

Abstract
The disease mechanisms underlying dystrophin-deficient muscular dystrophy are complex, involving not only muscle membrane fragility, but also dysregulated calcium homeostasis. Specifically, it has been proposed that calcium channels directly initiate a cascade of pathological events by allowing calcium ions to enter the cell. The objective of this study was to investigate the effect of chronically blocking calcium channels with the aminoglycoside antibiotic streptomycin from onset of disease in the mdx mouse model of Duchenne muscular dystrophy (DMD).
Treatment in utero onwards delayed onset of dystrophic symptoms in the limb muscle of young mdx mice, but did not prevent degeneration and regeneration events occurring later in the disease course. Long-term treatment had a positive effect on limb muscle pathology, reduced fibrosis, increased sarcolemmal stability, and promoted muscle regeneration in older mice. However, streptomycin treatment did not show positive effects in diaphragm or heart muscle, and heart pathology was worsened. Thus, blocking calcium channels even before disease onset does not prevent dystrophy, making this an unlikely treatment for DMD. These findings highlight the importance of analyzing several time points throughout the life of the treated mice, as well as analyzing many tissues, to get a complete picture of treatment efficacy.
Duchenne muscular dystrophy (DMD) is a devastating neuromuscular disease affecting 1 in 3500 live male births. The disease is caused by mutations in the gene encoding dystrophin, a protein localized to the cytoplasmic surface of the sarcolemma. Here, dystrophin is believed to exert a dual role, both as structural protein important for membrane integrity and as a scaffolding protein for ion channels and various signaling pathways.1 Loss of dystrophin results in sarcolemmal instability, which causes the muscle to undergo continuous cycles of degeneration and regeneration. The regenerative capacity is not maintained, however, and this results in early, progressive weakening of skeletal muscle leading to loss of ambulation and respiratory failure.2 Dystrophin is also important in cardiac muscle, and patients frequently develop a dilated cardiomyopathy. One key finding in the pathology of DMD is an elevated intracellular calcium level,3 and several studies have explored the potential of pharmacological modulation of calcium influx as a therapeutic option in this disease.4, 5, 6, 7, 8
One of the earliest suggestions that calcium is important in DMD was based on cytosolic calcium accumulation in nonnecrotic fibers from DMD patient biopsies not observed in other dystrophies or myopathies.3 Calcium accumulation has also been reported in cultures of dystrophin-deficient myotubes from DMD patients and dystrophic mdx mice.9, 10, 11
The simplest explanation for excessive calcium influx is entry through microtears in the sarcolemma, which are more abundant in the dystrophic muscle.2 However, calcium influx may also occur through the non-voltage gated calcium channels, such as the calcium leak channels12 or transient receptor potential (TRP) channel family members.13, 14 The TRP channels have been proposed to be implicated in regulated calcium entry of skeletal muscle. Specifically, members of this family have been suggested to either directly form calcium channels or indirectly regulate calcium influx. It has been proposed that the nature of these channels could constitute store-operated calcium entry and/or stretch-activated calcium entry.15 Of these, the best characterized in skeletal muscle are TRPC1 and TRPC3. TRPC1 associates with α-syntrophin, a protein connected to the dystrophin-glycoprotein complex (DGC) of the sarcolemma.16 Disruption of the DGC complex in Duchenne muscular dystrophy could therefore alter these protein complexes and change the activity level of TRPC1, causing excessive calcium influx into dystrophic myofibers. TRPC3 has been shown to associate with the type 1 ryanodine receptor and other triadic proteins.17 This channel protein is not involved in store-operated calcium entry, but it has been suggested that TRPC3 could form a link between calcium entry and gene regulation in skeletal muscle via the calcineurin-nuclear factor of activated T cells signaling pathway.18
Recent studies have suggested a direct relationship between calcium dysregulation and dystrophin-deficiency. Transgenic overexpression of TRPC3 in mice is sufficient to induce muscular dystrophy without concomitant muscle membrane fragility,19 and inhibition of TRP channels in dystrophin-deficient mdx mice drastically reduces both calcium influx and dystrophy.19, 20 Dystrophin suppresses spontaneous sarcoplasmic reticulum elementary Ca2+ release events, and this inhibitory control, mediated via mechanosensitive pathways, is disrupted in mdx mice.21 In addition, myofiber degeneration alone, as exemplified in the α-dystrobrevin null mouse, does not cause functional muscle defects, and so the development of the DMD/mdx dystrophic pathology cannot be explained solely by membrane fragility.22
The precise molecular nature of the channel or channels involved is currently unclear, and there is controversy as to whether the non-voltage gated calcium channels constituted by TRPC family members initiate the disorder by triggering a cascade of pathological events.23
Regardless of entry mechanisms, calcium overload in skeletal muscle fibers results in activation of calcium-dependent proteolytic enzymes (calpains), which cause necrosis and degeneration24, 25 and which even degrade the compensatory protein utrophin.26 In addition, calcium overload has a detrimental impact on mitochondrial structure and function, resulting in apoptosis, decreased activity and mislocalization.27 This chronic damage eventually exhausts the normal muscle repair mechanisms mediated by the resident satellite cells28 and leads to end-stage disease.
DMD fetuses display both ultrastructural disturbances and increased calcium content of the skeletal muscles.29, 30 In the mdx mouse model of DMD, the dystrophic process also begins early in embryonic development and includes stem cell loss and structural muscle changes.31 Knockdown of dystrophin in adult mice does not cause any evident dystrophy,32 indicating that the disease has a developmental onset. Based on the involvement of calcium homeostasis and stretch-activated calcium channels in dystrophic pathology and the embryonic/fetal prepathological disturbances, the objective of the present study was to test whether blocking calcium influx from embryonic development can prevent initiation of pathology or alleviate the pathology to a greater extent than has been shown previously using short-term treatment regimes.5, 6, 8 We started our treatment regimen when female mice were fertilized, as detected by a vaginal plug, and continued the treatment for up to 6 months after birth. The strategy adopted was to nonspecifically block calcium channels33, 34 by treating our mice with the aminoglycoside antibiotic streptomycin in the drinking water, as previously described.8
Long-term treatment starting in utero of mdx mice did alleviate limb muscle pathology, but had adverse effects on cardiac and diaphragm tissue. This discordant tissue effectiveness could relate to variations in pathological severity of the analyzed tissues and suggests that the pathology is more complex than previously believed. Any preclinical drug trial conducted in the mdx mouse should therefore include analysis of a range of time points and a variety of tissues. Specifically, because DMD patients succumb to respiratory and cardiac failure, cardiac tissue and diaphragm should be included in any preclinical studies on treatments for DMD.
Materials and Methods
Animal Experiments
C57BL/10-mdx (mdx) and C57BL/10 (BL/10) mouse breeding pairs were plug-checked every day. When a vaginal plug was observed, the mother was treated with streptomycin (Sigma-Aldrich, St. Louis, MO) in the drinking water (4 g/L); the bottles were changed twice per week. Streptomycin is known to cross the placental membrane.35 On giving birth, the mother was continued on streptomycin in the water; the pups, when weaned, were continued on the same dose in their drinking water. The treated pups were analyzed after 6 weeks (n = 3–5), 10 weeks (n = 4–6), and 6 months (n = 3–5 for histology and Western blots, n = 5–11 for magnetic resonance imaging [MRI]) of continued postnatal treatment. Male mice were used for MRI scanning and Evans Blue dye uptake, because extensive control data were available in male mice for these measurements and to be able to correlate heart membrane damage with heart function. To minimize animal usage, females were used for all other analyses.
Toxicity of streptomycin was tested on mouse urine samples. Samples were collected by scruffing the mice and allowing them to urinate into an Eppendorf tube. The samples were then analyzed using a dipstick test for proteinuria and ketonuria (Bayer Multistix GP diagnostic strips; MediSupplies, Dorset, UK). Urine samples were additionally analyzed for proteinuria using a bicinchoninic acid kit for protein determination (Sigma-Aldrich).
All animal experiments were in accordance with the Home Office Guidelines, United Kingdom, and were performed under license no. PPL60/3445.
Human Biopsy Material
Human fetal tissue was obtained during autopsy of aborted fetuses aged 13 weeks (DMD) and 15 weeks (control). Diagnosis of DMD was made on the basis of DNA analysis. Biopsies were obtained and used according to the guidelines of and with permission from the Regional Ethics Committee for Southern Denmark #15879.
