3− anions by a factor of 30–100. This result was rationalized in terms of preorganization of the urea groups into a face-to-face arrangement as the result of cation binding, as well as additional interactions between the bound cations and the incoming anions. In the presence of both Na+ and Ag+, the cation-induced enhancement in the association constants (Ka) of 30 reaches a factor of 1500 and 2000 for NO3− and CF3SO3−, respectively (Table 5).45
Fig. 15.

Tritopic receptor 30 and the binding modes that are thought to pertain in the presence of Na+, Ag+ and various anions.
Table 5.
Association constants, log Ka, for the 1 : 1 host–guest complexes formed from the neutral and cation-bound forms of receptor 30 and selected anionsa
| Host | NO3− | CF3SO3− | BF4− |
|---|---|---|---|
| 30 | 1.88 ± 0.03 | 1.40 ± 0.20 | b |
| 30·Ag+ | 3.31 ± 0.07 | 3.40 ± 0.07 | b |
| 30·Na+ | 3.82 ± 0.15 | 3.32 ± 0.11 | 3.46 ± 0.11 |
| 30·Ag+·Na+ | 5.07 ± 0.17 | 4.70 ± 0.20 | 4.28 ± 0.11 |
Determined by 1H NMR spectroscopy (400 MHz, CDCl3/CD3CN (9 : 1), [host] = 2.0 × 10−3 M).
Not determined due to the small chemical shift change.
In 2005, Pappalardo and coworkers reported the synthesis of receptor 31 (Fig. 16).46 This system was designed for organic ion pair recognition. It contains two convergent and conformationally fixed calix[5]arene subunits for cation recognition and a 1,4-bis(ureido)phenylene moiety capable of binding two anions.46 Proton NMR and TROESY (transverse rotating-frame Overhauser effect) NMR spectroscopy, and electrospray mass spectrometry provided support for the conclusion that the heterotetratopic ion pair receptor 31 is able to bind long-chained and ion-paired α,ω-alkanediyldiammonium salts efficiently (Ka > 106 M−1 for 1,12-dodeca-through 1,16-hexadecanediammonium dichloride salts in (CDCl2)2/CD3OD (2 : 1 v/v)). Three different binding modes were suggested depending on the length of alkyl chains and the number of guest equivalents added. Large upfield shifts in the signals for the methylene protons of the guest molecule and significant downfield shifts of the NH resonances of the host molecule were seen in the 1H NMR spectra. This was taken as evidence that receptor 31 recognizes the constituent ions of the ion pair concurrently and that the ammonium cations and chloride anions are bound to the host through π–cation and hydrogen bonding interactions, respectively. These spectroscopic analyses also revealed that long-chained α,ω-alkane-diyldiammonium acetate and chloride salts induce remarkable downfield shifts in the NH resonance of the urea moiety. These shifts are more substantial than what is seen for TBACl (tetrabutylammonium chloride), whose cation is thought to be too big to be accommodated by the calix[5]arene unit (i.e., Δδ = 0.75 and 0.77 ppm for TBACl and Δδ = 2.37–2.72 ppm for α,ω-alkanediyldiammonium salts). These findings were taken as evidence that receptor 31 forms stronger complexes with alkanediyldiammonium salts than with TBA+. Presumably, this reflects a cooperative interaction with the ion pairs, which are eventually separated by the host.46 This chemistry is summarized in Fig. 16.
Fig. 16.

Proposed recognition of long-chain, ion-paired α,ω-alkane-diyldiammonium salts by calix[5]arene-based heterotetratopic ion pair receptor 31.
In 2009, Secchi and coworkers described the ion pair receptors 32 and 33, where the upper rim of a cation recognizing cone-calix[4]arene moiety is covalently connected with an anion recognizing urea group through a methylene spacer (cf. Fig. 17).47 These workers demonstrated that these receptors display a binding ability that is enhanced by up to two orders of magnitude for organic salts, such as N-methylpyridinium and N,N′-dimethylviologen, with respect to the simple cation receptors 34 and 35.47 Proton NMR and UV/Vis spectroscopic and single crystal X-ray diffraction analyses provided support for the conclusion that ditopic receptors 32 and 33 form cooperative 2 : 1 (host : guest) complexes with such organic ion pairs. In the resulting complexes, the cations are bound to the electron-rich arene rings of the calix[4]arene moiety via cooperative CH–π and cation–π interactions, while the anions interact with the urea moiety via hydrogen bonds. By contrast, the monotopic receptors (34 and 35) recognize these organic salts via only CH–π and cation–π interactions (i.e., without the benefit of additional anion–host interactions) and form 1 : 1 (host : guest) complexes.47
Fig. 17.

Calix[4]arene-based monotopic and heteroditopic receptors 32–35 and depictions of the N-methylpyridinium and N,N′-dimethyl-viologen salts used as guest molecules.
Similarly, as reported by Ballistreri et al., a cone calix[5]- arene derivative bearing a urea subunit and four tert-butyl groups on the upper rim, as well as five isohexyl groups on the lower rim, was found to bind zwitterionic γ- and ω-amino acids very efficiently as compared with its calix[5]arene derivative without a urea group.48 In this case, 1H NMR spectra measured in a solution of C2D2Cl2/CD3OD (2/1) or neat C2D2Cl2 revealed that the observed strong interaction between the ureidocalix[5]arene receptor in question and the targeted amino acids is achieved as the result of the synergic action of the cation binding and anion binding sites; specifically, it was inferred that the carboxylate moiety of amino acids is hydrogen-bonded to the NH’s of the urea group, while the ammonium moiety is bound to the calix[5]arene cavity as the result of hydrogen bonding interactions involving the phenolic oxygen atoms, as well as via π–cation interactions.48
Also in 2009, Gargiulli et al. reported the synthesis of the ion pair receptor 36 (Fig. 18) which consists of a cation recognizing calix[5]arene crown-3 in the cone conformation tethered to three anion recognizing urea units.49 It was shown that this system is capable of capturing n-butylammonium chloride and 2-phenylethylamine hydrochloride ion pairs. The binding mode was inferred from 1H NMR spectroscopic analyses and semiempirical calculations carried out at the PM3 level. On this basis, it was proposed that the ammonium cation is bound to the calix[5]arene cavity, while the chloride anion is complexed by the three facing urea groups, as shown in Fig. 18. The net result is a receptor-separated ion pair complex.
Fig. 18.
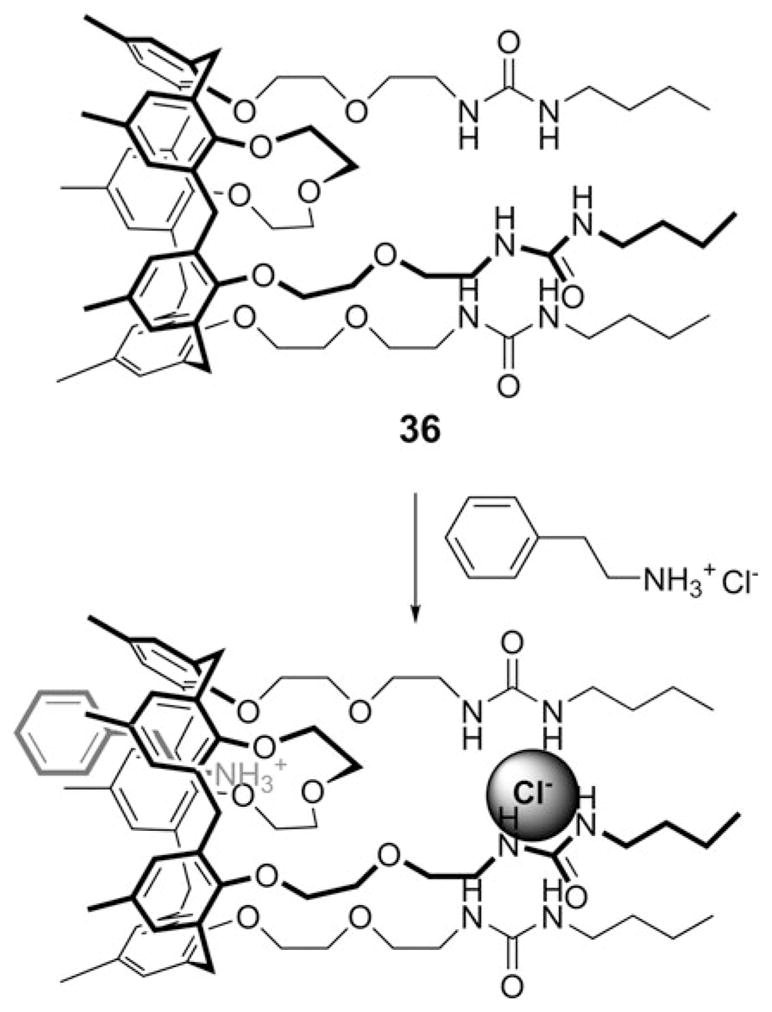
Proposed recognition of an alkyl ammonium salt (2-phenyl-ethylamine hydrochloride) by receptor 36.
Receptor 37 (Fig. 19) reported by Kubo and coworkers in 2000 antedates the above systems. It also works as an ion pair receptor and is thought to operate on the basis of an allosteric effect.50 In the presence of the K+ cation, the dibenzo-30-crown-10 portion of this receptor wraps around the K+ ion. This brings the two thiourea moieties into close proximity and provides a preorganized binding cavity for the phosphate anion. Compared to the cation-free form of receptor 37 (Ka1 = 490 M−1), the association constants are significantly enhanced for the phosphate anion in the presence of both K+ (Ka1 = 9200 M−1) and Cs+ (Ka1 = 3200 M−1). Prior to this work, it was demonstrated by Nishizawa and coworkers that the thiourea-functionalized benzo-15-crown-5 (38; Fig. 19) binds a series of anions via presumed cooperative interactions with a pre-bound Na+ cation.51 Specifically, in the presence of 2.0 molar equiv. of Na(BPh4) (conditions where the Na+ complexation is >95%), receptor 38 in CD3CN exhibits an approximate ten-fold increase in its affinity for NO3− (KNO3 = 6.0 M−1 vs. KNO3 (Na+) = 66 M−1) and Br− (KBr = 25 M−1 vs. KBr (Na+) = 260 M−1), and a five-fold increase for I− (KI = 4.3 M−1 vs. KI (Na+) = 20 M−1).
Fig. 19.

