

Abstract
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) regenerates active glucocorticoids (GCs) from intrinsically inert 11-keto substrates inside cells, including neurons, thus amplifying steroid action. Excess GC action exerts deleterious effects on the hippocampus and causes impaired spatial memory, a key feature of age-related cognitive dysfunction. Mice with complete deficiency of 11β-HSD1 are protected from spatial memory impairments with aging. Here, we tested whether lifelong or short-term decreases in 11β-HSD1 activity are sufficient to alter cognitive function in aged mice. Aged (24 months old) heterozygous male 11β-HSD1 knock-out mice, with ∼60% reduction in hippocampal 11β-reductase activity throughout life, were protected against spatial memory impairments in the Y-maze compared to age-matched congenic C57BL/6J controls. Pharmacological treatment of aged C57BL/6J mice with a selective 11β-HSD1 inhibitor (UE1961) for 10 d improved spatial memory performance in the Y-maze (59% greater time in novel arm than vehicle control). These data support the use of selective 11β-HSD1 inhibitors in the treatment of age-related cognitive impairments.
Introduction
Age-related cognitive deficits in humans and rodents are often associated with chronically elevated levels of the stress glucocorticoid (GC) hormones (Issa et al., 1990; Lupien et al., 1998). GCs appear causal since their prolonged elevation exerts deleterious effects on the hippocampus, a brain region particularly vulnerable to aging (McEwen et al., 1993), while manipulations that keep GC levels low throughout life prevent the emergence of cognitive deficits with aging (Landfield et al., 1981; Meaney et al., 1988). The exposure of specific tissues to GCs is normally tightly controlled by hypothalamic–pituitary–adrenal (HPA) axis negative feedback regulation of circulating hormone levels, the density of intracellular corticosteroid receptors, and cellular metabolism by 11β-hydroxysteroid dehydrogenases (11β-HSDs). There are two isozymes that control the intracellular concentration of active GCs (Seckl et al., 2002). In the adult forebrain, 11β-HSD type 1 (11β-HSD1) is the predominant or sole isoform (Holmes et al., 2003) and acts predominately as a ketoreductase in vivo, catalyzing conversion of inert cortisone (humans) or 11-dehydrocorticosterone (rodents) into active cortisol or corticosterone, thus amplifying intracellular GC action.
11β-HSD1 amplification of intracellular GCs shows particular impact with aging. Thus, aged 11β-HSD1 knock-out (11β-HSD1−/−) mice have better spatial memory, lower intrahippocampal levels of corticosterone, and better maintenance of hippocampal long-term potentiation than age-matched controls (Yau et al., 2001, 2007). While 11β-HSD1−/− mice are completely deficient in the enzyme for their lifespan, some evidence suggests that the cognitive protection is not a developmental phenotype. Thus, 11β-HSD1 is little expressed in the rodent brain until birth (Diaz et al., 1998). Moreover, short-term administration of nonselective (licorice-based) 11β-HSD inhibitors improves memory in young mice with scopolamine-induced amnesia (Dhingra et al., 2004) and key aspects of cognitive function in healthy elderly humans and patients with type 2 diabetes mellitus (Sandeep et al., 2004). However, licorice derivatives inhibit both isozymes of 11β-HSD, other short-chain dehydrogenases, and even gap junctions (Sagar and Larson, 2006).
We have recently developed potent selective 11β-HSD1 inhibitors that cross the blood–brain barrier and that do not inhibit 11β-HSD2 (Webster et al., 2007). Since drug-mediated inhibition of 11β-HSD1 in the brain is unlikely to be complete, we first examined 11β-HSD1+/− “heterozygous” mice to determine whether partial loss of the enzyme can exert cognitive effects with aging. In parallel, we tested whether short-term administration of our selective 11β-HSD1 inhibitor (UE1961) affects spatial memory in already aged C57BL/6J mice.
Materials and Methods
Animals
11β-HSD1 knock-out mice congenic on a C57BL/6J background (Carter et al., 2009) were bred after a minimum of 10 generations backcrossing from the original MF1/129 background (Kotelevtsev et al., 1997). Male 11β-HSD1−/−, 11β-HSD1+/−, and C57BL/6J littermates were generated from 11β-HSD1+/− crosses. For pharmacology studies, C57BL/6J mice were purchased from Harlan. Mice were genotyped by RT-PCR and housed 4 to 5 per cage under standard conditions on a 12 h light/dark cycle (lights on at 7:00 A.M.) with ad libitum access to food (standard chow) and water. All procedures were performed to the highest standards under the aegis of the UK Animals (Scientific Procedures) Act, 1986, and with local ethical committee approval.
11β-HSD1 inhibitor
The novel compound UE1961 (N-[3-[4-[4-[(4aR,8aR)-3,4,4a,5,6,7,8,8a-octahydro-2H-quinoline-1-carbonyl]-2-thienyl]-1-piperidyl]-3-oxo-propyl]methanesulfonamide) was synthesized by Argenta according to the published synthetic method (Webster et al., 2009). In vitro screening of UE1961 potency for the median inhibitory concentration (IC50) was determined in HEK293 cells as previously described (Webster et al., 2007).