Histology
Diaphragm, tibialis anterior (TA), and heart ventricular tissue was harvested and processed for histology by snap-freezing in isopentane cooled in liquid nitrogen and stored at −80°C. Cryosections were cut (10 μm thick) and were stained with hematoxylin and eosin (H&E) for general pathology, Sirius Red for detecting fibrosis, and Alizarin Red for detecting large accumulations of calcium/necrotic fibers, using standard procedures.
Muscle regeneration was assessed by counting the number of myofibers with centrally localized nuclei. Two random areas covering a total of 300–500 fibers were counted from 3 to 5 animals at 6 weeks and at 10 weeks of age. Sections were analyzed and photographed using a Zeiss Axioplan microscope.
Immunohistochemistry/Immunofluorescence
The 10-μm cryosections were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 minutes. Sections were washed and endogenous peroxidase blocked in 1.5% H2O2/Tris-buffered saline (TBS) followed by washing and blocking of endogenous biotin using avidin-biotin (DakoCytomation, Glostrup, Denmark). This was followed by three washes in TBS-Tween20 (TBS-T); finally, the slides were blocked with 4% bovine serum albumin (BSA)/TBS. Primary antibodies used were mouse-anti-human desmin 1:25 (DakoCytomation, D33) labeled with biotinylated-F(ab′) fragments using the ARK research kit (DakoCytomation) as previously described,36 rat-anti-CD45 1:50 (MCA1388; AbD Serotec, Kidlington, UK) as previously described,37 and rabbit-anti-TRPC3 (Alomone Labs, Jerusalem, Israel) 1:500. TRPC3 was detected using donkey anti-rabbit Alexa594 1:200 (Invitrogen, Carlsbad, CA) and mounted using Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) for detection of nuclei (Vector Laboratories, Burlingame, CA). CD45 was detected using rabbit anti-rat biotinylated antibody 1:200, followed by streptavidin-horseradish peroxidase and development using the DAB+ kit (Vector Laboratories), nuclei were counterstained in hematoxylin and slides were mounted with Aquatex (Merck Biosciences, Darmstadt, Germany). Desmin was detected similarly to CD45, except for omitting the secondary antibody step.
Desmin- and CD45-positive areas marking regenerating and inflamed areas, respectively, were semiquantified using a scoring system as follows: no staining = 0, weak staining with few cells/structures/areas = 1, moderate expression with small number of areas/structures = 2, strong expression, vast areas, many cells/structures = 3, and massive expression all over the tissue = 4. Sections were analyzed and photographed using a Zeiss Axioplan microscope.
Western Blotting
Two different preparations were used, either crude muscle extracts, used for the 6-week time point (10 mmol/L Tris-HCl, 1% Triton X-100, 0.5 μL/ml phenylmethanesulfonylfluoride [Roche], and protease inhibitor cocktail [Roche]) or the total membrane fraction isolated using a plasma membrane protein extraction kit (BioVision, Mountain View, CA), used for the 10-week and 6-month time points. Only total membrane fractions were extracted and analyzed using the kit.
The preparations (200–500 ng total membrane fraction or 25 μg crude extracts) were mixed in a 30-μL volume containing loading buffer (Invitrogen), reducing agent (Invitrogen) and water. Samples were incubated at 95–100°C for 5 minutes, briefly spun down and loaded onto Bis-Tris 4–12% gradient gels (Invitrogen). Gels were run for 45 minutes at 200V. Proteins were transferred to a PVDF membrane (GE Healthcare, Waukesha, WI) for 1 hour at 350 mA. Loading and transfer were analyzed using Ponceau staining (Sigma-Aldrich). Membranes were washed in TBS-T and blocked for 1 hour at room temperature or overnight at 4°C in TBS-T with 5% nonfat milk. Primary antibodies were incubated overnight at 4°C except for neural cell adhesion molecule (NCAM), which was incubated for 2 hours at room temperature, in the following concentrations: rabbit-anti-TRPC3 1:4000 (Sigma-Aldrich), rabbit-anti-TRPC3 1:5000 (Alomone Labs), mouse-anti-β-dystroglycan 1:50 (43DAG; Novocastra, Newcastle upon Tyne), mouse anti-dystrophin 1:100 (DY8; Novocastra), and rabbit anti-NCAM 1:8000 (Millipore Bioscience Research Reagents, Temecula, CA). Membranes were washed three times for 10 minutes with TBS-T and incubated with secondary goat anti-mouse-horseradish peroxidase (HRP) 1:5000 or goat anti-rabbit-HRP 1:5000 for 1 hour at room temperature. Immunoreactive bands were detected using an enhanced chemiluminescence kit (GE Healthcare).
Semiquantification of Western blots (TRPC3 and β-dystroglycan) was performed using Adobe Photoshop, CS5, and standardized to the ponceau stain band of approximately 80–90 kDa.
Evans Blue Dye Uptake
Evans Blue dye (0.25 mg per 10 g body weight in PBS) was injected intraperitoneally 16–24 hours before MRI. After in vivo cardiac measurements by MRI, muscles were dissected out for histological analysis. Atrial tissue was removed from the hearts, which were then washed in PBS and snap-frozen in isopentane cooled in liquid nitrogen along with TA and diaphragm muscle. Transverse sections (8–10 μm thick) were cut and incubated in ice-cold acetone at −20°C for 10 minutes, washed three times for 10 minutes with PBS, and mounted with Vectashield mounting medium (Vector Laboratories).
All sections were examined and photographed using a Zeiss Axioplan fluorescence microscope. Evans Blue dye staining shows a bright red emission. An Evans Blue dye uptake score was obtained as follows, performed blinded by two individual observers who scored all sections in parallel and decided on an agreed scoring: 0, no uptake; 1, fewer than three areas of uptake; 2, greater than three areas of uptake; 3, greater than three areas of uptake and at least one area >100 μm in diameter; 4, greater than three areas of uptake and with all areas showing massive uptake of >100 μm in diameter.
Magnetic Resonance Imaging
Mice were anesthetized using 5% isoflurane, and anesthesia was maintained at 1.5% in oxygen with a flow rate of 0.5 L/min. The tail vein was cannulated and the mouse was placed on a sled with cutaneous ECG electrodes, respiration pillow, and temperature detector (Dazai Research Instruments, Toronto, ON) and connected to SA instrument monitoring equipment (SA Instruments, Stony Brook, NY).
Mice were scanned in a 39-mm-diameter quadrature birdcage volume coil (Rapid Biomedical, Rimpar, Germany).
Images were acquired on a horizontal 7-T microimaging system (Varian; Agilent Technologies, Santa Clara, CA) equipped with a 12-cm microimaging gradient insert (maximum gradient, 40 mT/m).
After power calibration and global shimming, a series of four pilot transverse images were acquired over the heart. Single-slice coronal and sagittal images were then obtained to view the apex and mitral valve planes. These images were used to plan for the true short-axis plane.
Up to 10 contiguous short-axis slices were acquired to cover the entire left ventricle using a spoiled gradient-echo cine sequence: TR = 5 ms, TE = 1.42 ms, flip angle = 15 degrees, field of view = 30 × 30 mm, data matrix = 128 × 128, slice thickness = 1 mm. Images were ECG-triggered to the R-wave with a cine delay of 15 ms, and typically 30 phases were acquired distributed through the cardiac cycle. Images were zero-filled to a matrix size of 256 × 256.
Scans were analyzed using the freely available analysis software program Segment v1.8 (http://segment.heiberg.se)38 to give left ventricular functional parameters.
Statistics
Student's unpaired two-tailed t-test with an α-value of 0.05 was performed using GraphPad Prism 5 (GraphPad, La Jolla, CA). Cardiac MRI data were compared using one-way analysis of variance.
Results
Prepathological Calcium Accumulation Indicates Involvement of Calcium Dysregulation in DMD/mdx Pathogenesis
Skeletal muscle from DMD fetuses displays ultrastructural changes including degenerative fibers,30 suggesting that onset of DMD occurs already in utero. It is unclear what comparison can be made between late gestation in humans and the early days after birth in the mouse; however, the onset of pathology is later in the mdx mouse, beginning about 3 weeks postpartum. We therefore wanted to analyze if we could detect calcium disturbances in relation to this early onset of DMD pathology and also extend the analysis to include detection of the earliest time point for accumulation of calcium in the mdx mouse model of DMD. This would prove our hypothesis that calcium accumulation immediately precedes the onset of histologically defined pathology.