Simple crown ether–urea and –thiourea based ion pair receptors 37–40.
Almost a decade after these early reports, Nam and coworkers synthesized the anion-dependant colorimetric ion pair receptor 39 (Fig. 19) and evaluated its binding affinity for anions in the presence and absence of the Na+ cation.52 This system contains a nitrophenyl group attached to the thiourea moiety. It displays a color change from colorless to yellow when treated with the fluoride, acetate, benzoate and dihydrogen phosphate anions in acetonitrile. However, the yellow color induced by fluoride anion disappears and returns to colorless upon the addition of the Na+ cation. This was interpreted in terms of the fluoride anion leaving the thiourea moiety to form a contact ion pair with the sodium cation bound to the crown ether. To the extent such an interpretation is correct, receptor 39 exemplifies a negative cooperative effect on anion binding, which is induced by Na+. Interestingly, the affinities towards other anions are enhanced in the presence of the Na+ cation (by 1.2 fold for the acetate anion, 1.1 fold for the chloride anion, 1.6 fold for the bromide anion, 2.0 fold for the iodide anion, and 2.3 fold for the hydrogen sulfate anion).
An ostensibly similar receptor, the crown–urea system 40 (Fig. 19), was reported by Barboiu and coworkers in 2003.53 On the basis of X-ray crystallographic analyses and 1H NMR spectra recorded at various concentrations, it was concluded that the free host 40 forms a tubular superstructure stabilized via head-to-tail urea hydrogen bonding, π–π, and CH–π interactions with an internal van der Waals diameter of 1.27 Å and a spacing of 4.83 Å between parallel off-centered macrocycles (Fig. 20a). These researchers also demonstrated that receptor 40 is able to extract solid sodium salts, such as NaF, NaCl, NaNO3, and NaCF3SO3, into CDCl3 solution. Membrane transport experiments provided support for a direct relationship between the ability to effect synergetic ion pair recognition and mediate ion transport. Interestingly, receptor 40 was found to bind sodium salts in two different modes depending on the nature of the counter anions. For example, a 2 : 1 (host : guest) complex is formed with NaCl in which the constituent ions are separated by the receptor (Fig. 20b). In contrast, a 2 : 2 contact ion pair complex is stabilized in the case of NaNO3, at least in the solid state (Fig. 20c).
Fig. 20.
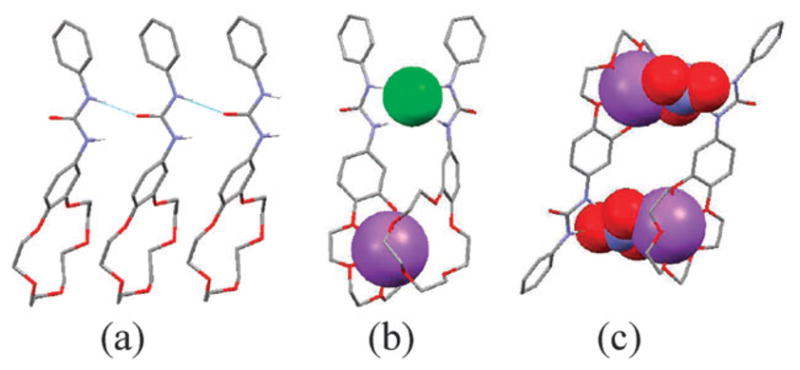
Crystal structures of (a) free receptor 40, (b) its NaCl complex, and (c) the corresponding NaNO3 complex. This figure was redrawn using coordinates that were originally published in ref. 53.
4. Ion pair receptors that contain amide groups for anion recognition
Several groups have designed and synthesized crown ether derivatives that contain one or more amide groups strapped via a short linker and used the resulting system as receptors for the recognition of contact ion pairs. The attraction of this approach is that the partial destabilization resulting from charge separation of ion pairs, the so-called Columbic penalty, can be avoided. In an early example, Kilburn and coworkers showed that compound 41 forms strong complexes with the mono-potassium salts of dicarboxylic acids and phenyl phosphonate, binding these guests as contact ion pairs (Fig. 21).54 A series of solid–liquid and liquid–liquid extraction experiments provided support for the suggestion that this ditopic receptor is able to effect the transfer of varying quantities of the mono-potassium salts of several dicarboxylic acids from water into CDCl3.
Fig. 21.
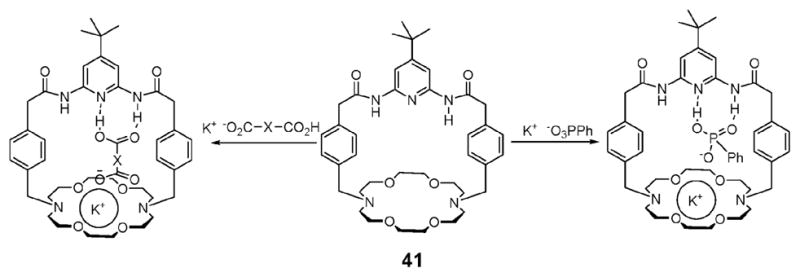
Chemical structure of the ion pair receptor 41 and its proposed binding mode with monopotassium salts of dicarboxylic acids (oxalic acid, malonic acid, methylmalonic acid, benzylmalonic acid, maleic acid, fumaric acid, succinic acid, Z-aminomalonic acid, Z-aspartic acid, and Z-glutamic acid) and phenyl phosphonate.
Seven years later, in 2000, Smith and coworkers prepared the preorganized bicyclic macromolecular ion pair receptor 42 by combining a dibenzo-18-crown-6 with a 1,3-phenylenedicarboxamide subunit (Fig. 22).55 Proton NMR spectroscopic studies carried out in pure DMSO-d6 and in a 3 : 1 mixture of DMSO-d6/CD3CN, combined with single crystal X-ray structural analyses, served to show that receptor 42 is capable of forming a complex containing both alkali metal and chloride ions. A series of complexation experiments carried out in DMSO-d6/CD3CN (3/1, v/v) provided support for the suggestion that 42 binds sodium chloride and potassium chloride in the form of ion pairs and in a highly cooperative fashion. In the presence of 1.0 molar equiv. of Na+ or K+ (added as their tetraphenylborate salts), the chloride anion affinity is enhanced relative to what is seen in the absence of a cation (Table 6). A single crystal X-ray structural analysis of the NaCl complex revealed that receptor 42 binds NaCl as a solvent-separated ion pair, at least in the solid-state. As expected, the Na+ cation is bound within the dibenzocrown unit, with an axial water molecule completing the coordination sphere, whereas the Cl− anion is hydrogen-bonded to the two NH residues as well as to the CH of a chloroform molecule that serves to bridge the two ions of the ion pair. The central cavity is occupied by a CHCl3 molecule or two molecules of water, with the ion–ion separation being 7.31 Å.55
Fig. 22.
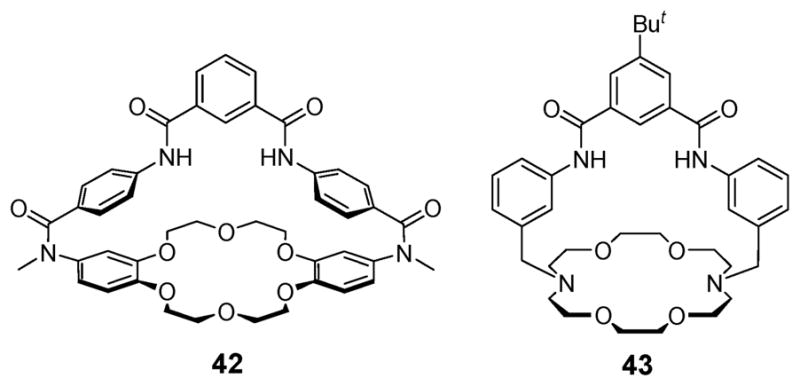
Amide-capped bicyclic ion pair receptors 42 and 43.
Table 6.
Association constants (M−1) for the binding of halide anions to the ion pair receptor 42 for halide anions in the presence or absence of alkali metal ions. The values in parentheses are the change in the chemical shift of the NH protons (Δδmax in ppm)a
| Anionsb | 42 | 42 + Na+c | 42 + K+c | 42 + Cs+c |
|---|---|---|---|---|
| Cl− | 50 (0.93) | 410 (1.16) | 470 (1.28) | 60 (0.95) |
| Br− | 9 (0.18) | 27 (0.38) | ||
| I− | <1 (<0.01) | 11 (0.03) |
T = 295 K, [42] = 10 mM in DMSO-d6/CD3CN (3/1, v/v) in the presence or absence of 1.0 molar equiv. of the designated metal cations. Association constants are the average of all receptor protons that exhibit significant complexation-induced shifts; uncertainty ±15%. The Δδmax values represent the change in the NH chemical shift after adding 10 molar equiv. of the indicated anion.
Anion added as the tetrabutylammonium salt.
Cation added as the tetraphenylborate salt.
A year later, the ion pair receptor 43 (Fig. 22), an analogue of 42, was reported by the same group.56 Receptor 43 was designed to have a smaller distance between the cation and anion binding sites and to recognize sodium and potassium halide salts as contact ion pairs. Single crystal X-ray diffraction structures of the NaCl and KCl complexes of 43 were solved, and revealed that this new receptor binds NaCl and KCl salts as contact ion pairs. This complexation mode stands in contrast to what is seen for 42, which binds NaCl as a solvent separated ion pair. The crystal structures of 43·NaCl and 43·KCl also show that, relative to the KCl complex, the effective cavity size of the diazacrown unit is reduced when the Na+ cation is bound (average K···O distance is 2.77 Å vs. 2.45 Å for Na···O). In addition, the average Cl···O distance in 43· NaCl is much shorter (4.20 Å) than that observed in 43·KCl (4.7 Å). These differences provide support for the appealing suggestion that once a K+ cation is bound, receptor 43·K+ is better able to bind the chloride counter anion to form a favorable contact ion-pair than in the case of 43·NaCl. Proton NMR spectroscopic titration experiments carried out in DMSO-d6 revealed cooperative effects. Specifically, in the presence of either Na+ and K+ or Cl−, the association constants for complexation of the corresponding counter ion increased significantly (Table 7).
Table 7.
Association constants, Ka (M−1), in DMSO at 295 Ka
|
Ka for Cl−b |
Ka for Na +c |
Ka for K +c |
||||
|---|---|---|---|---|---|---|
| 43 | 43 + Na+ | 43 + K+ | 43 | 43 + Cl− | 43 | 43 + Cl− |
| 35 | 50 | 460 | 5 | 25 | 8 | 340 |
Association constants are the average of all receptor protons which exhibit significant complexation-induced shifts. The initial [43]=10 mM; uncertainty ±15%.
43/Cl− association constant in the presence of 1 molar equiv. of metal tetraphenylborate.
43/M+ association constant in the presence of 1 molar equiv. of tetrabutylammonium chloride.
It was also shown by single crystal X-ray diffraction analysis and 1H NMR spectroscopy that receptor 43 can extract LiCl and LiBr from the solid state and bind these salts as water-separated ion pairs. On the other hand, it was found that NaCl, KCl, NaBr and KBr are bound as contact ion pairs.57 In the solid state, the complexes formed between 43 and LiCl and LiCl contain the Li+ cation coordinated to three oxygen atoms and one nitrogen atom of the six heteroatoms in the diazacrown ether moiety, as well as to an additional oxygen atom provided by the water molecule (Fig. 23). In contrast, the Cl− and Br− anions are hydrogen-bonded to the two amide NH protons and one OH proton from a water molecule. The average Li···Ocrown distance in both cases is 2.21 Å for the three coordinating oxygen atoms. The average Li···Owater distances are 1.90 Å for the LiCl complex and 1.91 Å for the LiBr complex. The Li–N distances for the coordinating crown nitrogen are 2.37 Å for the LiCl complex and 2.33 Å for the LiBr complex. In the case of the chloride complexes, the average Namide–Cl distances are 3.30 Å with the average N–H–Cl angle being 177.5°. The Cl–Owater distance is 3.10 Å. In the case of the LiBr complex, the Br− anion is bound to the receptor with distances of 3.41 Å and 3.29 Å being observed for the Namide–Cl and Br–Owater separations, respectively. The average N–H–Br angle is 176.9° (Fig. 23).
Fig. 23.
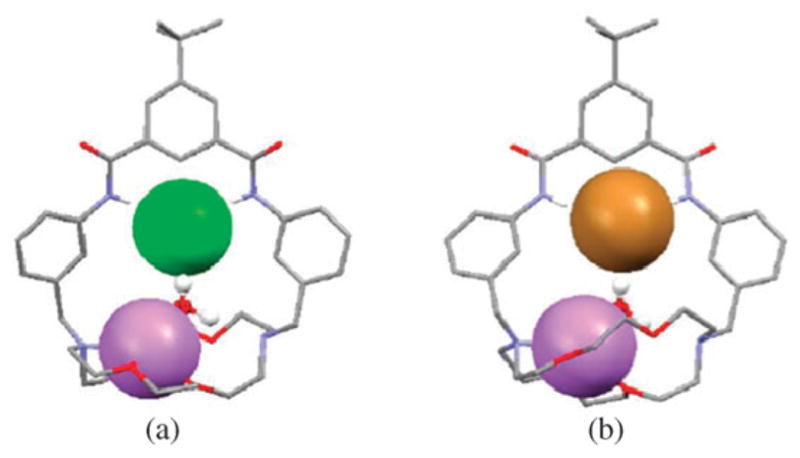
Crystal structures of (a) 43·LiCl·H2O and (b) 43·LiBr·H2O. The lithium cation is bound in the crown ether subunit in both cases. These structures were replotted using coordinates that were originally reported in ref. 57.
Using a supported liquid membrane and a high salt concentration in the source phase, Smith and coworkers carried out transport experiments that involve the use of ion pair receptor 43 as a carrier for alkali halide salt.58 On the basis of these studies, it was concluded that the ditopic receptor (43) can transport alkali halide salts up to 10-fold more quickly than does the monotopic cation receptor 44 or the anion receptor 45 (Fig. 24) and twice as fast as does a 1 : 1 mixture of 44 and 45 (Tables 8 and 9). The same qualitative order of ion selectivity is observed in all transport systems; that is, for a constant anion, the cation selectivity order is K+ > Na+ > Li+, and for a constant cation, the anion transport selectivity order is I− > Br− > Cl−.
Fig. 24.
Structures of control monotopic receptors 44 and 45.
Table 8.
Initial transport fluxes (×10−8 mol m−2 s−1)a
| Carrier | 43 | 44 |
|---|---|---|
| LiCl | 6 ± 1 | 6 ± 1 |
| NaCl | 37 ± 2 | 5 ± 1 |
| KCl | 90 ± 3 | 12 ± 1 |
| LiBr | 10 ± 1 | 6 ± 1 |
| NaBr | 32 ± 2 | 3 ± 1 |
| KBr | 111 ± 3 | 20 ± 1 |
| LiI | 7 ± 1 | 7 ± 1 |
| NaI | 43 ± 2 | 27 ± 2 |
| KI | 160 ± 5 | 40 ± 2 |
Source phase, 1 M salt; membrane, 50 mM receptor in NOPE (o-nitrophenyl n-octyl ether); receiving phase, water. T = 25 °C.
Table 9.
Initial transport fluxes for SLMs containing different receptorsa
| 43 | 44 | 45 | 44 + 45 | |
|---|---|---|---|---|
| Flux (×10−8 mol m−2 s−1) | 90 ± 3 | 12 ± 1 | 18 ± 1 | 50 ± 2 |
Source phase, 1 M KCl; membrane, 50 mM receptor in NOPE; receiving phase, water. T = 25 °C.
Recent reports from the Smith group also show that the ditopic receptor 43 forms strong complexes with trigonal oxyanions, such as NO3− and OAc−, as well as with the halide anions.59 Proton NMR spectral studies provided confirmation that compound 43 is able to extract solid NaNO3 and KCl into chloroform. Single crystal X-ray diffraction analyses further revealed that receptor 43 binds KOAc, NaNO3, KNO3, and NaNO2 as contact ion pairs and LiNO3 as a water-bridged ion pair (Fig. 25).
Fig. 25.
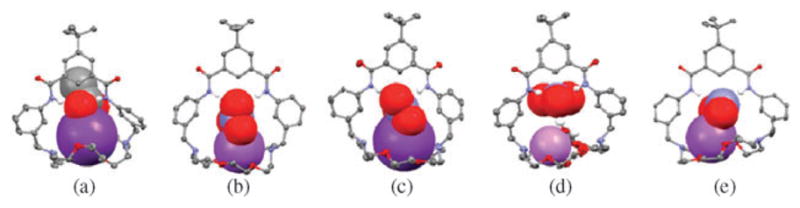
Crystal structures of (a) 43·KOAc, (b) 43·NaNO3, (c) 43· KNO3, (d) 43·LiNO3·2H2O, and (e) 43·NaNO2. The cation is bound in the crown subunit in all cases. This figure was produced using coordinates that originally appeared in ref. 59.
A more elaborated analogue of compound 43 was introduced by Gale, Smith and coworkers in 2001. These researchers used a 2,5-diamidopyrrole strap but otherwise retained the same basic receptor design. This gave rise to the macrobicyclic receptor 46, which has an additional pyrrole-derived hydrogen bond donor site. Proton NMR spectroscopic titrations revealed that the affinity of ditopic receptor 46 for Cl− is three times higher than 43.60 Moreover, it was found that the addition of one equivalent of the Na+ cation fails to increase appreciably the affinity for Cl−. However, adding one equivalent of K+ to 46 enhances the binding affinity for halide anions substantially. In the context of this work it was also confirmed via an X-ray crystallographic analysis of the NaCl complex (46·NaCl) that the receptor coordinates NaCl as a contact ion pair. Such a coordination mode is consistent with the enhanced binding affinity for Cl− observed for 46 as compared with 43. This same crystal structure also revealed that not only are the two amide NH’s interacting with the anion but also that the pyrrolic NH proton participates in chloride recognition via hydrogen bonding interactions.
A somewhat analogous ion pair receptor, the ferrocene-strapped system 47 having an electrochemically-active sensing unit, was reported by Tuntulani et al. in 2005 (Fig. 26).61 In this case, 1H NMR spectroscopic titration of ions coupled with cyclic voltammetric analyses confirmed that receptor 47 is unable to interact with the Br− anion. These same analyses also revealed that 47 binds the Br− anion (Ka = 16 096) selectively and efficiently in the presence of the Na+ cation. Cyclic voltammetric studies of 47 revealed reversible redox behavior and a value of E1/2 = 0.473 mV for the ferrocene/ferrocinium couple. The addition of cations, such as Na+ and K+, results in a slight anodic shift of the CV wave. Conversely, the addition of the Cl− induces a cathodic shift in the Fc/Fc+ redox couple with the wave disappearing as the Cl− concentration increases. It is thought that the reversibility of the Fc/Fc+ redox wave is lost as a consequence of ion pair association occurring within the receptor.61
Fig. 26.
Ion pair receptors 46 and 47.
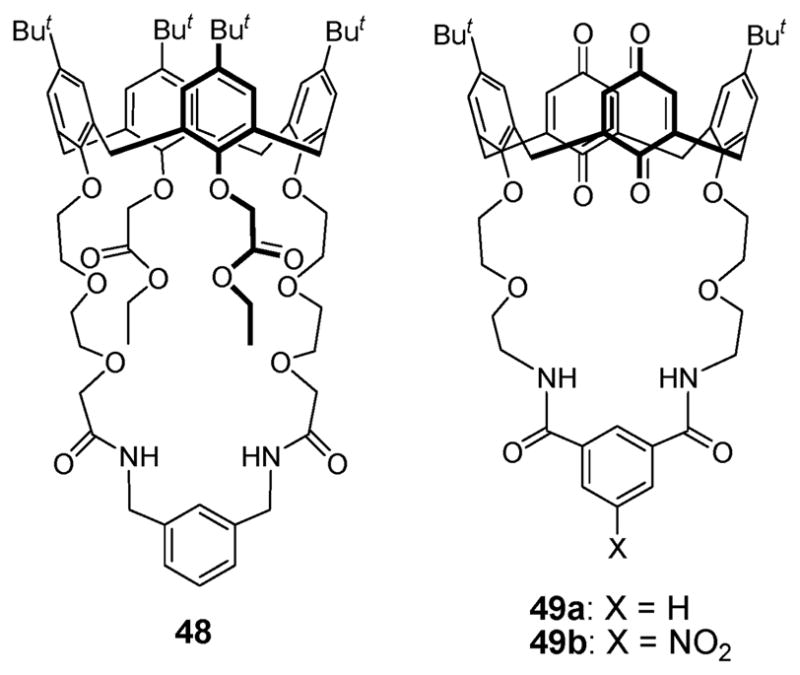
For the recognition of more specific ion pairs, Beer and coworkers synthesized the ion pair receptor 48 (Fig. 27).62 This system, reported in 2007, contains a cone calix[4]arene diester, a motif that is well known to bind the Li+ and Na+ cations strongly and selectively.62 It also has a strap with amide functionality for anion recognition. As expected, in the presence of the Br− anion, receptor 48 binds the Li+ cation (Ka > 10 000 M−1) roughly five times effectively than in its absence (Ka = 2840 M−1), as determined by 1H NMR spectroscopic titrations carried out in acetone-d6. This finding was considered indicative of a positive cooperative interaction between the co-bound ions and the receptor. Interestingly, complexation of Br− anion reduces the propensity of receptor 48 to bind the Na+ cation exhibiting Ka value that is reduced by ca. a factor of 4.5 relative to what is seen for free 48. Conversely, pre-complexation of Li+ and Na+ cations (using salts of the non-coordinating anion, perchlorate) serves to boost by a factor of 6.4–9.3 the binding affinity of 48 for the Br− and I− anions (Table 10).
Fig. 27.
Structures of ion pair receptors 48 and 49a,b.
Table 10.
Association constants (Ka) corresponding to the binding of halide anions by receptor 48 and its metal complexesa
|
Ka/M−1 |
||||
|---|---|---|---|---|
| Free 48 | [48·LiClO4] | [48·NaClO4] | [48·KPF6] | |
| TBACl | 1550 | —b | —b | —b |
| TBABr | 250 | 2320 | 2150 | —b |
| TBAI | 45 | 420 | 290 | —b |
Solvent: acetone-d6, 298 K, association constant errors<10%.
No association constant could be determined due to ion-pairing that takes place upon the addition of first equivalent of TBA+ salt.
Similar binding behavior was observed for the ion pair receptors 49a,b (Fig. 27).63,64 In this case, binding studies carried out in acetonitrile using 1H NMR and UV spectroscopy, as well as supporting calculations, revealed that receptors 49a,b are able to bind an anion and cation simultaneously by stabilizing a contact ion pair within the receptors. Ditopic receptors 49a,b do not bind small cations (Na+, K+, and NH4+) effectively in the absence of the Cl− anion. However, they form strong complexes with such cations in the presence of the Cl− anion and do so in a highly cooperative fashion. In the solid state, X-ray structural analyses confirmed that compound 49a binds KCl and NH4Cl as contact ion pairs. Interestingly, the resulting complexes display 1 : 1 and 2 : 2 stoichiometry, respectively. Also noteworthy is that receptor 49a binds KCl and NH4Cl in accord with AND logic. That is, compound 49a, unable to bind either the individual K+ or NH4+ cations, nor the individual Cl− anion, is able to bind the ion pairs, KCl and NH4Cl, very strongly in acetonitrile but only in the presence of both the constituent cations and anions.63,64
Quite early on (mid-1990’s), the Beer group detailed the synthesis of the calix[4]arene derivatives containing benzo[15]-crown-5 moieties via amide spacers (50 and 51; Fig. 28).65 These systems were expected to recognize ion pairs on the basis of a positive allosteric effect.65 Proton NMR spectroscopic binding studies carried out in acetonitrile-d3 using various Na+ and K+ salts revealed that receptor 50 forms 1 : 5 (host : guest) complexes with these cations, wherein the tetraamide NH’s do not cooperate to bind an anion. On the other hand, receptor 51 was found to form 1 : 1 complexes with the K+, Ba2+, and NH4+ cations in the same solvent. Here, it was proposed that the cations are sandwiched between the two benzo[15]crown-5 subunits, a recognition event that brings the amide NH’s into the closer proximity. This, in turn, provides a preorganized cavity for anion recognition (Fig. 28). Although receptor 51 in its free form was not found to bind anions, in the presence of potassium and ammonium cations co-complexes with anions, such as Cl−, NO3−, HSO4−, and H2PO4−, are stabilized.65
Fig. 28.