Spatial memory testing of 11β-HSD1-deficient mice in Y-maze
Young (6 months old) and aged (24 months old) C57BL/6J, 11β-HSD1+/−, and 11β-HSD1−/− mice were tested in the Y-maze. At least 1 week before testing, basal morning blood samples were taken by tail venesection for corticosterone levels. The Y-maze apparatus, made up of three enclosed black Plexiglas arms (50 cm long, 11 cm wide, and 10 cm high) with extramaze visual cues around the maze, was used to assess hippocampal-dependent spatial recognition memory (Yau et al., 2007). The test consisted of two trials separated by an intertrial interval (ITI). All mice were transported to the behavioral testing room in their home cages at least 1 h before testing. In the first training (acquisition) trial, mice were placed at the end of a pseudorandomly chosen start arm and allowed to explore the maze for 5 min with one of the arms closed (novel arm). Mice were returned to their home cage until the second (retrieval) trial, during which they could explore freely all three arms of the maze. The time spent in each arm was measured and analyzed from video recordings using a computer tracking system (Limelight, ActiMetrics). The time spent in the novel arm was calculated as a percentage of the total time in all three arms during the 2 min retrieval trial (Dellu et al., 1992; Conrad et al., 1999). A 1 min ITI was first used to control for spontaneous novelty exploration and also to test that the mice were able to see the spatial cues. Mice were retested 7 d later to measure spatial memory performance with a 2 h ITI. Following Y-maze testing, the mice were culled by cervical dislocation, brains dissected and stored frozen for later 11β-HSD1 (11β-reductase) activity assays.
Treatment of aged mice with UE1961
UE1961 was first tested in vivo in 12-month-old C57BL/6J mice (n = 9/group) to access the effectiveness of inhibition of 11β-HSD1 in brain following intraperitoneal administration (10 mg/kg, 12 hourly for 3 d); control mice received vehicle injections [38% PEG 400, 2% DMSO (Sigma) in 0.9% NaCl]. The mice were culled 1 h after the final dose, and brain tissue was dissected and processed for 11β-reductase activity. Following confirmation that peripheral administration of UE1961 effectively inhibited hippocampal 11β-HSD1 activity, aged (24 months old) C57BL/6J mice were treated with UE1961 (10 mg/kg, i.p.) or vehicle twice daily for 10 d. The mice were tested on the Y-maze with a 2 h ITI on day 10 of UE1961 treatment 1 h after the morning injection (7:00 A.M.). Tail venesection blood samples were taken in the morning 12 h after the last injection of UE1961.
11β-HSD enzyme activity assays
11β-HSD1 activity ex vivo.
Brain samples were homogenized and assayed for 11β-ketosteroid reductase activity essentially as described previously (Kotelevtsev et al., 1997), but instead of adding NADPH cosubstrate directly, glucose-6-phosphate, a substrate for hexose-6-phosphate dehydrogenase (H6PDH), which is colocalized in the lumen of the endoplasmic reticulum with 11β-HSD1 and acts to generate the cosubstrate, was added (Clarke and Mason, 2003). Briefly, the tissues were homogenized (0.5 mg/ml for hippocampus, 0.2 mg/ml for cerebellum and cortex) and incubated in C buffer (10% glycerol, 50 mm sodium acetate, 1 mm EDTA; pH 6.0) containing 10 mm d-glucose-6-phosphate dipotassium salt hydrate and 10 nm [3H]-11-dehydrocorticosterone ([3H]-11DHC) at 37°C for 60 min. The [3H]-11DHC was prepared as described previously (Brown et al., 1993), and the incubation time and protein concentration were predetermined from initial time course incubations with varying concentrations of protein. The steroids were extracted with ethyl acetate and analyzed by thin-layer chromatography.
11β-HSD1 activity in vitro.
Full-length 11β-HSD1 cDNAs were amplified by PCR and ligated into the vector pCDNA5/FRT/V5-His TOPO TA (Invitrogen). Flp-In HEK293 cells (Invitrogen) were cotransfected with vector and plasmid pOG44 to generate stably expressing cells. HEK-293 cells stably transfected with human, mouse, or rat 11β-HSD1 were seeded in 96-well plates at a density of 20,000 cells per well. Sixteen hours after seeding, the cells were incubated with 20 nm 3H-cortisone (GE Healthcare) in serum-free media (DMEM, Invitrogen) and compound in DMSO at a final concentration of 1%. Following incubation for 1–2 h, 50 μl of media was removed, and liberated 3H-cortisol was captured on anti-cortisol (HyTest Ltd)-coated scintillation proximity assay beads (protein A-coated YSi, GE Healthcare). Data were fitted to the four-parameter logistic equation using GraphPad Prism software.
11β-HSD2 activity in vitro.