We analyzed human DMD (13 weeks) and control (15 weeks) fetal skeletal muscle biopsies in comparison with mdx and C57/BL10 control mouse (BL/10) embryos at embryonic age 9.5, 11.5, and 18.5 days (data not shown) and mouse postnatal muscle at 12–13 days (data not shown) and 3 weeks old (Figure 1, A and B). We detected calcium accumulation in the human DMD fetal biopsy (Figure 1A), compared with the control fetal muscle. We did not detect any calcium accumulation in mdx muscles until age of known onset, which is around 3 weeks postnatally (Figure 1B).39 Centrally located nuclei indicative of ongoing muscle development were detected in both dystrophic and control mouse muscle, whereas in the dystrophic muscle an inflammatory infiltrate in the area positive for calcium uptake was observed as well (Figure 1B, H&E stain).
Figure 1.
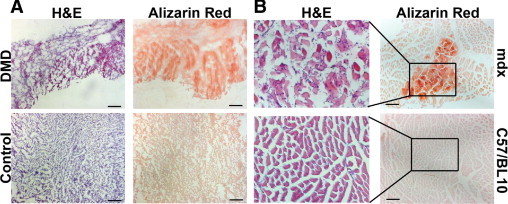
Pre-onset accumulation of calcium in dystrophin-deficient skeletal muscle suggests early implication of calcium dysregulation in DMD pathology. A: Calcium uptake (Alizarin Red positive fibers, red) in skeletal muscle from a fetus (13 weeks old) with DMD, compared with skeletal muscle from a control fetus (15 weeks old), which shows no uptake. B: Calcium uptake in tibialis anterior (TA) of C57/BL10-mdx (mdx) mice was not detectable until postnatal week 3. Central nuclei were detectable in both C57/BL10 control (BL/10) and mdx skeletal muscle (TA), confirming ongoing muscle development at this stage. Insets correspond to the higher-magnification image to the left. Scale bars = 100 μm.
Short-Term Treatment with Streptomycin Improves mdx Limb Skeletal Muscle Pathology
Streptomycin treatment was started as soon as female mice were plugged, to expose the offspring in utero. At 6 weeks of age, an improvement in pathology in the TA muscle was detectable (Figure 2), consistent with previous short-term streptomycin treatments of mdx mice.6, 8 A significant drop in the percentage of centrally localized nuclei (Figure 2A) (P < 0.05) and less inflammation (CD45 semiquantification 1.6 ± 0.55 in mdx S vs. 2.7 ± 0.57 in mdx C, P < 0.05) was observed in the treated animals (Figure 2D).
Figure 2.
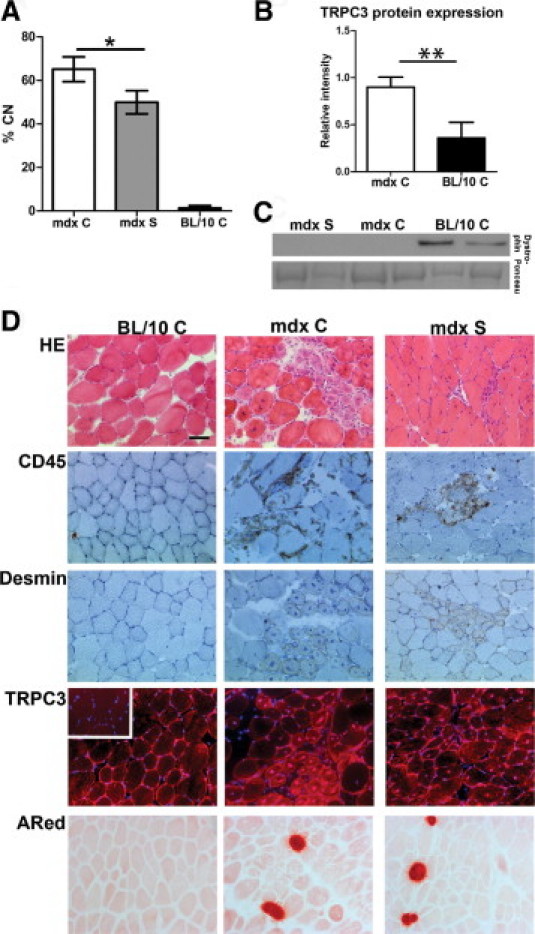
Treatment with streptomycin improves central nuclei counts and pathology in TA of mdx mice in utero and 6 weeks postnatal. A: Central nuclei counts (% CN) in C57/BL10 (BL/10 C), C57/BL10-mdx control (mdx C), and streptomycin-treated (mdx S) mice. Data are expressed as means ± SD of 3–5 mice. There is a statistically significant reduction in %CN in mdx S, compared with mdx C (*P < 0.05). B: Western blot detection of TRPC3 (mol. wt. ∼97 kDa) in crude muscle extracts. Ponceau stain band mol. wt. 80–90 kDa was used as standard and loading control. We detected significantly increased TRPC3 protein in mdx skeletal muscle, compared with BL/10 control (**P < 0.01). C: Western blot results of dystrophin (mol. wt. ∼427 kDa) expression in TA from 6-week-old mdx S, mdx C, and BL/10 C. There is no dystrophin protein expressed in mdx mice, regardless of treatment, indicating that read-through of the dystrophin stop codon mutation has not occurred. Representative results from two mice per group are shown. D: Representative images of histological examination of BL/10 C, mdx C, and mdx S mice. The H&E staining shows general histology; CD45 detects all lymphocytes and is a marker of inflammation; and desmin is expressed by regenerating and newly forming fibers and is a marker for immature fibers and ongoing regeneration. Overall histology points toward less necrosis and inflammation in mdx S, compared with mdx C. TRPC3 protein expression is detectable in membrane, cytoplasm, and nuclei of both mdx C and mdx S mice and the expression appears to be increased in newly formed myotubes. This TRPC3 expression pattern in mdx control and treated mice, compared with BL/10 C, correlates with the increased expression seen on Western blots (B). Inset: Negative control image for TRPC3 immunohistochemical staining for BL/10 C. Nuclei are counterstained with DAPI in blue. Alizarin Red (ARed) staining shows necrotic foci with calcium accumulations and there is no detectable difference between mdx C and mdx S. Scale bar = 50 μm.
Active regeneration was observed as desmin-positive myotubes in dystrophic mice, regardless of treatment, suggesting that streptomycin could not completely prevent onset and progression of pathology. This was supported by the presence of necrotic foci regardless of treatment as judged by Alizarin Red staining (Figure 2D). TRPC3 expression overall was very patchy, even in C57BL/10 control muscle (BL/10 C), with localization to the membrane and cytosol without any obvious difference in expression level. However, Western blot confirmed that 6-week-old mdx C mice showed increased expression of TRPC3, compared with BL/10 C (Figure 2B).
Intriguingly, TRPC3 was detected in the nuclei of newly regenerating fibers (Figure 2D). This staining pattern was generally not observed in nuclei of BL/10 C muscle, uninjured mdx muscle, or nuclei in heart transverse sections from both mdx and BL/10 mice. A negative control staining, omitting primary antibody, showed no nuclear staining (Figure 2D, inset); staining was abolished by competition with the appropriate peptide (data not shown), and the antibody specificity has been verified previously.40 An identical staining pattern was observed with an alternative antibody raised against TRPC3 (data not shown). The increased expression of TRPC3 observed in mdx mouse muscle (Figure 2B) could therefore be a consequence of ongoing regeneration, rather than a primary pathological feature.
Streptomycin is an aminoglycoside, and some aminoglycosides read through premature stop codons.41 Because the mdx mutation is a stop codon in exon 23,42 it appeared possible that read-through had occurred and caused any improvement seen in our study. However, the improvement observed at 6 weeks of age was not caused by read-through of the mdx stop-codon mutation, because dystrophin protein was not detected (Figure 2C).