Receptors 50 and 51 and their proposed ion binding modes for anions or/and cations.
In 2003, Evans and Beer described the ion pair receptors 52 and 53 that contain two benzo[15]crown-5 linked to the upper rim of a calix[4]arene via amide groups (Fig. 29).66 A 1H NMR spectroscopic titration study carried out in DMSO-d6/CD3CN (1/1, v/v) shows that the K+ cation significantly enhances the binding ability of 52 and 53 for Cl−, OBz−, and H2PO4−. It is thought that this enhancement reflects the formation of a sandwich complex with the two benzo[15]crown-5 that, in turn, serves to provide a cavity appropriate for anion recognition. The result is ion pair binding that benefits from a positive allosteric effect. On the contrary, the Na+ cation exerts a negative allosteric effect on anion binding, as can be seen from the affinity constants listed in Table 11. The effect of the Na+ cation was rationalized in terms of these two receptors (52 and 53) forming 1 : 2 (host : guest) complexes wherein two Na+ cations are bound to the receptor (i.e. one in each crown ether). Such a complexation mode, and the resulting electrostatic repulsion between the bound Na+ cations, serves to separate the two amide NH protons that link each crown ring to the calix[4]arene core. As a result, these protons cannot bind the associated counter anions in a cooperative manner.66
Fig. 29.

Chemical structures of ion pair receptors 52–54.
Table 11.
Anion association constants (Ka) for receptors 52 and 53 and that of their corresponding Na+ and K+ complexes (errors <10%)
|
Ka/M−1 |
|||||
|---|---|---|---|---|---|
| 52 | [52·2Na+] | [52·K+] | [53·2Na+] | [53·K+] | |
| Cl−a | 5 | <5 | 15 | <5 | 150 |
| OBz−a | 25 | <5 | 270 | 20 | 205 |
| H2PO4−b | 10 | <5 | 60 | <5 | c |
Solvent: DMSO-d6/CD3CN (1/1, v/v).
Solvent: 100% DMSO-d6.
No satisfactory fit to the data could be obtained using EQNMR.
Receptor 54 (Fig. 29), reported by Stibor et al. in 1997, was found to form a complex with the Na+ cation and to bind various anions in a positive cooperative manner.67 Specifically, in the presence of the Na+ cation, the affinities of receptor 54 towards Br−, Cl−, and HSO4− were found to be increased by a factor of 5.5, 8, and 14, respectively.
In the late 1900’s, the heteroditopic rhenium(I)-bipyridyl-bis( benzo[18]crown-6) receptor 55 (Fig. 30) was synthesized by Beer and coworkers with the goal of exploiting positive binding cooperative effects to bind ion pairs.68,69 In the presence of 2.0 molar equiv. of the K+ cation (added as the hexafluorophosphate salt), a cation that forms 1 : 1 complexes with the benzo[18]crown-6 moiety, the Cl− anion affinity was enhanced by 5-fold in DMSO. This enhancement was ascribed to favorable electrostatic and conformational effects. Analogous ion pair receptors, namely 56 and 57 (Fig. 30), were prepared by linking two benzo[15]crown-5 subunits to ruthenium(II)- and rhenium(I)-bipyridyl groups via four amide groups. In this case, the crown ether moieties were expected to form 2 : 1 (crown : K+) sandwich complexes with the K+ cation. Binding of K+ serves not only to induce an allosteric effect via preorganization of the four amide groups into an orientation that favors anion binding, but also to cause a switch in the inherent anion selectivity.
Fig. 30.

Chemical structures of ion pair receptors 55–58.
In the absence of the K+ cation, both receptors bind the H2PO4− anion more selectively than the Cl− anion in a 1 : 1 stoichiometry (for 56, Ka = 205 M−1 for H2PO4− vs. Ka = 55 M−1 for Cl− in DMSO-d6). However, the anion selectivity of both ion pair receptors is reversed after addition of K+, presumably because the two pendant benzo[15]crown-5 subunits form a sandwich complex with the K+ cation. In this instance, the association constants of both receptors for H2PO4− decrease while those for Cl− increase (for 56, in the presence of K+, Ka = 35 M−1 for H2PO4− vs. Ka = 300 M−1 for Cl− in DMSO-d6). In a similar manner, adding K+ to 58 serves to preorganize the four amide groups for anion recognition via formation of a crown-based sandwich complex. This gives rise to the result of enhancing the association constants for the acetate and chloride anions by a factor of 1.4 and 1.8, respectively.70
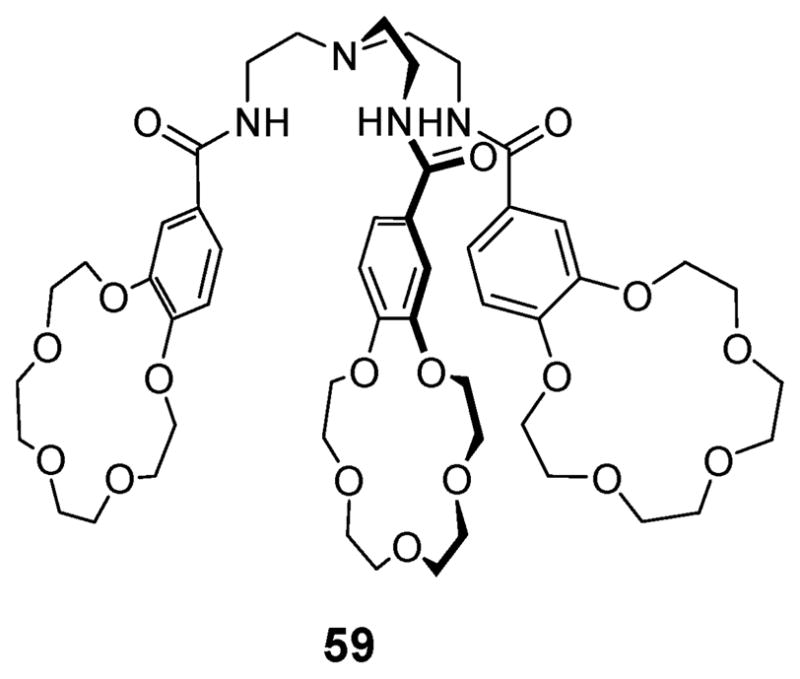
Beer and coworkers also synthesized the ion pair receptor 59 (Fig. 31), a tripodal tris(amido benzo[15]crown-5).71 This system, reported in 1999, was studied for its ability to extract and transport sodium halide salts and sodium perrhenate. In the presence of 1.0 equivalent of sodium picrate, compound 59 acts as a ditopic receptor, exhibiting enhanced affinities for Cl−, I− and ReO4− (Table 12). This result is ascribed to the cooperative binding of these anions via co-bound sodium cations complexed by the crown ether subunits. It was also found that receptor 59 can extract sodium pertechnetate (NaTcO4) from water and transport it efficiently under mixed organic aqueous conditions designed to simulate conditions that might be applicable to treatment of aqueous waste streams containing this radioactive salt.
Fig. 31.
Tripodal tris(amido benzo[15]crown-5) ion pair receptor 59.
Table 12.
Binding constants recorded for 59 in the presence and absence of sodium picrate in CDCl3
Determined at 298 K, errors estimated to be ≤10%.
Titration carried out in the presence of 1.0 molar equiv. of sodium picrate.
Determined at 298 K, errors estimated to be ≤15%.
In 2001, Beer and coworkers also demonstrated positive binding cooperativity in the case of the bis(calix[4]arene) rhenium(I)–bipyridyl receptors 60a,b (Fig. 32).72 In the presence of 2.0 molar equiv. of alkali metal ions, such as Li+, Na+, and K+, the binding affinities for iodide were observed to increase by a factor of 4.5, 3.1, and 1.5, respectively. This finding was attributed to the complexation of the metal cations with the ester groups in the lower rim of the calix[4]arene. This complexation fixes the calixarene scaffold in a more rigid conformation, thereby preorganizing the central cavity for iodide recognition. It was found that calix[4]arene tetraester 60b shows the greatest enhancement in the I− binding affinity in the presence of Na+ (in the presence of NaClO4, Ka=320 M−1 for TBAI vs. Ka = 40 M−1 in its absence, for I− in CD3CN). Ion pair receptors 61a,b, in which calix[4]arene esters are linked to a single ruthenium(II)– or rhenium(I)–bipyridyl metal site, were also reported by Beer et al. in 2000.73 It was found by separate 1H NMR titration experiments, carried out in acetonitrile- d3, that receptors 61a,b form 1 : 1 complexes with the Br− and I− anions (as the tetrabutylammonium salts), and the Li+ and Na+ cations (as the perchlorate salts). The presence of 1.0 equiv. of the alkali metal cations significantly increases the Br− and I− affinities of receptors 61a,b. The enhancement in these anion affinities seen in the presence of the pre-bound cation is ascribed to (i) the attraction between co-bound opposite charged-ions, (ii) the preorganized cavity, and (iii) the increased strength of the hydrogen bond interactions with the bound anion. This latter benefit is thought to result from cation complexation that serves to disrupt the intramolecular hydrogen bonds between the proximal amide NH proton and one or more of the calix[4]arene ester oxygen atoms.
Fig. 32.

Calix[4]arene-based ion pair receptors 60a,b and 61a,b.
In 2008, Le Gac and Jabin reported the synthesis of calix[6]cryptamide 62 (Fig. 33), a receptor that was put forward as a host for neutral molecules and alkylammonium chloride ion pairs (alkyl = ethyl and propyl).74 Receptor 62 contains a calix[6]arene subunit that is designed to act as a cation recognition site. This core is linked to a cyclotriveratrylene skeleton via three diamide groups, thereby providing an anion recognition site. Upon addition of excess (up to 15 molar equiv.) of ammonium salts RNH3 +Cl− (R = ethyl or propyl) to a chloroform solution of 62, the resonance of the amide NH protons nearest to the calix[6]arene backbone undergoes a significant downfield shift (Δδ > 0.5 ppm), whereas the peaks of alkyl protons of the ammonium cations appear in abnormally low field (δ ≈ −1.0 and −2.0 ppm for the methylene protons of propylammonium chloride). Such findings provide support for the notion that the alkylammonium chloride ion pairs are included in the receptor. That is, the chloride anion is hydrogen bonded to the amide groups and the alkylammonium cation is bound to the calix[6]arene cup via hydrogen bonds to the ethereal oxygen atoms, as well as CH–π interactions. Interestingly, neither the chloride anion nor the propylammonium cation with non-coordinating counterions, such as the tetrabutylammonium cation or the picrate anion, was bound to the receptor. This selectivity is consistent with the conclusion that this system is highly cooperative, and that interaction between the co-bound ions is essential for the formation of a stable host–guest ion pair complex.74
Fig. 33.

Calix[6]arene-based ion pair receptor 62 and its proposed mode of binding for alkylammonium chloride contact ion pairs.
Ditopic receptors 63a–c (Fig. 34) were also synthesized by the Jabin group in 2003.75 These systems contain three urea or thiourea groups attached to the calix[6]arene skeleton. In the presence of various anion salts containing the non-coordinating countercation, tetrabutylammonium (TBA+), a significant down-field shift of the urea or thiourea NH resonances is seen in the 1H NMR spectrum. This observation is consistent with the receptors being able to recognize directly the Br−, I−, AcO−, and HSO4− anions. In contrast, no chemical shift change is observed in the presence of an excess of the propylammonium picrate, a finding interpreted in terms of these receptors being unable to bind alkyl ammonium cations as independent substrates. On the other hand, the addition of tetrabutylammonium chloride to a mixture of receptors 63a–c and propylammonium picrate produces a remarkable upfield shift in the propylammonium resonance (to δ ≈ −1.0 and −2.0 ppm for the propyl CH2CH2 signals) as well as large downfield shifts in the urea or thiourea NH signals. Such large, correlated changes in the 1H NMR spectrum were taken as evidence that both the chloride anion and the propylammonium cation are bound concurrently to the receptors. To the extent this interpretation is correct, it implies that the chloride anion plays a crucial role in cation binding and that the system benefits from a positive cooperative interaction. Support for this appealing conclusion comes from studies of receptor 64 (Fig. 34), a system that bears an independent recognition site for ammonium cations. As expected, this latter receptor was found to form a 1 : 1 complex with propylammonium chloride and to do so with a positive cooperativity.76
Fig. 34.

Calix[6]arene–urea based ion pair receptors 63a–c and 64.
A different type of receptor, namely 65a–e (Fig. 35), containing amides and designed for organic ion pair recognition, was reported by Atwood et al. First reported in 2002, these systems are based on resorcin[4]arene frameworks that are linked to additional aromatic rings via a Mannich reaction to provide a hydrophobic cavity.77,78 Four amide groups on the upper rim of the capsule-like molecule then complete the structure. In a mixture of 65a–c in chloroform containing an excess of TMACl (tetramethylammonium chloride), the proton peak of the TMA+ cation appears at δ = −0.57 in the 1H NMR spectrum, while the NH signals appear at 9.6 ppm. These correspond to upfield and downfield shifts of +1.6 and −1.6 ppm, respectively. These findings provide support for the conclusion that the TMA+ cation is encapsulated in the cavity while the chloride anion is hydrogen bonded to the amide NH protons. The X-ray crystal structure of 65a·TMACl was solved (Fig. 36). It revealed that the receptor forms a simultaneous complex with both the cation and the anion as a contact ion pair. In the presence of excess TMACl, the existence of two sets of distinguishable proton peaks for the TMA cation (one for the complex, the other for the free) are seen in the 1H NMR spectrum. This is consistent with the equilibrium between the complexed and the uncomplexed TMA+ cation being slow on the NMR time scale and that the complex is kinetically and thermodynamically stable. Compounds 65d,e, having more acidic NH’s, were thought on the basis of 1H NMR spectroscopic studies to form an ion pair complex with other tetramethylammonium halides, such as TMABr and TMAI, as well as TMACl.77,78
Fig. 35.