Continued Treatment up to 10 Weeks of Age Failed to Prevent Progressive Pathology in Limb, Heart, and Diaphragm Muscle
Although the 6-week time-point data were supportive for unspecific calcium channel blockade as a potential treatment for Duchenne muscular dystrophy, continuation of the treatment up to the age of 10 weeks did not maintain the improvement. We no longer observed a significant difference in the number of central nuclei (Figure 3A) between treated and untreated dystrophic mice. On histological examination of limb and diaphragm muscles, there were no observable differences in overall histology (Figure 3, C and G). Semiquantitative analysis of the immunohistochemistry could not detect any significant difference in inflammation (CD45 in TA: mdx C 2 ± 0.04 vs. mdx S 2.2 ± 0.4, P > 0.05; CD45 in diaphragm: mdx C 2.25 ± 0.5 vs. mdx S 3.2 ± 0.2, P > 0.05) or in ongoing regeneration (desmin in TA: mdx C 1 ± 0.4 vs. mdx S 1.2 ± 0.2, P > 0.05; desmin in diaphragm: mdx C 3.25 ± 0.2 vs. mdx S 2.8 ± 0.2, P > 0.05). The ongoing regeneration in TA was confirmed by high expression levels of NCAM43 (Figure 3B). Necrotic foci detected by Alizarin Red staining were equally present in both control and treated mdx mice.
Figure 3.
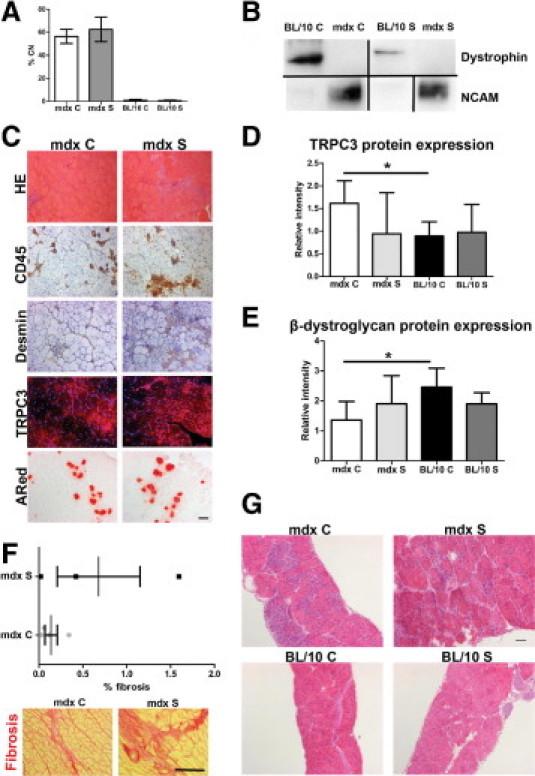
Continued treatment with streptomycin up to 10 weeks of age does not prevent pathological progression in TA (A–E), heart (F), and diaphragm (G). A: Central nuclei counts (% CN) in BL/10 C, mdx C, BL/10 S (C57BL/10 treated), and mdx S mice show no difference between mdx C and mdx S (P > 0.05) at 10 weeks of age. Data are expressed as means ± SD of 3–6 mice. B: Western blot detection of dystrophin (∼427 kDa) and neural cell adhesion molecule (NCAM; ∼160 kDa breakdown product) in total membrane fractions from TA muscles. Blots not run on same gels or reorganized from different parts of the same gels are all separated by a black line. C: Representative images of histological examination of TA muscles from mdx C and mdx S. Top to bottom: H&E, CD45, desmin, TRPC3, and Alizarin Red staining. Overall there appears to be no difference in pathology between mdx C and mdx S. Scale bar = 100 μm. D: TRPC3 protein expression is significantly increased in mdx C, compared with BL/10 C (*P < 0.05). Based on Western blot detection of TRPC3 (∼97 kDa). Ponceau stain band mol. wt. 80–90 kDa was used as standard and loading control. E: β-dystroglycan protein expression is significantly decreased in mdx C, compared with BL/10 C (*P < 0.05). Based on Western blot detection of β-dystroglycan (∼43 kDa). Ponceau stain band Mw 80–90 kDa was used as standard and loading control. F: Top panel: Quantification of fibrosis in 10-week-old mdx C and mdx S hearts. Data are actual measurements, expressed as mean ± SD of 3–4 mice. Bottom panel: Increased fibrosis (red) in heart muscle from mdx S, compared with mdx C, detected using Sirius Red. Scale bar = 100 μm. G: Histological analysis of diaphragm muscle from 10-week-old mice of each group using H&E shows no obvious difference in pathology between mdx C and mdx S. Scale bar = 100 μm.
Absence of dystrophin in both control and treated mdx mice confirmed that there was no read-through activity at 10 weeks of age (Figure 3B).
TRPC3 expression showed the same overall features as the 6-week time point, with patchy expression and presence of TRPC3 protein in the nuclei of regenerating fibers as well (Figure 3C). Semiquantification of TRPC3 Western blot (Figure 3D) showed that TRPC3 protein was increased in mdx C (1.62 ± 0.24) vs. BL/10 C (0.89 ± 0.59) (P < 0.05). After treatment with streptomycin, TRPC3 protein expression level did not change in BL/10 mice (0.97 ± 0.31, P > 0.05 BL/10 C vs. BL/10 S), but appeared to decrease in treated mdx mice (0.95 ± 0.45); however, this could not be confirmed by semiquantification (P > 0.05, mdx C vs. mdx S).
Semiquantification of β-dystroglycan protein expression in mdx C (1.36 ± 0.31) vs. BL/10 C (2.46 ± 0.31) (P < 0.05) showed down-regulation in dystrophic mice, as expected (Figure 3E).44 Expression of β-dystroglycan did not appear to change in BL/10 mice after streptomycin treatment (Bl/10 S 1.90 ± 0.19, P > 0.05 vs. BL/10 C). In dystrophic mice, treatment appeared to increase expression level of β-dystroglycan, but this could not be confirmed statistically (mdx S 1.90 ± 0.47, P > 0.05 vs. mdx C).
We also examined heart muscle pathology at this time point, using Sirius Red staining for fibrosis followed by semiquantitative analysis of the histological images (Figure 3F). There was a trend toward increased fibrosis in the hearts of treated mdx mice (mdx S % fibrosis 0.68 ± 0.47), compared with controls (mdx C % fibrosis 0.14 ± 0.071), but this did not reach statistical significance (P > 0.05). No fibrosis was detected in hearts from BL/10 C or BL/10 S mice (data not shown).
Long-Term Treatment Alleviates Limb Muscle Pathology but Cardiac and Diaphragm Pathology Is Worsened
The final time point to be analyzed was after continued treatment until the age of 6 months. To examine whether long-term treatment with streptomycin induced kidney damage, total protein urine content was analyzed. All mice had similar levels (0.3 g/L). In addition, we analyzed the urine samples for ketones, and all were negative. Histological analysis of kidneys and livers in H&E-stained sections from paraffin-embedded tissue did not demonstrate histological changes in any of the groups (data not shown), suggesting that there were no adverse side effects from the treatment. Ototoxicity was tested by performing a simple hearing test in which fingers were snapped behind the mice and the Preyer reflex observed.45 All groups behaved similarly.
On routine histological examination of H&E stainings, an improvement in limb muscle pathology was observable in treated mdx mice. Specifically, large areas with ongoing necrosis were still detectable in mdx C (Figure 4A), whereas in mdx S mice (Figure 4A) the only sign of pathology was occasional small repaired areas with myotubes and centrally located nuclei without any necrosis. However, in both treated and control mdx mice >95% of the limb muscle fibers had centrally located nuclei (data not shown), suggesting that most muscle fibers had gone through at least one round of degeneration and regeneration. Diaphragm muscles presented with severe necrosis combined with large areas of fibrosis and regenerative fibers, suggesting that streptomycin treatment had no effect in this more severely affected tissue46 (Figure 4A). These histological observations were confirmed by Sirius Red staining for fibrosis (Figure 4B) showing that TA muscles from treated mdx mice (Figure 4B) appeared to have reduced thickening of the endo- and perimysium, compared with control mice (Figure 4B), whereas there was no obvious difference in the diaphragm muscles (Figure 4B).
Figure 4.
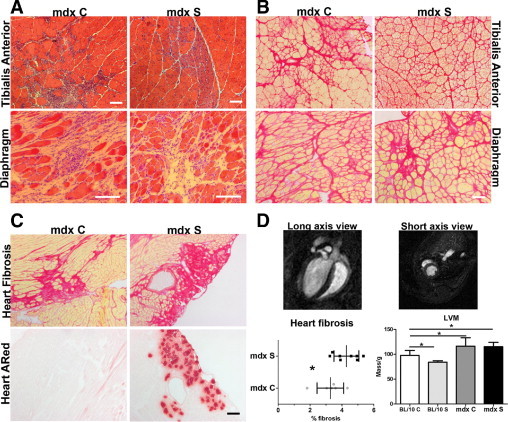
Improvement of limb muscle pathology combined with aggravation of cardiac pathology after continued treatment for 6 months. A: Representative images for H&E histological examination of TA and diaphragm from mdx C and mdx S. There is an overall improvement in limb muscle pathology in 6-month-old mdx S mice, but no obvious difference in diaphragm pathology, compared with mdx C. B: Sirius Red staining detecting fibrosis (red) in TA and diaphragm from 6-month-old mdx C and mdx S confirms the H&E histological findings of improvement in limb muscle pathology observed as reduced fibrosis. No difference in diaphragm fibrosis between mdx C and mdx S. C: Examination of fibrosis in 6-month-old heart muscle of mdx C and mdx S and presence of necrotic foci (Alizarin Red) in 6-month-old heart muscle of mdx C and mdx S mice. There is increased fibrosis combined with necrosis in mdx S hearts, compared with controls, showing a worsening of the pathology. D: Top panels: Representative images of a heart MRI scan from a 6-month-old C57BL/10 control mouse showing both short-axis and long-axis views. Bottom panels: Quantification of fibrosis in 6-month-old hearts shows a statistically significant increase in fibrosis in mdx S hearts, compared with mdx C. Data are actual measurements from 6–8 mice, expressed as means ± SD. *P < 0.05. Left ventricular mass (LVM) measurements normalized to body weight performed using MRI scanning and analysis using the software program Segment (as described under Materials and Methods). There were significant differences in LVM when comparing BL/10 to all other groups using one-way analysis of variance analysis (P < 0.0001), although streptomycin had no significant effects on LVM. Data are expressed as means of 5–11 mice ± SD.
We also examined the effect on cardiac pathology. Fibrosis was observed (Figure 4C) in hearts from both treated and control mdx mice, and on quantification the percentage of fibrosis was significantly increased in treated hearts (mdx C 3.27 ± 0.34 vs. mdx S 4.28 ± 0.28, P < 0.05) (Figure 4D). In addition, there was severe necrosis in the treated hearts, which was not observed in control dystrophic hearts (Figure 4C).
Magnetic resonance imaging scanning of treated and control BL/10 and mdx mice did not reveal any difference in left ventricular function between dystrophic mice, regardless of treatment, and both groups demonstrated well-compensated function, as previously reported using the conductance catheter technique.47 Cardiac pathology in dystrophic mice has previously been characterized by increased left ventricular mass.48 Magnetic resonance imaging confirmed that there was a significant increase in left ventricular mass in mdx C, compared with BL/10 C and that this was not improved in mdx S mice (Figure 4D). Overviews from the heart scans are presented in Figure 4D (long-axis and short-axis images taken from a healthy BL/10 C mouse).
No damage was observed in TA, diaphragm, or heart from BL/10 S, compared with controls, as judged from histological analysis (data not shown).
Membrane Damage Is Reduced in Limb Muscle after Long-Term Treatment with No Effect on Diaphragm and Cardiac Muscle
Membrane damage, as detected by Evans Blue dye uptake, was significantly reduced in TA muscle after 6 months treatment in dystrophic mice (Figure 5, A and B) (P < 0.01), compared with controls; however, there was no detectable difference in membrane damage in diaphragm and cardiac muscle (Figure 5, A and B).
Figure 5.
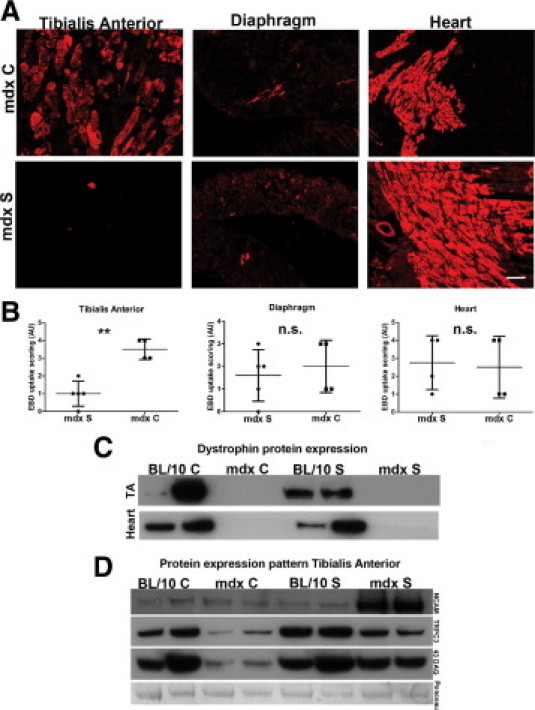
Membrane fragility is improved in limb muscle of mdx mice after 6 months treatment with streptomycin, but heart and diaphragm pathology is not improved. A: Representative images of Evans Blue Dye (EBD) uptake in TA, diaphragm, and heart tissues harvested from the same mouse for mdx C and mdx S, respectively. There is great variability in uptake both in different tissues examined from the same mouse and in the same tissue types examined from different mice. B: Quantification of EBD uptake in TA, diaphragm, and heart tissue from mdx C and mdx S. Data are from 4–5 mice, expressed as means ± SD. **P < 0.01 for EBD uptake in TA in mdx C vs. mdx S. There were no significant differences in diaphragm and heart tissue, which both show a highly variable uptake pattern. C: Western blot detection of dystrophin (∼427 kDa) expression in TA and heart muscle from 6-month-old BL/10 C, mdx C, BL/10 S, and mdx S mice. There is no dystrophin protein expressed in TA or heart muscle from mdx mice regardless of treatment, indicating that read-through of the dystrophin stop codon mutation has not occurred even after 6 months of treatment. Representative results from two mice per group are shown. D: Western blot detection of NCAM (∼160 kDa), TRPC3 (∼97 kDa), and β-dystroglycan (∼43 kDa) protein expression in total membrane fractions from 6-month-old TA muscle from BL/10 C, mdx C, BL/10 S, and mdx S. Representative results from two mice per group are shown. Ponceau staining was used as loading control (protein band of ∼80–90 kDa is shown).
Using Western blotting analysis of both heart tissue and TA, we confirmed that there was still no detectable read-through activity in the dystrophin gene by the aminoglycoside that could account for the positive effect streptomycin treatment (Figure 5C). There was increased protein expression of NCAM in TA of treated mdx mice, compared with all other groups, suggesting that there was increased muscle regeneration or stalled myofiber maturation in these mice at this time point (Figure 5D). Both treated and control mdx mice appeared to have a reduced expression of TRPC3 compared with the BL/10 groups. However, TRPC3 protein expression level appeared to be higher in treated mdx mice, compared with controls, again indicative of TRPC3 protein being actively transcribed and expressed by regenerating fibers, because increased NCAM was detected in the treated mdx mice as well (Figure 5D). Finally, there appeared to be increased expression of β-dystroglycan in the treated dystrophic mice, which appeared to have normalized toward wild-type levels (Figure 5D).
Discussion
The understanding of the pathophysiological mechanisms of DMD has become increasingly complex since the discovery of the underlying genetic defect,49 and still no cure exists, more than 20 years later. The only available treatment to ameliorate skeletal muscle weakness and wasting is glucocorticoids, but this treatment is associated with potentially severe side-effects, such as weight gain and osteoporosis.50 It is therefore essential to continue testing new treatment strategies.
In the present study, we focused our attention on the hypothesis that calcium dysregulation could be a primary event in developmental onset of pathology, based on several recent studies suggesting that calcium channel proteins might be involved in disease initiation.13, 16, 19 Our confirmation of early calcium accumulation in DMD fetal muscle is important for determination of the pathophysiological mechanisms, and also for defining the optimal time-point for initiation of treatment. This is supported by a previous study in which transgenic expression of utrophin in mdx mice from developmental onset reduced dystrophic pathology to a much greater extent than expression initiated at birth or postnatally,23 indicating that it is important when a therapeutic intervention is started. Based on the defects observed before histopathological onset in DMD/mdx, we wanted to analyze whether blocking calcium channels in the dystrophic mice from embryonic development and throughout adulthood could prevent pathological onset or progression. We observed a complex pathological pattern in which treatment outcome varied with age, ongoing disease progression, and tissue. Long-term treatment for 6 months, which may be the most relevant time point for comparison with the human condition, resulted in improved limb muscle pathology, but worsened cardiac pathology.
Previous studies have suggested a direct role for the TRP channel proteins in DMD pathology, and recent data show that transgenic expression of TRPC3 could induce a dystrophic pathology without concomitant membrane fragility.19 During this study we therefore recorded expression and localization pattern of TRPC3 to see if calcium channel blockade induced changes in expression pattern and whether this correlated with pathology.