Chemical structures of molecular capsules 65a–e.
Fig. 36.
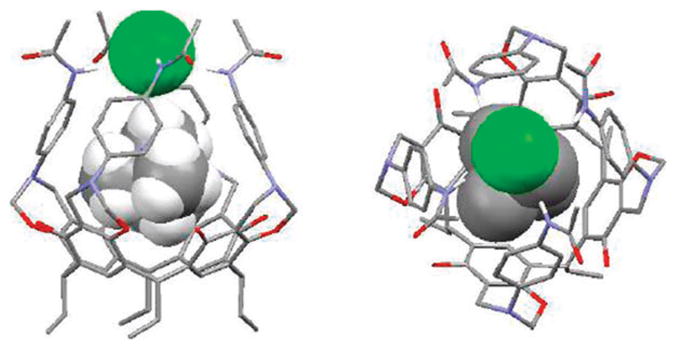
Two different views of the X-ray crystal structure of 65a· TMACl (TMA+=tetramethylammonium). This figure was produced using coordinates that originally appeared in ref. 77.
In 2005, Frontera et al. reported that a squaramide moiety connected to a [18]crown-6 subunit (receptor 66; Fig. 37) is able to bind both the K+ or Na+ cation and the acetate (OAc−) anion concurrently, as evidenced by 1H NMR spectroscopy and supporting PB86/optimized structures.79 Receptor 66 was also found to dissolve otherwise insoluble carboxylate salts, such as sodium benzoate and sodium acetate in chloroform.
Fig. 37.

Chemical structure of the crowned squaramide ion pair receptor 66 and its predicted binding mode for NaOAc or KOAc contact ion pairs.
Another approach to binding ion pairs involves the use of cyclopeptides. These systems are multi-amides and can thus interact with anions via hydrogen bond donating NH residues and cations via carbonyl lone pair electrons. A pioneer in this area is Kubik, who synthesized the cyclopeptide 67 (Fig. 38).80 This receptor contains L-glutamic acid and 3-aminobenzoic acid subunits linked in an alternating sequence. It was examined for its ability to bind both anions and cations. For instance, 1H NMR spectroscopic analyses revealed that receptor 67 forms a 1 : 1 complex with n-butyltrimethylammonium iodide (BTMAI). An upfield shift in the proton resonance of the BTMA+ cation in conjunction with an unchanged NH peak in the 1H NMR spectrum was taken as evidence that macrocycle 66 is able to recognize the ammonium cation via a π–cation interaction (Ka = 300 M−1), but not the I− anion.
Fig. 38.
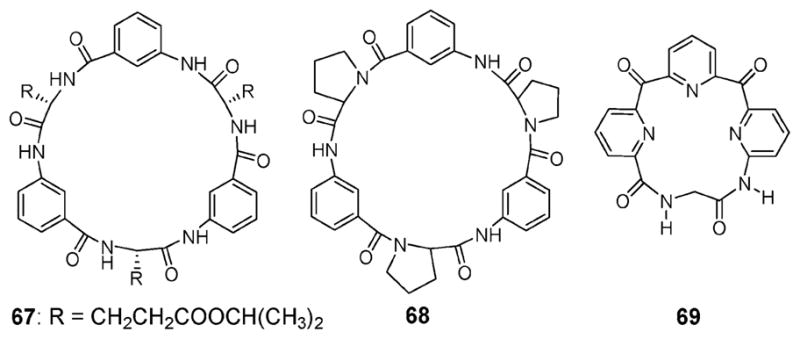
Cyclopeptide-based ditopic receptors 67–69.
In addition to the BTMA+ cation, receptor 67 was found to bind certain anions, including tosylates and phosphates via hydrogen bonding interactions involving the amide NH protons and the bound anions. It was also noted that in the presence of the tosylate anion, the affinity of cyclopeptide 67 for the BTMA+, Na+, and K+ cations was significantly enhanced. This increase (up to a factor of 103–104) was ascribed to an increase in the preorganization of the cyclopeptide by the bound anion, as well as to an enhanced electrostatic interaction between the anions and cationic substrates in the final complex. That is, the binding of the tosylate anion via interaction with the six amide NH protons serves to enforce a calix[4]arene-like cone conformation, i.e., one, in which all the carbonyl oxygen atoms, as well as aromatic groups, point in the same direction. The net result is the stabilization of a bowl-shaped cavity suitable for cation binding. Accordingly, substrate binding is highly cooperative.
Kubik, together with Goddard, also synthesized cyclopeptide 68 (Fig. 38), which contains L-proline and 3-aminobenzoic acid subunits linked in an alternating sequence.81 With most ion pairs tested, compound 68 was found to display binding behavior similar to that of compound 67. An exception to this generalization is BTMAI. Receptor 68 was found to recognize BTMAI in a highly cooperative fashion, as evidenced by the observation of a significant upfield shift in the proton resonance (Δδ = −1.1 ppm for 68 and Δδ = −0.02 for 67) of the ammonium cation (in both cases the chemical shifts of the N-methyl protons were monitored). The stability constant of cyclopeptide 68 (Ka = 21 100 M−1) for BTMAI is almost two orders of magnitude higher than that of 67 (Ka = 300 M−1).
In 2005, Gong and Gibb reported that the macrocyclic compound 69 (Fig. 38), composed of a tripyridyl unit cyclized with a diamide linker, acts as an ion pair receptor for monoalkylammonium salts, being especially effective for salts of L-phenylalanine methyl ester.82 Little or no change in the 1H NMR spectrum was seen in the presence of anions paired with the non-coordinating counter cation, tetrabutylammonium. This observation led these authors to suggest that the anions were not bound strongly to the receptor. In contrast, when salts of smaller mono-alkyl ammonium cations were used, relatively large downfield shifts was seen for both the amide (Δδ > 0.5 ppm) and pyridyl (Δδ = 0.1–0.2 ppm) signals. These spectroscopic changes were interpreted in terms of both the anion and the cation being bound to the receptor. Compared to what was seen for anions studied using noncoordinating counter cations, the enhancement in the association constants seen when these ion pairs were used with 69 is remarkable. Not surprisingly, this increase was ascribed to cooperative interactions between the co-bound ions (see Table 13). To the extent this interpretation is true, it shows that when both components of ion pairs are bound concurrently, much stronger complexes can be attained.
Table 13.
Association constants between 69 and various ammonium saltsa
| + NBu4 salt | Ka/M−1 | + NH3CH(Bn)–CO2Me salt | Ka/M−1 | Cooperativity factorb |
|---|---|---|---|---|
| F− | 110 | F− | —c | —c |
| Cl− | 63 | Cl− | 12 100 | 190 |
| Br− | 40 | Br− | 1900 | 48 |
| I− | 32 | I− | 400 | 13 |
| PF6− | —d | PF6− | —c | —c |
| NO3− | 70 | NO3− | 18 400 | 257 |
| CF3CO2− | 52 | CF3CO2− | 6300 | 121 |
| TsO− | 42 | TsO− | 1500 | 36 |
At 298 K, initial [69] = 1.0 mM in CDCl3. Errors are ±10%.
Ka(amino acid)/Ka(TBA).
Guest insoluble in CDCl3.
No binding observed.
Efforts to apply ion pair recognition principles to the synthesis of more elaborate systems, such as rotaxanes and catenanes, were made by Beer and coworkers.83,84 In what is arguably the first example of work along these lines, the pseudorotaxane 72 (Fig. 39) was made by mixing macrocyclic compound 70, acting as an ion pair receptor, with the organic ion pair 71. In acetone solution, this organic ion pair (71) threads through the ion pair receptor (70). Presumably, this threading is driven by complementary chloride binding by the NH protons of 70 and 71, as well as by additional hydrogen bonding interactions involving the positively charged N-methyl protons and the crown ether ring (Fig. 39). In addition, a charge transfer complex, stabilized by π donor– acceptor interactions between the pyridinium cation and the two hydroquinone moieties of 70, provides another driving force for the formation of pseudorotaxane 72. The formation of this latter product is evidenced by the appearance of a broad band at approximately 370 nm in the UV/Vis spectrum, features that correspond to color change from colorless to pale yellow. Consistent with this proposed mechanism, it was found that pseudorotaxane 72 is not formed in protic solvents because of competition from the solvents.83,84 In contrast, the permanently interlocked [2]- and [3]-catenanes 74 and 75 can be formed from the ion pair receptor 70 and the organic ion pair 73 by ring-closing metathesis (Fig. 39).85 In dichloromethane, the addition of 10 wt% Grubbs’ catalyst to the pre-formed pseudorotaxane between 70 and 73 gives [2]- and [3]-catenanes 74 and 75 in 45% and <5% yield, respectively (Fig. 40). Furthermore, removal of the Cl− template from catenane 74 by AgPF6 provides the anion-free catenane, which is able to act as anion receptor for Cl−, H2PO4−, and −OAc anions.85
Fig. 39.

Formation of pseudorotaxane 72 via cooperative ion pair (71) recognition by 70.
Fig. 40.

Formation of [2]- and [3]-catenanes 74 and 75 by ring-closing metathesis using Grubbs’ catalyst.
In 2007, the Beer group further reported the use of calix[4]arenes strapped with a diamide ring (e.g., 74) for ion paired thread recognition instead of compound 70.86 The oxygen atoms of this calix[4]arene in the cone conformation bind the positively charged N-methyl group of appropriately chosen substrates via hydrogen bonds, whereas the amides recognize the chloride anion to form pseudorotaxanes, such as 76 (Fig. 41). Finally, catenane 77 could be formed from the starting diamide calixarene receptor via the combined use of a functionalized substrate and Grubbs’ catalyst (Fig. 41).
Fig. 41.
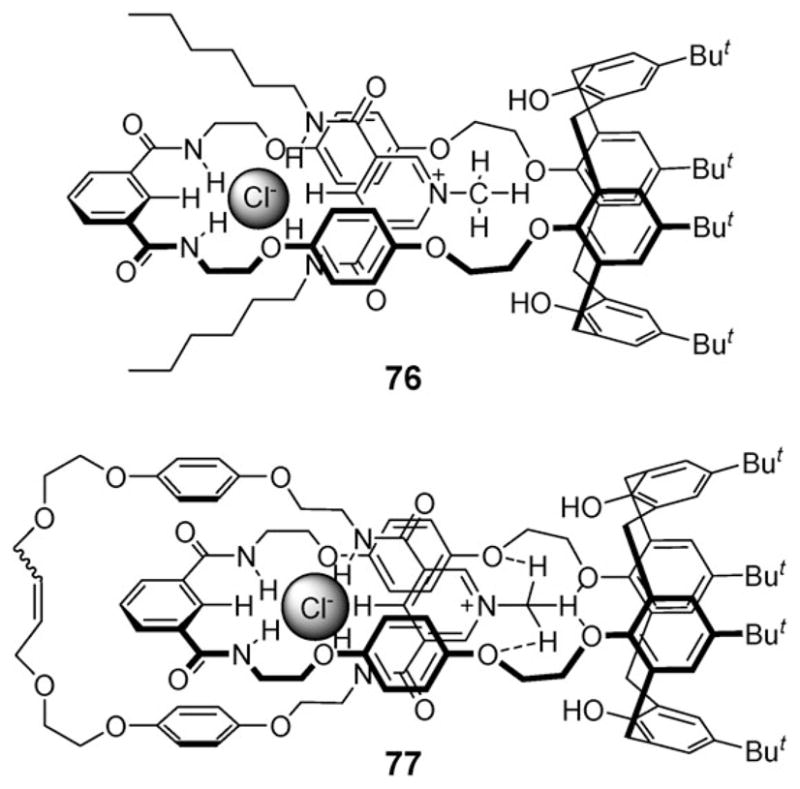
Pseudorotaxane 76 and catenane 77 formed using a calix[4]-arene-based ion pair receptor and appropriately chosen pairs of anions and cations.
5. Ion pair receptors based on positively charged components for anion recognition
Subunits that provide positively charged centers have proved effective for the construction of ion pair receptors. Several that rely on protonated ammoniums have proved useful at low pH. An early example, dating from 1992, was reported by Lockhart et al. These researchers synthesized compound 78 (Fig. 42), consisting of two benzo[5]crown-15 moieties linked via a tetraammonium spacer, and examined its ability to bind KCl (Fig. 42).87 Detailed 35Cl NMR and 39K NMR spectroscopic analyses provided support for the notion that receptor 78 forms a complex with the KCl, wherein the central tetraammonium linker wraps around the Cl− anion so as to bring the two benzo[5]crown-15 ether ring into closer proximity. This wrapping allows the K+ cation to be bound in a “sandwich-like” fashion. In this case, ion pair binding to receptor 78 takes place on the basis of a positive allosteric effect.
Fig. 42.
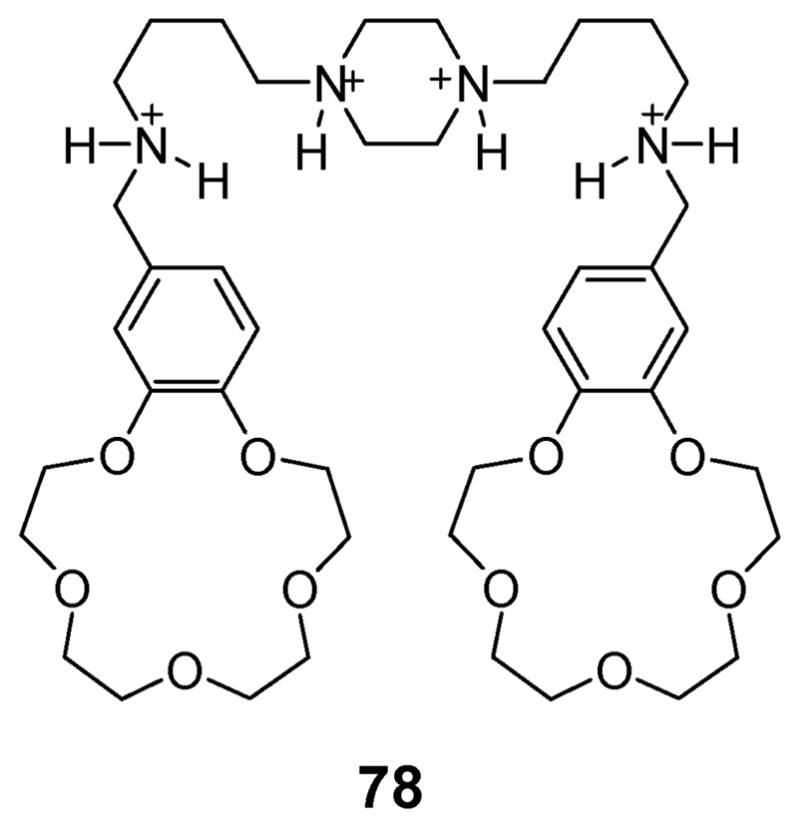
An ion pair receptor for KCl, 78, that operates at low or neutral pH.
In 1999, White and coworkers reported an ion pair receptor that can be used for the separation of transition metal salts [M2+X2−] through solvent extraction. This system, receptor 79 (Fig. 43), incorporates into a lipophilic framework a dianionic binding site for transition metal cations and a dicationic binding site for anions.88 Transition metal cations, such as Cu2+ and Ni2+, are bound to the salen-based cation-recognition site, inducing proton transfer from the phenolic ring to the nitrogen atoms of the morpholine moieties. This provides positively charged ammonium species and also serves to bring the two ammonium units into proximity for cooperative anion recognition (Fig. 43).
Fig. 43.
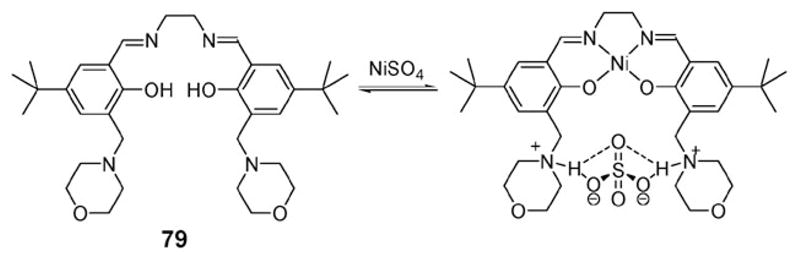
Ion pair receptor 79 and its binding mode for NiSO4.
As evidenced by the X-ray crystal structure of 79·NiSO4, in the solid state the sulfate dianion and Ni2+ cation are spatially separated, being bound to the protonated central amines via two hydrogen bonds and the deprotonated phenol units of the salen subunit, respectively. Sulfate anion binding is presumably reinforced by electrostatic interactions involving the salen-bound Ni2+ cation. Liquid–liquid extraction experiments were also carried out and served to demonstrate that ion pair receptor 79 is able to extract CuSO4 into chloroform from an aqueous solution of CuSO4 at pH 3.8.
In 2001, Tuntulani et al. reported the synthesis of the ion pair receptor 80 (Fig. 44), a tripodal aza crown ether calix[4]-arene, and its interaction with halide anions and alkali metal cations.89 In this case, 1H NMR spectroscopic analyses revealed that compound 80 can form complexes with anions such as Br− and I− but not F−, as well as with cations, such as Na+ and K+ in 100% DMSO-d6 and in a mixture of CDCl3 and CD3OD. In the presence of the K+ cation, the association constant of compound 80 for the Br− anion was found to be enhanced by nearly 1.5 fold.
Fig. 44.
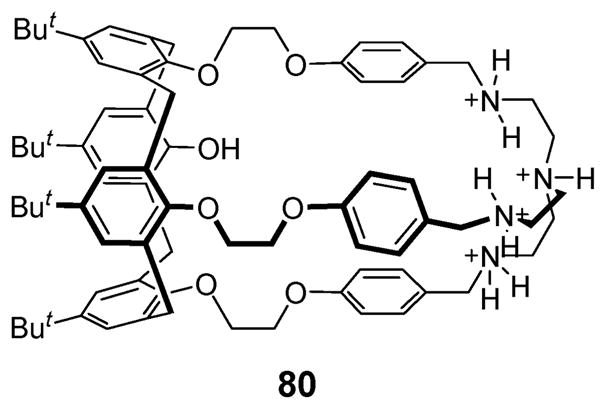
Tripodal aza crown ether calix[4]arene 80 containing both cation and anion binding sites.
For recognition of zwitterionic amino acids, Schmidtchen synthesized the ditopic receptor 81 (Fig. 45).90 This system, reported in 1986, contains a triaza[18]crown-6 subunit covalently linked to a tetraammonium macrocylic anion recognition motif via a p-xylene tether. In this case, a 1H NMR spectroscopic binding study revealed that receptor 81 is able to bind certain zwitterionic amino acids, such as γ-aminobutyric acid and 6-aminohexanoic acid in 90% aqueous methanol. The association constants (Ka) of compound 81 for some primary ammonium guests as determined from competition experiments were found to be reduced as compared to those obtained with the monotopic control compound 82. The selectivity of ditopic receptor 81 for zwitterionic amino acids is, however, increased by a factor of 2.5 as compared to the monotopic receptor 82.
Fig. 45.
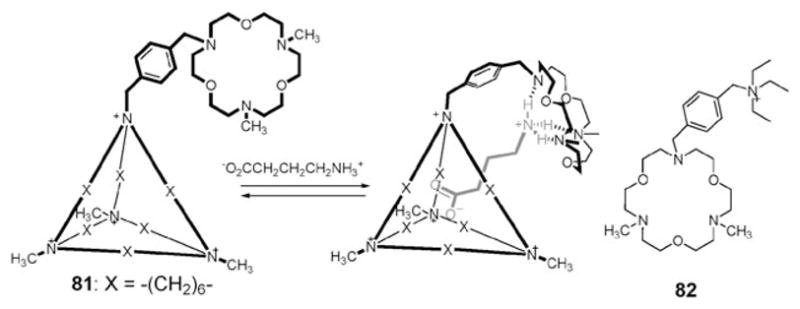
Ditopic receptor 81 and its predicted binding mode for a zwitterionic ω-amino carboxylate. Also shown is the control monotopic receptor 82.
In an effort to produce a more sophisticated receptor for amino acids, de Mendoza and coworkers prepared the chiral receptor 83 (Fig. 46).91 This system, reported in 1992, contains a guanidinium group for carboxylate anion recognition and a monoaza[18]crown-6 for ammonium cation recognition.91 The appended aromatic naphthalene ring was designed to provide an additional interaction with the side chain of aromatic amino acids via π–π interactions. The chiral nature of receptor 83 was expected to provide for the enantioselective recognition of phenylalanine and tryptophan. The affinity of compound 83 towards amino acids was determined by liquid–liquid single extraction experiments using water and CH2Cl2. The extraction efficiencies (i.e., fraction of receptor molecules occupied by substrate) in the organic phase, determined by integration of the NMR spectra, were ca. 40% for both L-Trp and L-Phe. On the other hand, L-Val without an aromatic side chain was not extracted to an appreciable extent. This led to the conclusion that the aromatic naphthalene pendant plays a critical role in recognizing aromatic amino acids. Enantioselective recognition of L-Trp and L-Phe by (S,S)-83 was confirmed by 1H NMR spectroscopic analysis. Conversely, the other antipode, receptor (R,R)-83, was found to extract selectively D-Trp and D-Phe, but not their corresponding L-enantiomers.
Fig. 46.
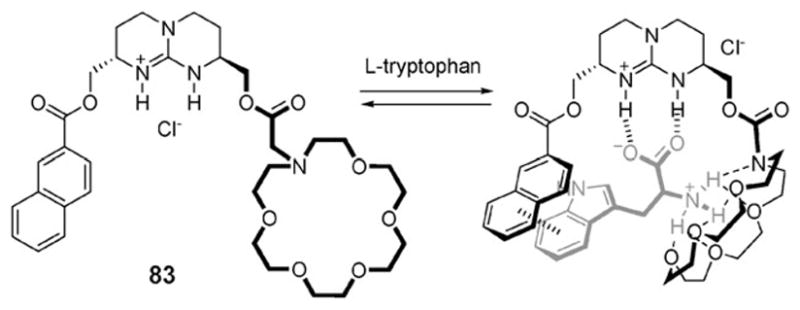
Ditopic receptor (S,S)-83 and its proposed binding mode for L-tryptophan.
Ion pair receptors, suitably functionalized with a fluorophore or chromophore, can act as optical sensors for ion pairs, exhibiting an easily detectable signal change in color or fluorescence intensity. An example of such a system comes from de Silva and coworkers who synthesized the fluorogenic compound 84 (Fig. 47).92 This system, reported in 2003, contains both anion and cation binding sites, namely a Na+-selective benzo[15]crown-5 moiety and a H2PO4−-selective polyammonium group linked via an anthracene fluorophore. In the absence of substrates (ions), the fluorescence intensity of receptor 84 is inherently low because of PET (photo-induced electron transfer) from both the benzo[15]crown-5 and the benzylic amine lone pair to the anthracene. The PET processes can be inhibited by complexation of the Na+ cation by the benzo[15]crown-5 and H2PO4− binding to the polyammonium moiety, respectively. Thus, in the presence of both ions the fluorescence is enhanced. However, in the presence of a single ion (either Na+ or H2PO4− studied using a non-coordinating counter ion), the fluorescence is still weak. Presumably, this is because PET from either the benzylic amine lone pair or the benzene ring of the crown ether suffices to quench the intensity of the anthracene fluorescence. Only the simultaneous complexation of both a Na+ cation and an H2PO4− anion gives rise to fluorescence enhancement. Therefore, receptor 84 acts as a rudimentary photoionic AND logic gate.
Fig. 47.
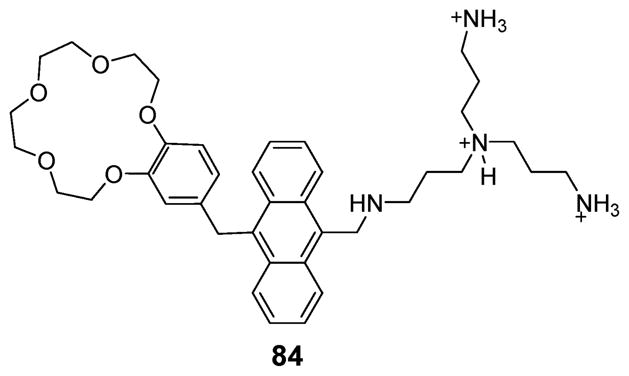
Fluorescent ion pair sensor 84 that functions as an AND logic system.
6. Phosphine oxide and sulfoxide-based ion pair receptors
An alternative approach to the design of ion pair receptors involves the use of phosphine oxide and sulfoxide groups as cation recognition sites. These are moieties that are known to be strong hydrogen bond acceptors, but weak Brønsted bases.93,94 Recognizing the utility of these motifs, Gellman et al. synthesized the macrocyclic phosphine oxide disulfoxide 85 (Fig. 48). This system, first reported in 1993, was found to bind monoalkylammonium cations and certain halide anions with non-coordinating counter ions via hydrogen bondings and through interactions with the positive ends of the S=O and P=O dipoles, respectively. In the case of monoalkylammonium halide ion pairs, 1H NMR spectroscopic studies revealed that receptor 85 is able to bind the monoalkylammonium cation via hydrogen bonding interactions with the three oxygen atoms present on the sulfur and phosphorus atoms. These interactions serve to induce polarization and to preorganize the receptor. The result is a cavity better suited to bind the Cl−, Br− or I− counter ion on the opposite face as shown in Fig. 48.93,94
Fig. 48.
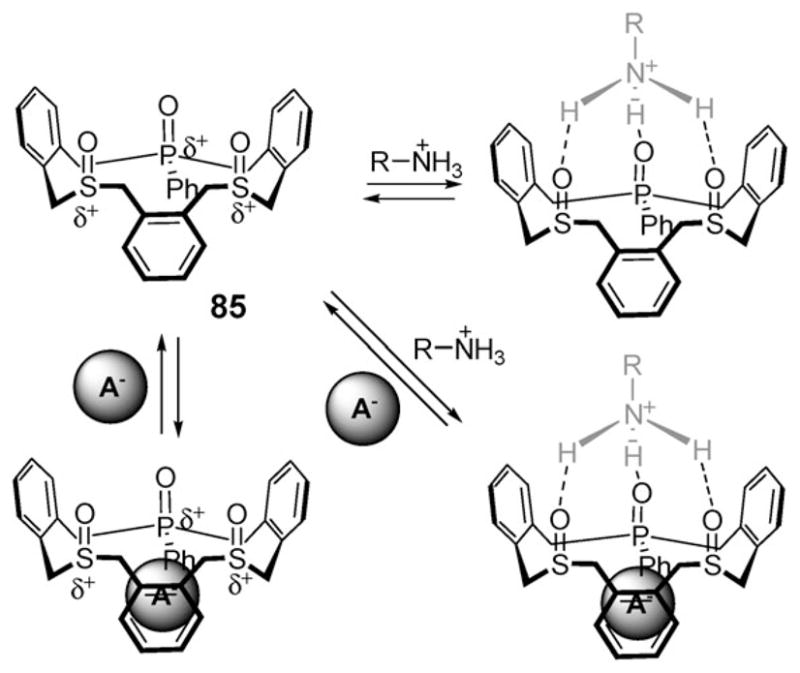
Macrocyclic phosphine oxide disulfoxide ion pair receptor 85 and its proposed interactions with monoalkylammonium cations, halide anions, and ion pairs containing these species.
7. An ion pair receptor that is based on halogen bonding to achieve anion recognition
In 2005, Resnati and coworkers reported the tripodal ion pair receptor 86 that relies on halogen bonding (XB) for the recognition of anions, such as halides (Fig. 49).95 The four strong electron-withdrawing fluorine atoms on the aromatic ring of receptor 86 make the iodine atom an effective XB donor. An X-ray crystal structure of the NaI complex revealed that in the solid state, receptor 86 binds both the Na+ cation and the I− anion concurrently. However, as can be inferred from an inspection of Fig. 50, these latter species are spatially separated by the receptor at a distance of ca. 5.59 Å. This structure also revealed that the Na+ cation is completely encompassed by the arms of the receptor and that the I− anion is bound to the iodine atom of the tetrafluoroaromatic ring via an XB interaction. Compared to that of compound 87, which lacks an XB donor (Ka = 1.3 × 104 M−1), the binding constant of receptor 86 was found to be 20 times larger (Ka = 2.6 × 105 M−1). On this basis, it was concluded that the I···I− XB interaction has a positive effect on the Na+ binding in the case of receptor 86.
Fig. 49.
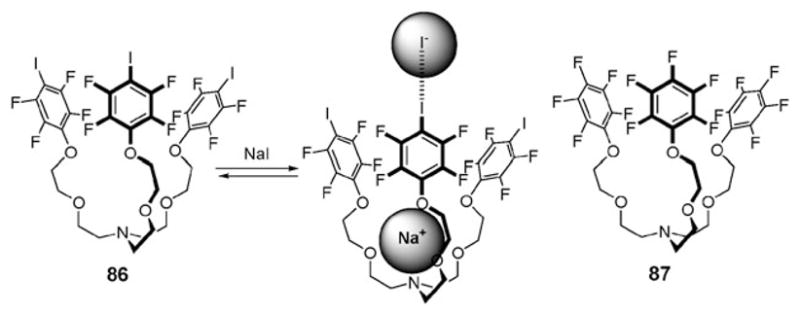
Ion pair receptor 86 and its proposed NaI binding mode. Also shown is control compound 87 that lacks an XB donor.
Fig. 50.
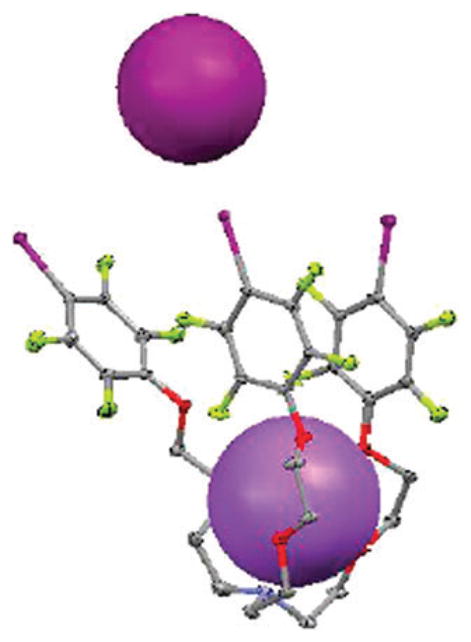
Crystal structure of 86·NaI. This figure was produced using coordinates that originally appeared in ref. 95.
8. An ion pair receptor based on hydroxyl groups for anion recognition
The hydroxyl group is also able to form hydrogen bonds with anions. This motif was exploited by Pochini and coworkers, who in 2001 reported that the flexible cone-calix[4]arene (88), bearing four 4-hydroxybenzyl groups on the upper rim, can act as an ion pair receptor for tetramethylammonium salts (Fig. 51).96 In this case, 1H NMR spectroscopic studies carried out in CDCl3 provided support for the notion that receptor 88 binds the tetramethylammonium cation via CH–π interactions, while anions such as OTs−, Cl−, OAc−, CF3COO−, and picrate are bound via hydrogen bonds involving the four hydroxy groups. In contrast to receptor 88, the corresponding analogue 89 showed no tendency to bind such ion pairs. These results are thus consistent with the suggestion that anion binding by the hydroxy groups present in 88 plays a crucial role in stabilizing the receptor-bound ion pair. While the simple ion receptor 34 was found to form strong complexes with loose ion paired tetramethylammonium salts, such as TMATFA or TMAPic, receptor 88 was found to form strong complexes with tightly bound ion pairs such as TMACl and TMAOTs. This observation led to the conclusion that receptor 88 binds such ion pairs via a cooperative interaction that involves the two binding sites and the co-bound ions. To obtain further support for this suggestion, a dual host system, consisting of a mixture of hosts 88 and 34, as well as the specific anion receptor (90) for the Cl− anion, was analyzed (Fig. 51). In the presence of the chloride receptor (90), the binding constant (Ka) of single ion receptor 34 for TMACl in chloroform is enhanced from 80 M−1 to 428 M−1. This increase is attributed to a breaking up of the ion pairing TMACl ion pair caused by complexation of the chloride anion by receptor 90. In contrast, compound 34 serves to decrease the binding constant of receptor 88 from 3526 M−1 to 440 M−1. Presumably, this latter finding reflects the fact that the Cl− anion is bound to the hydroxy groups of 88 less strongly as the result of competition.
Fig. 51.
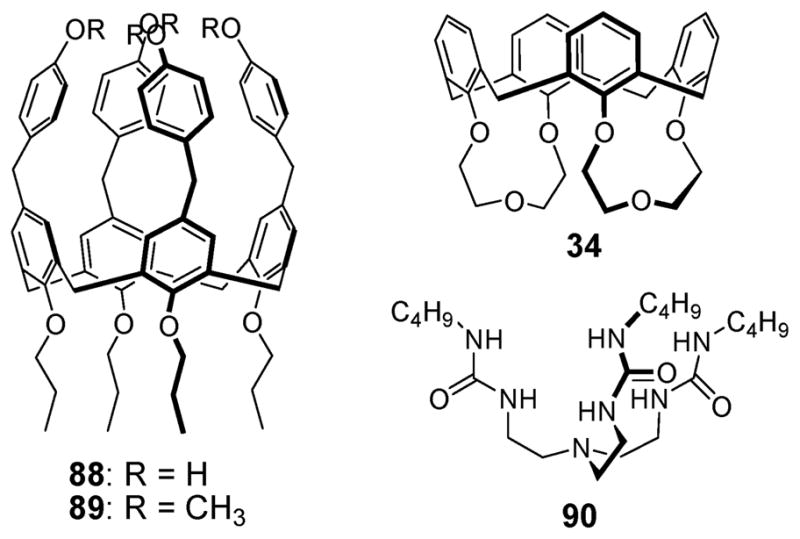
Ion pair receptor 88 and control single ion receptors 34, 89 and 90.
9. Ion pair receptors based on G-quartets
Guanine nucleotides are known to bind alkali metal cations in water via formation of inter alia G-quartets, wherein the cations are bound to four nucleotide-derived carbonyl oxygen atoms oriented towards the center of what is a self-assembled core. These cation-containing G-quartets stack to form octamers, dodecamers, hexadecamers, and higher aggregates. In 2001, Davis and coworkers reported that the guanosine nucleotide 91 forms G-quadruplex-based hexadecamers in the presence of BaPic2 and SrPic2. The resulting systems were proposed to consist of two C4-symmetric 918·M2+ octamers as shown in Fig. 52.97 An X-ray crystal structure provided support for this conclusion and revealed that the G-quartets within each octamer are stacked head-to-tail with a 30° rotation between the layers. As a result, the divalent Ba2+ and Sr2+ cations are sandwiched between the G-quartets and separated from their picrate counterion at a distance of >8.5 Å. In contrast, the four picrate counter anions are involved in hydrogen bonding interactions involving the outer NHb of the amino group. The net result is two metal-filled octamers of formal stoichiometry 9116·2M2+·4Pic−. Proton NMR spectroscopic analyses provided support for the conclusion that the picrate anions play a critical role in maintaining the G-quadruplex in solution. The kinetic stability of the hexadecamer was confirmed by a cross-over experiment. In this experiment, crystalline 9116·2Sr2+·4Pic− and 9116·2Ba2+·4Pic− were mixed in a 1 : 1 ratio in CD2Cl2 to give a new G-quadruplex, namely the mixed hexadecamer 918·2Sr2+·918·2Ba2+·4Pic−. 1H NMR spectroscopy revealed the slow formation of the mixed hexadecamer (t1/2 = 42 h), as would be expected for a structure wherein the four picrate anions serve to hold the G-quadruplex together tightly in solution. By contrast, when the thiocyanate anion, a species incapable of bridging the 918·2M2+ octamers as effectively as the picrate, was used, the mixed hexadecamer, 918·2Sr2+·918·2Ba2+·4SCN−, is formed significantly faster (t1/2 = 0.5 h). These findings lend credence to the proposal that the strength of the anion binding interaction, as well as the nature of the cation, have a remarkable effect on the kinetic stability of the hexadecamers.97