TRPC3 expression was increased in mdx mouse muscle of young mice, but appeared to be reduced in older mice. Expression appeared stronger and localized both to the membrane and to cytoplasm and nuclei in regenerating fibers, compared with uninjured fibers in younger mice. The intracellular localization is in agreement with a study showing colocalization of TRPC3 with GLUT4 in the transverse tubular system.51 The nuclear localization of TRPC3 is unexplained, but has also been observed in a recent study.14 and regulated expression of TRPC3 during myotube development in vitro17 supports a correlation between TRPC3 expression and regeneration.
In the present study, localization of TRPC3 to the nuclei was observed only in regenerating myofibers of skeletal muscle; we did not detect this nuclear pattern in uninjured skeletal muscle or ventricular myocytes (data not shown). Our observations are in accordance with a recent study in which localization of TRPC3 to the intercalated disks and intracellular membranes of ventricular myocytes was shown.52 Because the function of TRPC3 appears to be different in adult muscle versus developing muscle,14 our observations lead us to propose that the up-regulation of TRPC3 protein in young dystrophic mouse muscle could relate to ongoing regeneration. Nonetheless, dysregulated TRPC3 in early development caused by lack of dystrophin could also be a specific pathological feature. This needs to be investigated further in specific models of muscle regeneration.36, 37
TRPC1 may form part of the putative calcium channels implicated in DMD pathology, because inactivating TRPC1 expression in mdx muscle fibers abrogated a calcium leak channel13 and because TRPC1 is directly associated with the dystrophin-glycoprotein complex.16 Specifically it has been proposed that the TRPC1 protein might constitute a stretch-activated calcium channel.53 However, recent observations in TRPC1 knockout mice suggest that this channel is not mechanosensitive, nor is it involved in store-operated calcium entry54; thus, the actual gating mechanism remains unknown. Nonetheless, TRPC1 appears to be indispensible for muscle activity, and TRPC1 knockout mice undergo muscle fatigue much faster than control mice.54 The role of TRPC3 and TRPC1 in relation to DMD remains elusive.
In the present study, reducing calcium influx in limb muscle via blockade of the calcium channels resulted in reduced signs of fiber damage in 6-week-old mice, in accordance with a previous study.8 The number of central nuclei then increased in both treated and untreated mdx mice with continued treatment and was no longer significantly different at 10 weeks of age. This suggests that there is an early delay in pathological progression after treatment.
Streptomycin has also been shown to have a directly harmful effect on mitochondrial ribosomes55; thus, cumulative damage on mitochondria may present later as accelerated degeneration (observed here as increased numbers of centrally localized nuclei). Another explanation for the transitory effect might be that the beneficial treatment effect diminishes over time.56
Our study on limb muscle pathology, however, actually indicated that blocking calcium influx results in ongoing regeneration and less necrosis in the muscles of older mdx mice, thus supporting the suggested mechanism of more efficient repair. In this context it is important to notice that the presence of centrally located nuclei indicates only that regeneration has occurred, not how many times each individual myofiber has undergone a round of necrosis followed by regeneration. It can therefore be speculated that, if the muscles have undergone fewer cycles of degeneration and regeneration, the satellite cell pool would benefit, because it will not become exhausted. The satellite cells will then still be available for repair of muscle fibers damaged by membrane fragility-induced events. This would result in the observed pattern of regeneration (increased NCAM expression) with reduced necrosis.
We observed a positive effect on membrane stability in the limb muscles of older, treated mdx mice, as ascertained by reduced Evans Blue dye uptake. The improvement, however, was not caused by read-through of the dystrophin stop codon mutation.5 Increased membrane stability in response to calcium blockade was also observed after blocking the L-type voltage-dependent calcium channels,6 indicating that the down-regulation of DGC proteins normally observed in DMD occurs via calcium-dependent proteolytic activity. This is consistent with the decrease in DGC proteins found in other types of muscle wasting not related to DGC protein expression, such as cancer.57 Our results, together with previous data,6 therefore demonstrate that drugs capable of blocking Ca2+ uptake are directly associated with stabilization of β-dystroglycan. We noted that increased β-dystroglycan did not protect TA muscles after treatment with verapamil and diltiazem,6 suggesting either that the mechanism of action for streptomycin is different from the L-type calcium channels blockers or that restoration of β-dystroglycan expression alone is not sufficient to stabilize the membrane. Aminoglycoside antibiotics can, however, displace Ca2+ from biomembranes and have a high affinity for phospholipids,58 suggesting that they may have an ability to intercalate into the fragile membranes of mdx muscle and stabilize them in this manner.
Most DMD patients develop cardiomyopathy characterized by left ventricular involvement, which is believed to be directly responsible for 20% of deaths.59 Older mdx mice present with left ventricular dilated cardiomyopathy and fibrosis, thus mimicking DMD-associated cardiomyopathy.48 In the present study, we detected a trend toward increased heart fibrosis in short-term treated dystrophic mice, which became significant with long-term treatment. The increased fibrosis was accompanied by substantial necrosis, suggesting a worsening of heart pathology in the treated mdx mice. Functionally, dystrophic control mice presented with a compensated cardiomyopathy, similar to previous studies,47, 48, 60 and this was not changed with treatment. We observed an increased left ventricular mass in both treated and untreated mdx mice, in agreement with previous studies.47, 47, 60
Calcium influx in dystrophic cardiomyocytes occurs via several pathways, including the stretch-activated channels, short-lived microruptures, and the Na+-Ca2+ exchanger.61 Calcium influx after stretch appears to depend on Na+ permeability and thus seems to be a secondary event.62 It has been suggested that, in old mdx hearts, increased activity and/or expression of TRPC1 might account for the increased calcium influx through stretch-activated channels.63 However, considering the report showing that TRPC1 does not constitute a mechanosensitive channel in skeletal muscle,54 lack of improvement in cardiac pathology after streptomycin treatment could be caused by other adverse effects of streptomycin on the heart. Specifically, it has been shown that streptomycin affects heart cell viability in vitro.64; thus, streptomycin could induce cardiotoxicity and this, in combination with mitochondrial dysfunction, could explain the lack of improvement observed in the hearts.
This study demonstrates opposite effects of streptomycin treatment on different tissues. Blocking calcium uptake might delay onset, but without concomitant correction of the membrane fragility defect, microtears, and nonspecific calcium uptake; eventually, necrosis, degeneration, and end-stage fibrosis will occur. This protection may be more pronounced in limb muscles, compared with diaphragm and heart, simply due to differences in physical activity. There are two possible scenarios to consider. First, the drug might simply reduce daily activity of the mice. This could relate to their behavior or to the mice feeling sick.65 This is important, because reduced or altered movements would directly reduce activity-induced necrosis of mdx mice. Therefore, introducing mechanical stress (eg, using forced tread-mill running) and hence inducing necrosis of the muscle might counter the early improvement observed. We did not notice any obvious behavioral changes, and the mice appeared to be active and well-groomed, but this possibility should be tested using specific behavior studies and exercise regimens. Second, the relative activity level in limb muscle versus diaphragm and heart may in part explain differential effects of the drug. Physical activity level of the mice is more relevant for limb muscle than for heart and diaphragm, as the latter are in constant motion throughout life and undergo the same level of stress in both control and treated mice. Based on the present findings, systemic calcium channel blockade may not be a useful therapeutic strategy for DMD, as it may result in an aggravation of heart pathology. Calcium accumulation and structural disturbances before clinical onset in DMD/mdx needs careful consideration in designing future preclinical drug trials in the mdx mouse, because the type of muscle changes present at the start of intervention might influence the outcome considerably.
Acknowledgments
We thank Dr. Johannes Riegler (University College London) for kindly providing Matlab scripts, and also thank all staff at the Functional Genomics Units, Newcastle University, for help and assistance with animal care.
Footnotes
Supported by the Lundbeck Foundation (R19-A2177) to L.H.J., Muscular Dystrophy Campaign UK (RA3/691) sponsored by the Big Lottery Fund (RG/1/010136302) to E.G., and the Medical Research Council UK through the Centre for Neuromuscular Diseases (G0601943) to K.B., H.L., and V.S.
References
- 1.Pilgram G.S., Potikanond S., Baines R.A., Fradkin L.G., Noordermeer J.N. The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol Neurobiol. 2010;41:1–21. doi: 10.1007/s12035-009-8089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deconinck N., Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 3.Bodensteiner J.B., Engel A.G. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology. 1978;28:439–446. doi: 10.1212/wnl.28.5.439. [DOI] [PubMed] [Google Scholar]
- 4.Bertorini T.E., Palmieri G.M., Griffin J.W., Igarashi M., McGee J., Brown R., Nutting D.F., Hinton A.B., Karas J.G. Effect of chronic treatment with the calcium antagonist diltiazem in Duchenne muscular dystrophy. Neurology. 1988;38:609–613. doi: 10.1212/wnl.38.4.609. [DOI] [PubMed] [Google Scholar]
- 5.De Luca A., Nico B., Rolland J.F., Cozzoli A., Burdi R., Mangieri D., Giannuzzi V., Liantonio A., Cippone V., De Bellis M., Nicchia G.P., Camerino G.M., Frigeri A., Svelto M., Camerino D.C. Gentamicin treatment in exercised mdx mice: identification of dystrophin-sensitive pathways and evaluation of efficacy in work-loaded dystrophic muscle. Neurobiol Dis. 2008;32:243–253. doi: 10.1016/j.nbd.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura C.Y., Pertille A., Albuquerque T.C., Santo Neto H., Marques M.J. Diltiazem and verapamil protect dystrophin-deficient muscle fibers of MDX mice from degeneration: a potential role in calcium buffering and sarcolemmal stability. Muscle Nerve. 2009;39:167–176. doi: 10.1002/mus.21188. [DOI] [PubMed] [Google Scholar]
- 7.Pernice W., Beckmann R., Ketelsen U.P., Frey M., Schmidt-Redemann B., Haap K.P., Roehren R., Sauer M. A double-blind placebo controlled trial of diltiazem in Duchenne dystrophy. Klin Wochenschr. 1988;66:565–570. doi: 10.1007/BF01720830. [DOI] [PubMed] [Google Scholar]
- 8.Yeung E.W., Whitehead N.P., Suchyna T.M., Gottlieb P.A., Sachs F., Allen D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J Physiol. 2005;562:367–380. doi: 10.1113/jphysiol.2004.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mongini T., Ghigo D., Doriguzzi C., Bussolino F., Pescarmona G., Pollo B., Schiffer D., Bosia A. Free cytoplasmic Ca++ at rest and after cholinergic stimulus is increased in cultured muscle cells from Duchenne muscular dystrophy patients. Neurology. 1988;38:476–480. doi: 10.1212/wnl.38.3.476. [DOI] [PubMed] [Google Scholar]
- 10.Bakker A.J., Head S.I., Williams D.A., Stephenson D.G. Ca2+ levels in myotubes grown from the skeletal muscle of dystrophic (mdx) and normal mice. J Physiol. 1993;460:1–13. doi: 10.1113/jphysiol.1993.sp019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner P.R., Fong P.Y., Denetclaw W.F., Steinhardt R.A. Increased calcium influx in dystrophic muscle. J Cell Biol. 1991;115:1701–1712. doi: 10.1083/jcb.115.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarter G.C., Steinhardt R.A. Increased activity of calcium leak channels caused by proteolysis near sarcolemmal ruptures. J Membr Biol. 2000;176:169–174. doi: 10.1007/s00232001086. [DOI] [PubMed] [Google Scholar]
- 13.Vandebrouck C., Martin D., Colson-Van Schoor M., Debaix H., Gailly P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol. 2002;158:1089–1096. doi: 10.1083/jcb.200203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krüger J., Kunert-Keil C., Bisping F., Brinkmeier H. Transient receptor potential cation channels in normal and dystrophic mdx muscle. Neuromuscul Disord. 2008;18:501–513. doi: 10.1016/j.nmd.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Whitehead N.P., Yeung E.W., Allen D.G. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol. 2006;33:657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- 16.Vandebrouck A., Sabourin J., Rivet J., Balghi H., Sebille S., Kitzis A., Raymond G., Cognard C., Bourmeyster N., Constantin B. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007;21:608–617. doi: 10.1096/fj.06-6683com. [DOI] [PubMed] [Google Scholar]
- 17.Lee E.H., Cherednichenko G., Pessah I.N., Allen P.D. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J Biol Chem. 2006;281:10042–10048. doi: 10.1074/jbc.M600981200. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg P., Hawkins A., Stiber J., Shelton J.M., Hutcheson K., Bassel-Duby R., Shin D.M., Yan Z., Williams R.S. TRPC3 channels confer cellular memory of recent neuromuscular activity. Proc Natl Acad Sci USA. 2004;101:9387–9392. doi: 10.1073/pnas.0308179101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millay D.P., Goonasekera S.A., Sargent M.A., Maillet M., Aronow B.J., Molkentin J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci USA. 2009;106:19023–19028. doi: 10.1073/pnas.0906591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwata Y., Katanosaka Y., Arai Y., Shigekawa M., Wakabayashi S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum Mol Genet. 2009;18:824–834. doi: 10.1093/hmg/ddn408. [DOI] [PubMed] [Google Scholar]
- 21.Teichmann M.D., Wegner F.V., Fink R.H., Chamberlain J.S., Launikonis B.S., Martinac B., Friedrich O. Inhibitory control over Ca(2+) sparks via mechanosensitive channels is disrupted in dystrophin deficient muscle but restored by mini-dystrophin expression. PLoS One. 2008;3:e3644. doi: 10.1371/journal.pone.0003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bunnell T.M., Jaeger M.A., Fitzsimons D.P., Prins K.W., Ervasti J.M. Destabilization of the dystrophin-glycoprotein complex without functional deficits in alpha-dystrobrevin null muscle. PLoS ONE. 2008;3:e2604. doi: 10.1371/journal.pone.0002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Squire S., Raymackers J.M., Vandebrouck C., Potter A., Tinsley J., Fisher R., Gillis J.M., Davies K.E. Prevention of pathology in mdx mice by expression of utrophin: analysis using an inducible transgenic expression system. Hum Mol Genet. 2002;11:3333–3344. doi: 10.1093/hmg/11.26.3333. [DOI] [PubMed] [Google Scholar]
- 24.Spencer M.J., Croall D.E., Tidball J.G. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995;270:10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- 25.Alderton J.M., Steinhardt R.A. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275:9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- 26.Courdier-Fruh I., Briguet A. Utrophin is a calpain substrate in muscle cells. Muscle Nerve. 2006;33:753–759. doi: 10.1002/mus.20549. [DOI] [PubMed] [Google Scholar]
- 27.Kawamata H., Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010;131:517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charge S.B., Rudnicki M.A. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 29.Bertorini T.E., Cornelio F., Bhattacharya S.K., Palmieri G.M., Dones I., Dworzak F., Brambati B. Calcium and magnesium content in fetuses at risk and prenecrotic Duchenne muscular dystrophy. Neurology. 1984;34:1436–1440. doi: 10.1212/wnl.34.11.1436. [DOI] [PubMed] [Google Scholar]
- 30.Turkel S.B., Howell R., Iseri A.L., Chui L. Ultrastructure of muscle in fetal Duchenne's dystrophy. Arch Pathol Lab Med. 1981;105:414–418. [PubMed] [Google Scholar]
- 31.Merrick D., Stadler L.K., Larner D., Smith J. Muscular dystrophy begins early in embryonic development deriving from stem cell loss and disrupted skeletal muscle formation. Dis Model Mech. 2009;2:374–388. doi: 10.1242/dmm.001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghahramani Seno M.M., Graham I.R., Athanasopoulos T., Trollet C., Pohlschmidt M., Crompton M.R., Dickson G. RNAi-mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum Mol Genet. 2008;17:2622–2632. doi: 10.1093/hmg/ddn162. [DOI] [PubMed] [Google Scholar]
- 33.Gannier F., White E., Lacampagne A., Garnier D., Le Guennec J.Y. Streptomycin reverses a large stretch induced increases in [Ca2+]i in isolated guinea pig ventricular myocytes. Cardiovasc Res. 1994;28:1193–1198. doi: 10.1093/cvr/28.8.1193. [DOI] [PubMed] [Google Scholar]