Fig. 52.
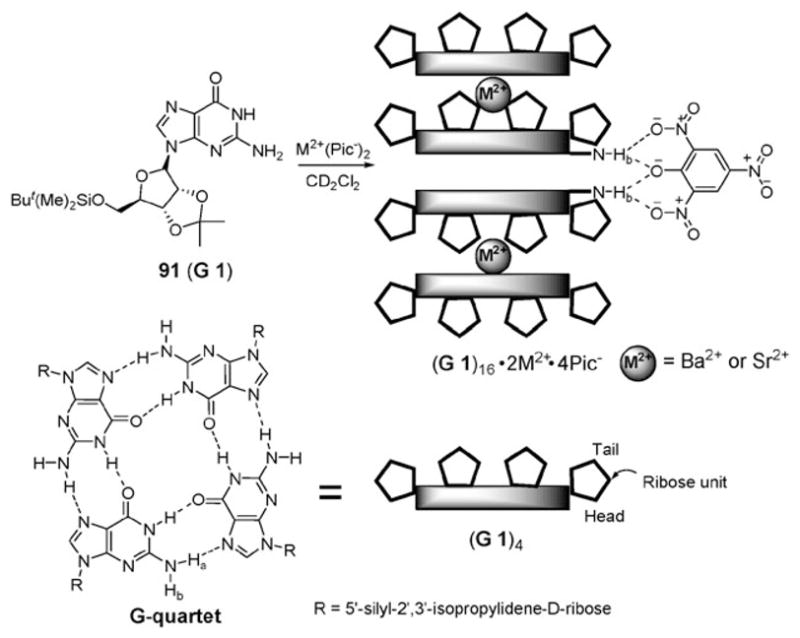
Guanosine 91 (G 1) and the proposed binding modes displayed by the G-quartet built up from this monomer in the presence of BaPic2 and SrPic2.
In 2003, this same group also synthesized the calix[4]arene– guanosine conjugate 92 (G 2) and investigated its properties of the formation of a G-quartet and its ability to effect ion pair recognition (Fig. 53).98 This calix[4]arene nucleotide (92 (G 2)) is poorly soluble in dry CDCl3 and gave an ill-resolved 1H NMR spectrum in this solvent. Such a finding is consistent with this nucleotide 92 (G 2) forming one or more nonspecific aggregates in the absence of water. Nucleotide 92 (G 2) is much more soluble in D2O-saturated CDCl3, and under these more polar solvent conditions gives two sets of well-resolved 1H NMR signals for the guanosine moieties. This latter observation led to the suggestion that the inherent D2 symmetry is destroyed and that 92 (G 2) is dimerized via the formation of a G-quartet (Fig. 53). On the basis of 1H NMR and 2-D NMR spectroscopic analyses, the resulting dimer, 922·(H2O)n was found to bind certain ion pairs and to extract them from an aqueous environment into a chloroform phase. The dimer is thought to recognize cations, such as K+ and Na+, via the carbonyl oxygen atoms of the G-quartet formed under these conditions, as well as anions, such as Br− and Cl−, via hydrogen bonds involving the amide NH protons and the anions. Selectivity for K+ over Na+ and Br− over Cl− was seen in these extractions.
Fig. 53.
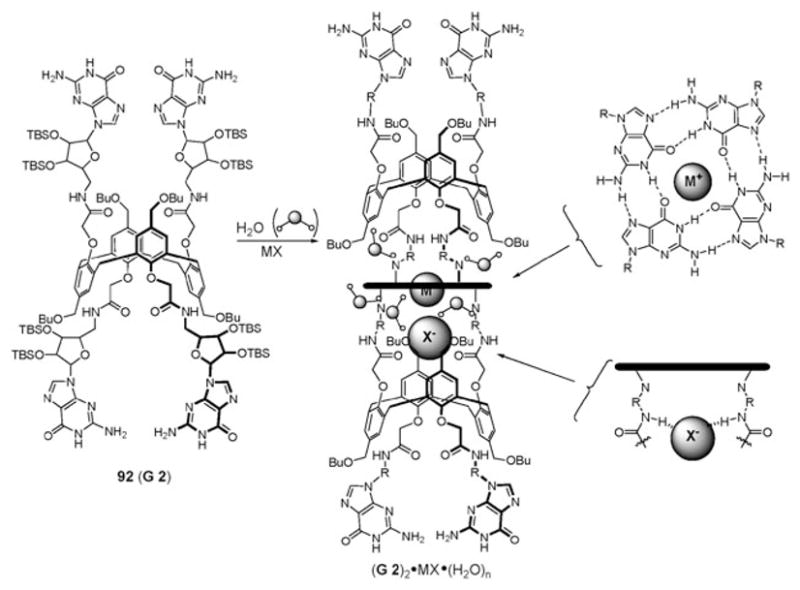
Calix[4]arene-based guanosine 92 (G 2) and its water-mediated ion pair recognition.
More recently, the Lippert group presented computational evidence that two stacked DNA-base quartets, namely a G-quartet (G 3)4 and an A-quartet (A 1)4, are able to bind the NaCl ion pair in a cooperative fashion (Fig. 54).99 Nucleo-bases 93 and 94 which combine to form a G-quartet (G 3)4 and an A-quartet (A 1)4, respectively, recognize the Na+ cation and the Cl− anion. The ion-filled DNA-base quartets are thought to stack so as to allow a direct interaction between the bound Na+ cation and Cl− anion (Fig. 54).
Fig. 54.
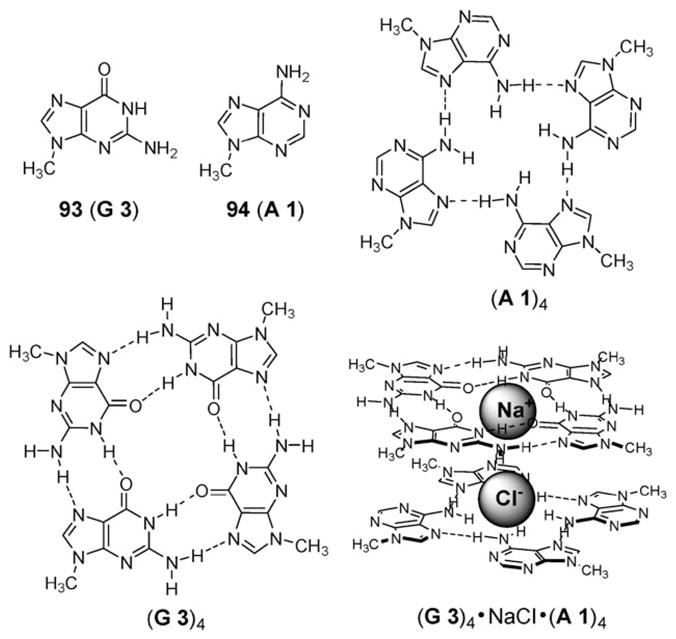
G-quartet (G 3)4 and A-quartet (A 1)4 and their proposed cooperative recognition of NaCl.
10. Ion pair receptors based on pyrroles and indoles for anion recognition
Pyrroles and indoles can also act as hydrogen bond donors for anion recognition. Not surprisingly, therefore, such motifs have been used to create ion pair receptors. One recent example comes from Jeong and coworkers. Specifically, in 2007 this group reported the preparation of receptor 95 (Fig. 55) that contains both a diazacrown ether for cation binding and an adjacent rigid biindole scaffold for anion recognition.100 Detailed 1H NMR titration experiments, carried out in DMSO-d6/CD3CN, provided evidence that the presence of alkali metal ions bound within the azacrown ether ring enhances the affinity of receptor 95 for anions. For example, the association constant of the complex of 95·Na+ for Cl− is ca. 2000 times higher than that of the metal-free form of 95 (Table 14). This increase in anion affinity was ascribed to the additional electrostatic interaction between the co-bound ions. An energy-minimized structure of 95·NaCl generated with a Macro Model 9.1, MMFFs force field, provided support for the proposal that NaCl is bound to the receptor as a contact ion pair.
Fig. 55.
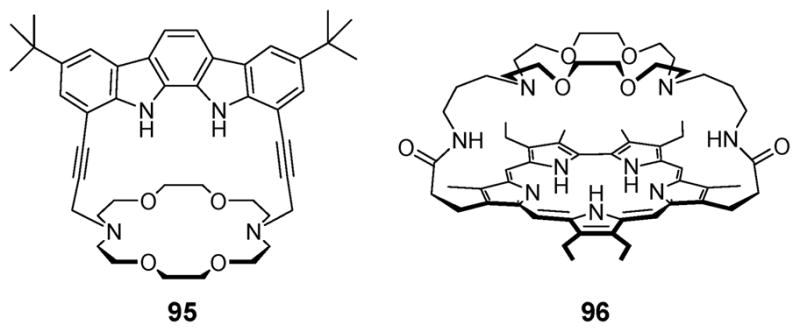
Biindole-diazacrown-based ion pair receptor 95 and diazacrown-capped sapphyrin 96.
Table 14.
Association constants (Ka/M−1) between ion pair receptor 95 and halides recorded in the presence and absence of alkali metal salts (1.0 equiv.) in 10% (v/v) DMSO-d6/CD3CN at 297 ± 1 K
| Halidea | Cation additive | Association constant/M−1 |
|---|---|---|
| Cl− | None | 7 |
| LiClO4 | 120 | |
| NaClO4 | 14 000 | |
| KPF6 | 6200 | |
| Br− | None | —b |
| LiClO4 | 24 | |
| NaClO4 | 600 | |
| KPF6 | 200 | |
| I− | None | —b |
| LiClO4 | 9 | |
| NaClO4 | 61 | |
| KPF6 | 45 |
Halide anions were studied as the corresponding tetrabutylammonium salts.
The chemical shift changes during the titration were too small to determine accurately an association constant.
Very early on (1995), Sessler and Brucker synthesized the diazacrown-capped sapphyrin 96 (Fig. 55) as a possible ion pair receptor.101 In this case, 1H NMR and UV/Vis spectroscopic analyses carried out in 10% CD3OD in CDCl3 revealed that the diprotonated form of compound 96 (bis-HCl salt) is able to bind an ammonium cation and a fluoride anion simultaneously.
First reported in 1886 by Baeyer,102 the tetrapyrrolic macrocycle 97 (Fig. 56), calix[4]pyrrole, was found in 1996 by Sessler et al. to be able to bind certain anions in organic solvents.103 In 2005, Moyer, Sessler, Gale and coworkers reported that calix[4]pyrrole 97 can act as an ion pair receptor for various cesium salts and certain organic halide salts in the solid state (Fig. 56).104 X-Ray crystal structures of several cesium and organic cation-containing anion complexes of calix[4]pyrrole 97 were solved. Taken in concert, they revealed that the anions are bound to the pyrrolic NH protons via hydrogen bonds. These interactions, which were expected on the basis of prior studies, serve to fix the calix[4]pyrrole in the cone conformation. This conformational locking, in turn, provides an electron-rich bowl-shaped cavity into which, e.g., cesium cation is bound via a combination of π–metal and dipole interactions.
Fig. 56.
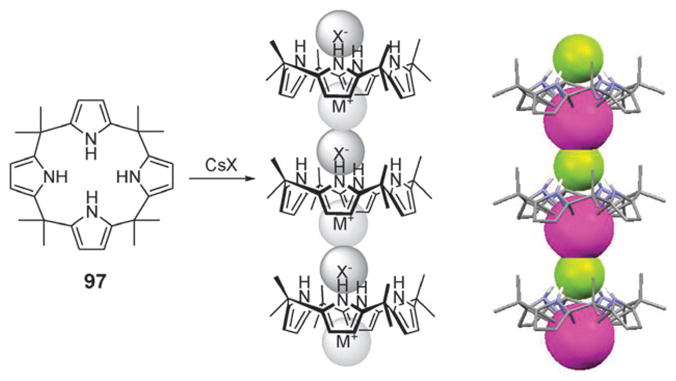
Calix[4]pyrrole 97, its binding mode for CsX and the crystal structure of 97·CsF. The X-ray structure shown was produced using coordinates that originally appeared in ref. 104.
Further evidence that calix[4]pyrrole 97 is effective for ion pair recognition came from liquid–liquid extraction studies carried out by Wintergerst et al.105 This study demonstrated that compound 97 can extract CsCl and CsBr, but not CsNO3, from an aqueous phase into nitrobenzene, a relatively polar organic phase. The solvent extraction process was modeled in terms of three thermochemical steps (Fig. 57). The first of these steps involves a partitioning of the cesium cations and the halide anions into the nitrobenzene phase from the water phase. The second step involves a conformational change of the calix[4]pyrrole such that it adopts the cone conformation, a geometry it maintains as the result of halide anion binding. The third step involves the cesium cation binding within the bowl-shaped calix cavity created as the result of the conformational change taking place in step 2. While thermodynamically equivalent in terms of the final state, it was appreciated that these steps could be taking place concurrently. The key point is that under this extraction, calix[4]pyrrole 97 binds both the cesium cation and a halide anion (Cl− or Br−).
Fig. 57.
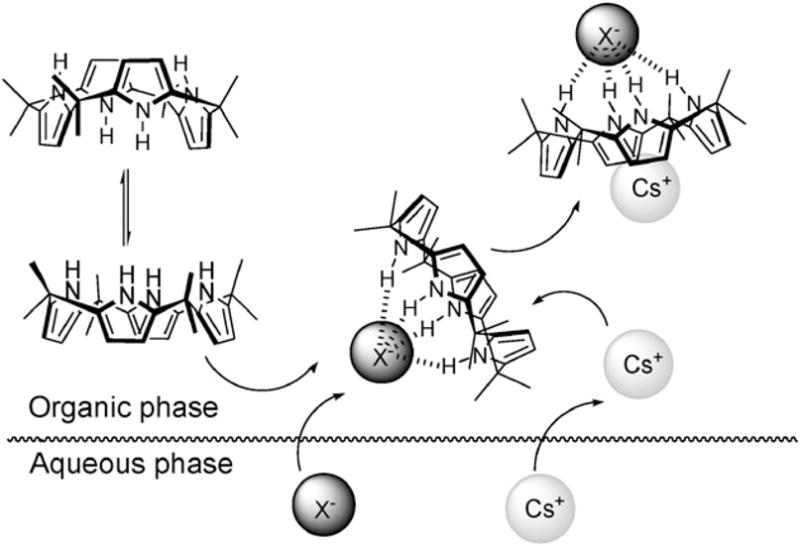
Proposed thermodynamic steps used to model cesium salt extraction by calix[4]pyrrole 97.
Very recently, transmembrane transport experiments were carried out with calix[4]pyrrole 97.106 It was found that this simple-to-obtain receptor transports CsCl across phospholipid bilayers with significant selectivity and with an efficiency that is enhanced relative to other alkaline chloride salts. Such findings provide further support for the conclusion that calix[4]pyrrole 97 can act as an ion pair receptor under appropriate conditions.
11. Ion pair recognition achieved using dual host receptors
A different approach to ion pair recognition, and one that is potentially simpler, than the creation of a ditopic receptor system, involves the use of an anion receptor in conjunction with a cation binding motif. This so-called dual-host approach was used by Cafeo et al. to bind organic ion pairs (Fig. 58).107 Here, it was found that upon addition of nBuNH3 +Cl− to a solution of anion receptor 98 in CD2Cl2, little in the way of discernible chemical shifts were seen in the 1H NMR spectrum. This led to the suggestion that the host–guest interaction between calix[6]pyrrole 98 and the Cl− anion is weak. On the other hand, it was found that when solutions of the three dimensional p-tert-butylcalix[5]arenes 99a–c, receptors that are known to form selective 1 : 1 complexes with linear alkylammonium cations,107 were treated with nBuNH3 +Cl−, two sets of distinguishable proton signals in the corresponding 1H NMR spectra were seen. These signals correspond to the uncomplexed and the nBuNH3 +complexed forms of these receptors, respectively.
Fig. 58.
Hosts and a guest used for binding studies involving so-called dual-host systems.
On the basis of these findings, the authors suggested that the binding of nBuNH3 + by 99a–c is slow on the NMR timescale. In the case of 99a, the cation-bound form makes up less than 2% of the equilibrium mixture. On the other hand, the addition of 1.0 molar equiv. of the putative anion receptor 98 to a 1 : 1 mixture of 99a and nBuNH3 +Cl− causes the proton signal of the free guest (nBuNH3 +) to disappear. Such a dramatic change is attributed to the complete complexation of the ions making up nBuNH3 +Cl− by this particular combination of anion and cation receptors. Evidence that the nBuNH3 + cation and the Cl− anion are bound to the calix[5]-arene (99a) and calix[6]pyrrole (98) subunits, respectively, came from 1H NMR spectral analyses. Specifically, after formation of the proposed complexes, the proton signals of the nBuNH3 + cation appear in the high field region (i.e., from δ = −0.4 ppm to δ = −2.0 ppm), characteristic of a cation inclusion complex. The NH proton signal of the calix[6]-pyrrole moiety 98 was found to be shifted to lower field (i.e., from δ = 7.74 ppm to δ = 10.91 ppm), as would be expected upon anion binding. These findings show that a weak anion or cation receptor can be transformed into a powerful one via the synergic action of a complementary receptor.
This same group also demonstrated that the combined action of the anion receptor 98 and the β-cyclodextrin-based cation receptor 100 (Fig. 58) is able to solubilize the otherwise sparingly soluble organic ion pair 101, paraquat chloride, in CD3CN and CD2Cl2.108 Furthermore, this dual-host system was found to extract paraquat chloride 101 from an aqueous phase into dichloromethane. Such a finding was also rationalized in terms of a synergic effect involving this particular set of paired anion and cation receptors.
12. Mixed calixarene–calixpyrrole ion pair receptors
More recently, Sessler and coworkers reported the synthesis of the calix[4]arene crown-6-strapped calix[4]pyrrole 102 (Fig. 59), as well as its interactions with the Cs+ cation, the F− anion, and the CsF ion pair.109 The 1,3-alternate crown-6 strapped calix[4]arene and strapped calix[4]pyrrole subunits are known to form strong complexes with the Cs+ cation and various halide anions, respectively. When combined to create receptor 102, the result is a system that binds and releases these ions slowly on the NMR timescale. Moreover, on the basis of 1H NMR spectral studies carried out in 10% methanol-d4 in chloroform-d, it was concluded that the cesium cation (studied using the non-coordinating perchlorate counteranion) is strongly bound to the crown-6 ring. The addition of the F− anion engenders no change in the 1H NMR spectrum, a result interpreted in terms of a lack of appreciable interaction between the ion pair receptor 102 and the F− anion, as might be expected given the strong solvation of this particular anion by the protic solvent, methanol. However, in the presence of the Cs+ cation, a species presumably bound to the crown-6, the F− anion is strongly complexed within the calix[4]pyrrole cavity (Fig. 59), as is evidenced by 1H NMR spectral studies. For instance, the proton signal of the pyrrolic NH protons was found to be shifted downfield by Δδ ≈ 6 ppm. Likewise, the β-protons of the pyrroles were found to be shifted upfield, presumably reflecting an increase in the anionic character of the pyrrole ring caused by hydrogen bonding between the NH protons and the F− anion. The NH proton peak, initially a singlet, was found to be split into a doublet (JFH = 44 Hz), a finding that is ascribable to the coupling between the bound fluoride anion and the NH protons.
Fig. 59.
Ion pair receptor 102 and its binding behavior towards the Cs+ cation and the F− anion.
An 1H NMR titration experiment involving 102 and CsF ion pair, carried out in 9 : 1 CDCl3/CD3OD, revealed that the complexation processes occurs stepwise, not concurrently; that is, only after the cesium cation is complexed within the calix[4]arene crown-6 ring, does binding of the fluoride anion by the calix[4]pyrrole moiety occur to an appreciable extent. A single crystal X-ray structure of the CsF complex revealed that ion pair receptor 102 forms a stable 1 : 1 complex with the CsF in spite of the large separation (≈10.92 Å ) enforced between the receptor-bound anion and cation (Fig. 60).109
Fig. 60.
Two different views of the crystal structure of 102·CsF· MeOH.
13. Ion pair recognition achieved using functionalized polymers
In 2008, a new approach to the recognition of ion pairs was introduced by Sessler, Bielawski, and coworkers. This approach relies on the use of polymer containing both anion and cation recognition groups. In their report, the synthesis, characterization, and extraction properties of the mixed MMA (methyl methacrylate) copolymer 103 (Fig. 61) were detailed. This system (i.e., copolymer 103) bears both pendant calix[4]-pyrrole and benzo-[15]crown-5 subunits, recognition motifs known to be capable of binding halide anions in organic media and forming a 2 : 1 sandwich complex with the K+ cation, respectively (Fig. 61).110 The control MMA copolymers 104 and 105 containing either the calix[4]pyrrole subunits or the benzo-[15]crown-5 were also synthesized. The monomeric simple ion receptors 106 and 107 were included in this study (Fig. 61).
Fig. 61.