- 34.Willems M.E., Stauber W.T. Streptomycin and EDTA decrease the number of desmin-negative fibers following stretch injury. Muscle Nerve. 2005;32:310–315. doi: 10.1002/mus.20370. [DOI] [PubMed] [Google Scholar]
- 35.Rubin A., Winston J., Rutledge M.L. Effects of streptomycin upon the human fetus. AMA Am J Dis Child. 1951;82:14–16. doi: 10.1001/archpedi.1951.02040040021003. [DOI] [PubMed] [Google Scholar]
- 36.Chiu Y.H., Hornsey M.A., Klinge L., Jorgensen L.H., Laval S.H., Charlton R., Barresi R., Straub V., Lochmüller H., Bushby K. Attenuated muscle regeneration is a key factor in dysferlin-deficient muscular dystrophy. Hum Mol Genet. 2009;18:1976–1989. doi: 10.1093/hmg/ddp121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jørgensen L.H., Jensen C.H., Wewer U.M., Schrøder H.D. Transgenic overexpression of ADAM12 suppresses muscle regeneration and aggravates dystrophy in aged mdx mice. Am J Pathol. 2007;171:1599–1607. doi: 10.2353/ajpath.2007.070435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heiberg E., Engblom H., Engvall J., Hedström E., Ugander M., Arheden H. Semi-automatic quantification of myocardial infarction from delayed contrast enhanced magnetic resonance imaging. Scand Cardiovasc J. 2005;39:267–275. doi: 10.1080/14017430500340543. [DOI] [PubMed] [Google Scholar]
- 39.McGeachie J.K., Grounds M.D., Partridge T.A., Morgan J.E. Age-related changes in replication of myogenic cells in mdx mice: quantitative autoradiographic studies. J Neurol Sci. 1993;119:169–179. doi: 10.1016/0022-510x(93)90130-q. [DOI] [PubMed] [Google Scholar]
- 40.Tadros S.F., Kim Y., Phan P.A., Birnbaumer L., Housley G.D. TRPC3 ion channel subunit immunolocalization in the cochlea. Histochem Cell Biol. 2010;133:137–147. doi: 10.1007/s00418-009-0653-6. [DOI] [PubMed] [Google Scholar]
- 41.Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S., Wilde R.G., Karp G., Takasugi J., Chen G., Jones S., Ren H., Moon Y.C., Corson D., Turpoff A.A., Campbell J.A., Conn M.M., Khan A., Almstead N.G., Hedrick J., Mollin A., Risher N., Weetall M., Yeh S., Branstrom A.A., Colacino J.M., Babiak J., Ju W.D., Hirawat S., Northcutt V.J., Miller L.L., Spatrick P., He F., Kawana M., Feng H., Jacobson A., Peltz S.W., Sweeney H.L. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 42.Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 43.Dubois C., Figarella-Branger D., Pastoret C., Rampini C., Karpati G., Rougon G. Expression of NCAM and its polysialylated isoforms during mdx mouse muscle regeneration and in vitro myogenesis. Neuromuscul Disord. 1994;4:171–182. doi: 10.1016/0960-8966(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 44.Turk R., Sterrenburg E., de Meijer E.J., van Ommen G.J., den Dunnen J.T., 't Hoen P.A. Muscle regeneration in dystrophin-deficient mdx mice studied by gene expression profiling. BMC Genomics. 2005;6:98. doi: 10.1186/1471-2164-6-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jero J., Coling D.E., Lalwani A.K. The use of Preyer's reflex in evaluation of hearing in mice. Acta Otolaryngol. 2001;121:585–589. [PubMed] [Google Scholar]
- 46.Grounds M.D., Radley H.G., Lynch G.S., Nagaraju K., De Luca A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol Dis. 2008;31:1–19. doi: 10.1016/j.nbd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bauer R., Straub V., Blain A., Bushby K., MacGowan G.A. Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur J Heart Fail. 2009;11:463–471. doi: 10.1093/eurjhf/hfp028. [DOI] [PubMed] [Google Scholar]
- 48.Quinlan J.G., Hahn H.S., Wong B.L., Lorenz J.N., Wenisch A.S., Levin L.S. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 50.Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve. 2007;36:424–435. doi: 10.1002/mus.20812. [DOI] [PubMed] [Google Scholar]
- 51.Lanner J.T., Bruton J.D., Assefaw-Redda Y., Andronache Z., Zhang S.J., Severa D., Zhang Z.B., Melzer W., Zhang S.L., Katz A., Westerblad H. Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB J. 2009;23:1728–1738. doi: 10.1096/fj.08-116814. [DOI] [PubMed] [Google Scholar]
- 52.Goel M., Zuo C.D., Sinkins W.G., Schilling W.P. TRPC3 channels colocalize with Na+/Ca2+ exchanger and Na+ pump in axial component of transverse-axial tubular system of rat ventricle. Am J Physiol Heart Circ Physiol. 2007;292:H874–H883. doi: 10.1152/ajpheart.00785.2006. [DOI] [PubMed] [Google Scholar]
- 53.Franco-Obregón A., Jr, Lansman J.B. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J Physiol. 1994;481:299–309. doi: 10.1113/jphysiol.1994.sp020440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zanou N., Shapovalov G., Louis M., Tajeddine N., Gallo C., Van Schoor M., Anguish I., Cao M.L., Schakman O., Dietrich A., Lebacq J., Ruegg U., Roulet E., Birnbaumer L., Gailly P. Role of TRPC1 channel in skeletal muscle function. Am J Physiol Cell Physiol. 2010;298:C149–C162. doi: 10.1152/ajpcell.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holliday R. Streptomycin, errors in mitochondria and ageing. Biogerontology. 2005;6:431–432. doi: 10.1007/s10522-005-4911-2. [DOI] [PubMed] [Google Scholar]
- 56.Grounds M.D., Torrisi J. Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004;18:676–682. doi: 10.1096/fj.03-1024com. [DOI] [PubMed] [Google Scholar]
- 57.Acharyya S., Butchbach M.E., Sahenk Z., Wang H., Saji M., Carathers M., Ringel M.D., Skipworth R.J., Fearon K.C., Hollingsworth M.A., Muscarella P., Burghes A.H., Rafael-Fortney J.A., Guttridge D.C. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421–432. doi: 10.1016/j.ccr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Lullmann H., Vollmer B. An interaction of aminoglycoside antibiotics with Ca binding to lipid monolayers and to biomembranes. Biochem Pharmacol. 1982;31:3769–3773. doi: 10.1016/0006-2952(82)90291-x. [DOI] [PubMed] [Google Scholar]
- 59.Fayssoil A., Nardi O., Orlikowski D., Annane D. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev. 2010;15:103–107. doi: 10.1007/s10741-009-9156-8. [DOI] [PubMed] [Google Scholar]
- 60.Zhang W., ten Hove M., Schneider J.E., Stuckey D.J., Sebag-Montefiore L., Bia B.L., Radda G.K., Davies K.E., Neubauer S., Clarke K. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: an MRI and MRS study. J Mol Cell Cardiol. 2008;45:754–760. doi: 10.1016/j.yjmcc.2008.09.125. [DOI] [PubMed] [Google Scholar]
- 61.Fanchaouy M., Polakova E., Jung C., Ogrodnik J., Shirokova N., Niggli E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009;46:114–121. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Youm J.B., Han J., Kim N., Zhang Y.H., Kim E., Joo H., Hun L.C., Joon K.S., Cha K.A., Earm Y.E. Role of stretch-activated channels on the stretch-induced changes of rat atrial myocytes. Prog Biophys Mol Biol. 2006;90:186–206. doi: 10.1016/j.pbiomolbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 63.Ward M.L., Williams I.A., Chu Y., Cooper P.J., Ju Y.K., Allen D.G. Stretch-activated channels in the heart: contributions to length-dependence and to cardiomyopathy. Prog Biophys Mol Biol. 2008;97:232–249. doi: 10.1016/j.pbiomolbio.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 64.Hu J.F., Gilmer L., Hopkins R., Wolfinbarger L., Jr Effects of antibiotics on cellular viability in porcine heart valve tissue [Erratum appeared in Cardiovasc Res 1990;24:168] Cardiovasc Res. 1989;23:960–964. doi: 10.1093/cvr/23.11.960. [DOI] [PubMed] [Google Scholar]
- 65.Schirmer M., Kaiser A., Lessenich A., Lindemann S., Fedrowitz M., Gernert M., Löscher W. Auditory and vestibular defects and behavioral alterations after neonatal administration of streptomycin to Lewis rats: similarities and differences to the circling (ci2/ci2) Lewis rat mutant. Brain Res. 2007;1155:179–195. doi: 10.1016/j.brainres.2007.04.012. [DOI] [PubMed] [Google Scholar]