Schematic representation of polymeric ion pair receptor 103 and various control systems.
On the basis of 1H NMR spectroscopic analyses, it was concluded that compounds 106 and 107 or a 1 : 1 mixture of 97 and 108 are unable to extract hard alkaline halide salts, such as KF and KCl, from D2O into a dichloromethane-d2. In marked contrast, polymer 103 containing both cation and anion receptors was found to extract KF and KCl under these same conditions. Further support for this conclusion came from 1H, 19F NMR, and flame emission spectroscopic analyses. It was thus proposed that the polymer backbone can play an important role in enhancing the ability of ion receptors to bind ion pairs, or at least pairs of ions. In addition, it was found that polymer 103 is capable of extracting both KF and KCl much more effectively than either polymers 104 and 105. This provides support for the notion that the presence of both cation and anion recognition sites is essential if efficient binding and extraction of ion pairs is to be achieved.110
14. Conclusion
In this review, we have attempted to classify the various ion pair receptors reported so far on the basis of the anion recognition motif used to effect their creation. While a review of this nature cannot be fully comprehensive, a strong effort has been made to describe the binding ability and properties of representative examples of ion pair receptors. Compared with simple ion receptors capable of binding either cations or anions, ion pair receptors, possessing both cation and anion recognition sites, have attracted relatively little attention. Presumably, this reflects their synthetic difficulties, as well as experimental complexities resulting from the more elaborate nature of the binding phenomena involved. Nevertheless, we believe that the design and study of ion pair receptors is worthwhile. As detailed herein, such systems often display an enhanced ability to solubilize otherwise insoluble ions, a sometimes remarkable capacity to extract certain ion pairs from water, and selectivities for targeted ions that are superior to that of simple ion receptors. We thus predict that this is an area of supramolecular chemistry that is ripe for future growth.
Acknowledgments
This work was supported by the Office of Basic Energy Sciences, U.S. Department of Energy (DOE) (DE-FG02-01ER15186), the National Institutes of Health (GM 58907), and the Robert A. Welch Foundation (F-1018). J.L.S. also thanks the Korean government for support under the WCU (World Class University) program (R32-2008-000-10217-0).
Biographies

Mr Sung Kuk Kim earned a MS degree from Konyang University in South Korea under the supervision of Prof. Jong Seung Kim. He is a currently a graduate student at The University of Texas at Austin working in the group of Professor Jonathan L. Sessler. His research interests are focused on the development of various pyrrole- and calix[4]-arene-based systems for the recognition and sensing of neutral guests and ions, including ion pairs.

Professor Jonathan L. Sessler received a BS degree in chemistry in 1977 from the University of California, Berkeley. He obtained a PhD from Stanford University in 1982 (supervisor: Professor James P. Collman). He was an NSF-CNRS and NSF-NATO Postdoctoral Fellow with Professor Jean-Marie Lehn at L’Université Louis Pasteur de Strasbourg, France. He was then a JSPS Visiting Scientist in Professor Iwao Tabushi’s group in Kyoto, Japan. In 1984 he accepted a position as an Assistant Professor of Chemistry at the University of Texas at Austin, where he is currently the Roland K. Pettit Chair. Dr Sessler is also a WCU Professor at Yonsei University in South Korea. He is a co-founder of two companies, Pharmacyclics, Inc. and Anionics, Inc.
Footnotes
Part of a themed issue on the supramolecular chemistry of anionic species.
References
- 1.Lehn JM. Supramolecular Chemistry: Concepts and Perspectives. VCH; Weinheim: 1995. [Google Scholar]
- 2.Steed JW, Atwood JL. Supramolecular Chemistry: An Introduction. Wiley; Chichester: 2000. [Google Scholar]
- 3.Gokel GW. Molecular Recognition Receptors for Cationic Guests. In: Lehn JM, Atwood JL, Davies JED, MacNicol DD, Vogtle F, editors. Comprehensive Supramolecular Chemistry. Pergamon; Oxford: 1996. p. 1. [Google Scholar]
- 4.Sessler JL, Gale PA, Cho WS. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]
- 5.Gale PA, García-Garrido SE, Garric J. Chem Soc Rev. 2008;37:151–190. doi: 10.1039/b715825d. [DOI] [PubMed] [Google Scholar]
- 6.Gale PA, Quesada R. Coord Chem Rev. 2006;250:3219–3244. [Google Scholar]
- 7.Bianchi A, Bowman-James K, García-España E, editors. Supramolecular Chemistry of Anions. Wiley-VCH; New York: 1997. [Google Scholar]
- 8.Gale PA. In: The Encyclopedia of Supramolecular Chemistry. Atwood JL, Steed JW, editors. Dekker; New York: 2004. pp. 31–41. [Google Scholar]
- 9.Beer PD, Gale PA. Angew Chem, Int Ed. 2001;40:486–516. [PubMed] [Google Scholar]
- 10.Caltagirone C, Gale PA. Chem Soc Rev. 2009;38:520–563. doi: 10.1039/b806422a. [DOI] [PubMed] [Google Scholar]
- 11.Martínez-Máñez R, Sancenán F. Chem Rev. 2003;103:4419–4476. doi: 10.1021/cr010421e. [DOI] [PubMed] [Google Scholar]
- 12.Smith BD. In: Ion Pair Recognition by Ditopic Receptors, Macrocyclic Chemistry: Current Trends and Future Prospectives. Gloe K, Antonioli B, editors. Kluwer; London: 2005. pp. 137–151. [Google Scholar]
- 13.Kirkovits GJ, Shriver JA, Gale PA, Sessler JL. J Inclusion Phenom Macrocyclic Chem. 2001;41:69–75. [Google Scholar]
- 14.Antonisse MMG, Reinhoudt DN. Chem Commun. 1998:443–448. [Google Scholar]
- 15.Gokel GW, Leevy WM, Weber ME. Chem Rev. 2004;104:2723–2750. doi: 10.1021/cr020080k. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda A, Shinkai S. Chem Rev. 1997;97:1713–1734. doi: 10.1021/cr960385x. [DOI] [PubMed] [Google Scholar]
- 17.Kim JS, Quang DT. Chem Rev. 2007;107:3780–3799. doi: 10.1021/cr068046j. [DOI] [PubMed] [Google Scholar]
- 18.Jones JW, Gibson HW. J Am Chem Soc. 2003;125:7001–7004. doi: 10.1021/ja034442x. [DOI] [PubMed] [Google Scholar]
- 19.Huang F, Jones JW, Slebodnick C, Gibson HW. J Am Chem Soc. 2003;125:14458–14464. doi: 10.1021/ja036606f. [DOI] [PubMed] [Google Scholar]
- 20.Huang F, Jones JW, Gibson HW. J Org Chem. 2007;72:6573–6576. doi: 10.1021/jo070792g. [DOI] [PubMed] [Google Scholar]
- 21.Reetz MT, Niemeyer CM, Harms K. Angew Chem, Int Ed Engl. 1991;30:1472–1474. [Google Scholar]
- 22.Reetz MT, Johnson BM, Harms K. Tetrahedron Lett. 1994;35:2525–2528. [Google Scholar]
- 23.Rudkevich DM, Brzozka Z, Palys M, Visser HC, Verboom W, Reinhoudt DN. Angew Chem, Int Ed Engl. 1994;33:467–468. [Google Scholar]
- 24.Rudkevich DM, Verboom W, Reinhoudt DN. J Org Chem. 1994;59:3683–3686. [Google Scholar]
- 25.Rudkevich DM, Mercer-Chalmers JD, Verboom W, Ungaro R, de Jong F, Reinhoudt DN. J Am Chem Soc. 1995;117:6124–6125. [Google Scholar]
- 26.Cametti M, Nissinen M, Cort AD, Mandolini L, Rissanen K. Chem Commun. 2003:2420–2421. doi: 10.1039/b307849c. [DOI] [PubMed] [Google Scholar]
- 27.Cametti M, Nissinen M, Cort AD, Mandolini L, Rissanen K. J Am Chem Soc. 2005;127:3831–3837. doi: 10.1021/ja042807n. [DOI] [PubMed] [Google Scholar]
- 28.Cametti M, Nissinen M, Cort AD, Mandolini L, Rissanen K. J Am Chem Soc. 2007;129:3641–3648. doi: 10.1021/ja068561z. [DOI] [PubMed] [Google Scholar]
- 29.Nagasaki T, Fujishima H, Takeuchi M, Shinkai S. J Chem Soc, Perkin Trans 1. 1995:1883–1888. [Google Scholar]
- 30.Rudkevich DM, Shivanyuk AN, Brzozka Z, Verboom W, Reinhoudt DN. Angew Chem, Int Ed Engl. 1995;34:2124–2126. [Google Scholar]
- 31.Kim Y-H, Hong J-I. Chem Commun. 2002:512–513. doi: 10.1039/b109596j. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Shao X-B, Jia M-X, Jiang X-K, Li Z-T, Chen G-J. Tetrahedron. 2005;61:8095–8100. [Google Scholar]
- 33.Gunning P, Benniston AC, Peacock RD. Chem Commun. 2004:2226–2227. doi: 10.1039/b409132a. [DOI] [PubMed] [Google Scholar]
- 34.Gunning PT. Org Biomol Chem. 2005;3:3877–3879. doi: 10.1039/b510262f. [DOI] [PubMed] [Google Scholar]
- 35.Kemmer M, Biesemans M, Gielen M, Martins JC, Gramlich V, Willem R. Chem–Eur J. 2001;7:4686–4695. doi: 10.1002/1521-3765(20011105)7:21<4686::aid-chem4686>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Reeske G, Bradtmöller G, Schürmann M, Jurkschat K. Chem–Eur J. 2007;13:10239–10245. doi: 10.1002/chem.200701141. [DOI] [PubMed] [Google Scholar]
- 37.Reeske G, Schürmann M, Costisella B, Jurkschat K. Organometallics. 2007;26:4170–4179. [Google Scholar]
- 38.Reeske G, Schürmann M, Jurkschat K. Dalton Trans. 2008:3398–3400. doi: 10.1039/b803552k. [DOI] [PubMed] [Google Scholar]
- 39.Scheerder J, van Duynhoven JPM, Engbersen JFJ, Reinhoudt DN. Angew Chem, Int Ed Engl. 1996;35:1090–1093. [Google Scholar]
- 40.Chrisstoffels LAJ, de Jong F, Reinhoudt DN, Sovelli S, Gazzola L, Casnati A, Ungaro R. J Am Chem Soc. 1999;121:10142–10151. [Google Scholar]
- 41.Kang SO, Nam KC. Bull Korean Chem Soc. 2002;23:640–642. [Google Scholar]
- 42.Oh JM, Cho EJ, Ryu BJ, Lee YJ, Nam KC. Bull Korean Chem Soc. 2003;24:1538–1540. [Google Scholar]
- 43.Webber PRA, Beer PD. Dalton Trans. 2003:2249–2252. [Google Scholar]
- 44.Tumcharern G, Tuntulani T, Coles SJ, Hursthouse MB, Kilburn JD. Org Lett. 2003;5:4971–4974. doi: 10.1021/ol035894h. [DOI] [PubMed] [Google Scholar]
- 45.Nabeshima T, Saiki T, Iwabuchi J, Akine S. J Am Chem Soc. 2005;127:5507–5511. doi: 10.1021/ja042882y. [DOI] [PubMed] [Google Scholar]
- 46.Garozzo D, Gattuso G, Notti A, Pappalardo A, Pappalardo S, Parisi MF, Perez M, Pisagatti I. Angew Chem, Int Ed. 2005;44:4892–3896. doi: 10.1002/anie.200500985. [DOI] [PubMed] [Google Scholar]
- 47.Pescatori L, Arduini A, Pochini A, Secchi A, Massera C, Ugozzoli F. Org Biomol Chem. 2009;7:3698–3708. doi: 10.1039/b906409e. [DOI] [PubMed] [Google Scholar]
- 48.Ballistreri FP, Notti A, Pappalardo S, Parisi MF, Pisagatti I. Org Lett. 2003;5:1071–1074. doi: 10.1021/ol034093b. [DOI] [PubMed] [Google Scholar]
- 49.Gargiulli C, Gattuso G, Liotta C, Notti A, Parisi MF, Pisagatti I, Pappalardo S. J Org Chem. 2009;74:4350–4353. doi: 10.1021/jo802777v. [DOI] [PubMed] [Google Scholar]
- 50.Tozawa T, Misawa Y, Tokita S, Kubo Y. Tetrahedron Lett. 2000;41:5219–5223. [Google Scholar]
- 51.Nishizawa S, Shigemori K, Teramae N. Chem Lett. 1999:1185–1186. [Google Scholar]
- 52.Jeon NJ, Yeo HM, Nam KC. Bull Korean Chem Soc. 2008;29:663–665. [Google Scholar]
- 53.Barboiu M, Vaughan G, van der Lee A. Org Lett. 2003;5:3073–3076. doi: 10.1021/ol035096r. [DOI] [PubMed] [Google Scholar]
- 54.Flack SS, Chaumette J-L, Kilburn JD, Langley GJ, Webster M. J Chem Soc, Chem Commun. 1993:399–401. [Google Scholar]
- 55.Deetz MJ, Shang M, Smith BD. J Am Chem Soc. 2000;122:6201–6207. [Google Scholar]
- 56.Mahoney JM, Beatty AM, Smith BD. J Am Chem Soc. 2001;123:5847–5858. doi: 10.1021/ja0156082. [DOI] [PubMed] [Google Scholar]
- 57.Mahoney JM, Beatty AM, Smith BD. Inorg Chem. 2004;43:7617–7621. doi: 10.1021/ic049066b. [DOI] [PubMed] [Google Scholar]
- 58.Mahoney JM, Nawaratna GU, Beatty AM, Duggan PJ, Smith BD. Inorg Chem. 2004;43:5902–5907. doi: 10.1021/ic0494859. [DOI] [PubMed] [Google Scholar]
- 59.Mahoney JM, Stucker KA, Jiang H, Carmichael I, Brinkmann NR, Beatty AM, Noll BC, Smith BD. J Am Chem Soc. 2005;127:2922–2928. doi: 10.1021/ja0440295. [DOI] [PubMed] [Google Scholar]
- 60.Mahoney JM, Marshall RA, Beatty AM, Smith BD, Camiolo S, Gale PA. J Supramol Chem. 2001;1:289–292. [Google Scholar]
- 61.Suksai C, Leeladee P, Jainuknan D, Tuntulani T, Muangsin N, Chailapakul O, Kongsaeree P, Pakavatchai C. Tetrahedron Lett. 2005;46:2765–2769. [Google Scholar]
- 62.Lankshear MD, Dudley IM, Chan K-M, Beer PD. New J Chem. 2007;31:684–690. [Google Scholar]
- 63.Lankshear MD, Cowley AR, Beer PD. Chem Commun. 2006:612–614. doi: 10.1039/b515634c. [DOI] [PubMed] [Google Scholar]
- 64.Lankshear MD, Dudley IM, Chan K-M, Cowley AR, Santos SM, Felix V, Beer PD. Chem–Eur J. 2008;14:2248–2263. doi: 10.1002/chem.200701347. [DOI] [PubMed] [Google Scholar]
- 65.Beer PD, Drew MGB, Knubley RJ, Ogden MI. J Chem Soc, Dalton Trans. 1995:3117–3123. [Google Scholar]
- 66.Evans J, Beer PD. Dalton Trans. 2003:4451–4456. [Google Scholar]
- 67.Stibor I, Hafeed DSM, Lhoták P, Hodačová J, Koča J, Cajan M. Gazz Chim Ital. 1997;127:673. [Google Scholar]
- 68.Redman E, Beer PD, Dent SW, Drew MGB. Chem Commun. 1998:231–232. [Google Scholar]
- 69.Beer PD, Dent SW. Chem Commun. 1998:825–826. [Google Scholar]
- 70.Uppadine H, Redman JE, Dent SW, Drew MGB, Beer PD. Inorg Chem. 2001;40:2860–2869. doi: 10.1021/ic001450v. [DOI] [PubMed] [Google Scholar]
- 71.Beer PD, Hopkins PK, Mckinney JD. Chem Commun. 1999:1253–1254. [Google Scholar]
- 72.Cooper JB, Drew MGB, Beer PD. J Chem Soc, Dalton Trans. 2001:392–401. [Google Scholar]
- 73.Cooper JB, Drew MGB, Beer PD. J Chem Soc, Dalton Trans. 2000:2721–2728. [Google Scholar]
- 74.Le Gac S, Jabin I. Chem–Eur J. 2008;14:548–557. doi: 10.1002/chem.200701051. [DOI] [PubMed] [Google Scholar]
- 75.Hamon M, Ménand M, Le Gac S, Luhmer M, Dalla V, Jabin I. J Org Chem. 2008;73:7067–7071. doi: 10.1021/jo800712q. [DOI] [PubMed] [Google Scholar]
- 76.Le Gac S, Ménand M, Jabin I. Org Lett. 2008;10:5195–5198. doi: 10.1021/ol8021726. [DOI] [PubMed] [Google Scholar]
- 77.Atwood JL, Szumna A. J Am Chem Soc. 2002;124:10646–10647. doi: 10.1021/ja027191l. [DOI] [PubMed] [Google Scholar]
- 78.Atwood JL, Szumna A. Chem Commun. 2003:940–941. doi: 10.1039/b301511d. [DOI] [PubMed] [Google Scholar]
- 79.Frontera A, Orell M, Garau C, Quiñonero D, Molins E, Mata I, Morey J. Org Lett. 2005;7:1437–1440. doi: 10.1021/ol0474608. [DOI] [PubMed] [Google Scholar]
- 80.Kubik S. J Am Chem Soc. 1999;121:5846–5855. [Google Scholar]
- 81.Kubik S, Goddard R. J Org Chem. 1999;64:9475–9486. [Google Scholar]
- 82.Gong J, Gibb BC. Chem Commun. 2005:1393–1395. doi: 10.1039/b414923h. [DOI] [PubMed] [Google Scholar]
- 83.Wisner JA, Beer PD, Berry NG, Tomapatanaget B. Proc Natl Acad Sci US A. 2002;99:4983–4986. doi: 10.1073/pnas.062637999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wisner JA, Beer PD, Drew MGB. Angew Chem, Int Ed. 2001;40:3606–3609. doi: 10.1002/1521-3773(20011001)40:19<3606::aid-anie3606>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 85.Sambrook R, Beer PD, Wisner JA, Paul RL, Cowley AR. J Am Chem Soc. 2004;126:15364–15365. doi: 10.1021/ja045080b. [DOI] [PubMed] [Google Scholar]
- 86.Lankshear D, Evans NH, Bayly SR, Beer PD. Chem–Eur J. 2007;13:3861–3870. doi: 10.1002/chem.200700041. [DOI] [PubMed] [Google Scholar]
- 87.Arafa EA, Kinnear KI, Lackhart JC. J Chem Soc, Chem Commun. 1992:61–64. [Google Scholar]
- 88.White DJ, Laing N, Miller H, Parsons S, Coles S, Tasker PA. Chem Commun. 1999:2077–2078. [Google Scholar]
- 89.Tuntulani T, Poompradub S, Thavornyutikarn P, Jaiboon N, Ruangpornvisuti V, Chaichit N, Asfari Z, Vicens J. Tetrahedron Lett. 2001;42:5541–5544. [Google Scholar]
- 90.Schmidtchen FP. J Org Chem. 1986;51:5161–5168. [Google Scholar]
- 91.Galán A, Andreu D, Echavarren AM, Prados P, de Mendoza J. J Am Chem Soc. 1992;114:1511–1512. [Google Scholar]
- 92.de Silva P, McClean GD, Pagliari S. Chem Commun. 2003:2010–2011. doi: 10.1039/b305262a. [DOI] [PubMed] [Google Scholar]
- 93.Savage PB, Holmgren SK, Gellman SH. J Am Chem Soc. 1993;115:7900–7901. [Google Scholar]
- 94.Savage PB, Holmgren SK, Gellman SH. J Am Chem Soc. 1994;116:4069–4070. [Google Scholar]
- 95.Mele A, Metrangolo P, Neukirch H, Pilati T, Resnati G. J Am Chem Soc. 2005;127:14972–14973. doi: 10.1021/ja054862h. [DOI] [PubMed] [Google Scholar]
- 96.Arduini A, Giorgi G, Pochini A, Secchi A, Ugozzoli F. J Org Chem. 2001;66:8302–8308. doi: 10.1021/jo016035e. [DOI] [PubMed] [Google Scholar]
- 97.Shi X, Fettinger JC, Davis JT. Angew Chem, Int Ed. 2001;40:2827–2831. [PubMed] [Google Scholar]
- 98.Kotch FW, Sidorov V, Lam Y-F, Kayser KJ, Li H, Kaucher MS, Davis JT. J Am Chem Soc. 2003;125:15140–15150. doi: 10.1021/ja0364165. [DOI] [PubMed] [Google Scholar]
- 99.van der Wijst T, Guerra CF, Swart M, Bickelhaupt FM, Lippert B. Angew Chem, Int Ed. 2009;48:3285–3287. doi: 10.1002/anie.200900057. [DOI] [PubMed] [Google Scholar]
- 100.Chae MK, Lee J-I, Kim N-K, Jeong K-S. Tetrahedron Lett. 2007;48:6624–6627. [Google Scholar]
- 101.Sessler JL, Brucker EA. Tetrahedron Lett. 1995;36:1175–1176. [Google Scholar]
- 102.Baeyer A. Ber Dtsch Chem Ges. 1886;19:2184–2185. [Google Scholar]
- 103.Gale A, Sessler JL, Král V, Lynch V. J Am Chem Soc. 1996;118:5140–5141. [Google Scholar]
- 104.Custelcean R, Delmau LH, Moyer BA, Sessler JL, Cho W-S, Gross D, Bates GW, Brooks SJ, Light ME, Gale PA. Angew Chem, Int Ed. 2005;44:2537–2542. doi: 10.1002/anie.200462945. [DOI] [PubMed] [Google Scholar]
- 105.Wintergerst MP, Levitskaia TG, Moyer BA, Sessler JL, Delmau LH. J Am Chem Soc. 2008;130:4129–4139. doi: 10.1021/ja7102179. [DOI] [PubMed] [Google Scholar]
- 106.Tong C, Quesada R, Sessler JL, Gale PA. Chem Commun. 2008:6321–6323. doi: 10.1039/b814988g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cafeo G, Gattuso G, Kohnke FH, Notti A, Occhipinti S, Pappalardo S, Parisi MF. Angew Chem, Int Ed. 2002;41:2122–2126. doi: 10.1002/1521-3773(20020617)41:12<2122::aid-anie2122>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 108.Cafeo G, Gargiulli C, Gattuso G, Kohnke FH, Notti A, Occhipinti S, Pappalardo S, Parisi MF. Tetrahedron Lett. 2002;43:8103–8106. [Google Scholar]
- 109.Sessler JL, Kim SK, Gross DE, Lee CH, Kim JS, Lynch VM. J Am Chem Soc. 2008;130:13162–13166. doi: 10.1021/ja804976f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aydogan A, Coady DJ, Kim SK, Akar A, Bielawski CW, Marquez M, Sessler JL. Angew Chem, Int Ed. 2008;47:9648–9652. doi: 10.1002/anie.200803970. [DOI] [PMC free article] [PubMed] [Google Scholar]