CHO cells stably transfected with 11β-HSD2 (Brown et al., 1996) were seeded in 96-well plates at a density of 20,000 cells per well. Sixteen hours after seeding, the cells were incubated with 20 nm 3H-cortisol (GE Healthcare) in serum-free media (DMEM, Invitrogen) and compound in DMSO at a final concentration of 1%. Following incubation for 16 h, 50 μl of media was removed and steroids were extracted in 2 volumes of ethyl acetate. The organic phase was then evaporated under nitrogen and the extracts resuspended in 20 μl of ethanol. Samples were analyzed by TLC in a mobile phase of 92:8 chloroform/ethanol, using a Fujifilm FLA-2000 phosphoimager to quantify tritium-labeled steroids following exposure to a tritium screen.
Measurement of GR binding in vitro
GR binding was measured using the Polar Screen Glucocorticoid Receptor competitor assay kit (Green) (Invitrogen), according to the manufacturer's instructions.
Corticosterone radioimmunoassay
Plasma corticosterone levels were measured using an in-house RIA (Al-Dujaili et al., 1981) modified for microtiter plate scintillation proximity assay (GE Healthcare UK). Corticosterone antiserum was kindly donated by Dr. C. Kenyon (University of Edinburgh, Edinburgh, UK). For brain hippocampal corticosterone levels, steroids were extracted by solvolysis from the dissected tissues as described previously (Ebner et al., 2006) with modifications. Tissue homogenates in phosphate buffer were added slowly to 95% ethanol at −30°C to avoid protein denaturation and steroid trapping. After incubation for 16 h at −30°C, extracts were centrifuged, dried down, reconstituted in 40% methanol, and further extracted using C18 Sep-Pak cartridges to remove conjugated and sulfated metabolites before RIA.
Statistical analysis
Data are expressed as mean ± SEM and were analyzed using a one-way ANOVA followed by Fischer PLSD and Scheffé F tests post hoc as appropriate for individual between-group comparisons. The percentage time in the novel arm comparison with the other arms of the Y-maze within a group was performed by Student's paired t test. Significance was set at p < 0.05.
Results
Both 11β-HSD1+/− and 11β-HSD1−/− mice are protected from spatial memory impairments with aging
All groups of mice [young (6 months old) and old (24 months old), C57BL/6J, 11β-HSD1+/−, and 11β-HSD1−/−] spent significantly (p < 0.05) more time in the novel arm than the previously visited arms (other and start) following a short 1 min ITI (Fig. 1). This immediate version of the Y-maze showed all mice groups were equally motivated to explore the novel environment. There was no significant effect of age or genotype on the time spent in the novel arm following the 1 min ITI (Fig. 1). For the hippocampus-dependent spatial memory test provided by the 2 h ITI, two-factor ANOVA revealed a significant genotype (F (2,39) = 3.7, p < 0.05) effect and interaction between age and genotype (F (2,39) = 3.4, p < 0.05). While young C57BL/6J mice spent more time in the novel arm of the Y-maze after a 2 h ITI (p < 0.05 compared to other arm or start arm), aged C57BL/6J mice could not distinguish the novel arm, thus showing impaired spatial recognition memory (Fig. 1). In contrast, both aged 11β-HSD1+/− and 11β-HSD1−/− mice spent more time in the novel arm than the other two arms (p < 0.05) and significantly more time in the novel arm than aged C57BL/6J controls (F (2,22) = 7.4, p < 0.01) (Fig. 1).
Figure 1.

Maintained spatial memory in the Y-maze for aged 11β-HSD1+/− and 11β-HSD1−/− mice but not aged C57BL/6J controls. A, Both young (6 months young; n = 6–8/genotype) and aged (24 months aged; n = 7–8/genotype) mice of C57BL/6J (CON) controls, 11β-HSD1+/− (KO+/−), and 11β-HSD1−/− (KO−/−) mice were able to perform the immediate version of the Y-maze with the 1 min ITI. B, Young mice of all three genotypes show intact spatial memory (with 2 h ITI) spending more time in the novel arm than the other two arms. Aged C57BL/6J (but not aged 11β-HSD1+/− and aged 11β-HSD1−/−) mice show impaired spatial memory with 2 h ITI. Values are means ± SEM. *p < 0.05 compared to novel arm. § p < 0.01 compared to novel arm of aged C57BL/6J mice.
11β-reductase activity and hippocampal corticosterone levels are decreased in the brains of aged 11β-HSD1+/− mice
No detectable 11β-reductase activity (conversion of [3H]-11DHC to [3H]-corticosterone) was observed in tissue homogenates from the hippocampus, cortex, and cerebellum of aged 11β-HSD1−/− mice (Fig. 2). Aged 11β-HSD1+/− mice showed intermediate 11β-reductase activity (hippocampus, 62 ± 6% decrease; cerebellum, 68 ± 6% decrease; cortex, 64 ± 8% decrease; compared to aged C57BL/6J controls) (Fig. 2). Hippocampal tissue corticosterone levels were decreased in aged 11β-HSD1+/− mice (16.1 ± 1.4 ng/g) and aged 11β-HSD1−/− mice (12.0 ± 1.6 ng/g) compared to aged C57BL/6J controls (28.2 ± 5.5 ng/g) [F (2,22) = 5.6, p < 0.05]; levels between aged 11β-HSD1+/− mice and aged 11β-HSD1−/− mice were not significantly different. These changes in tissue corticosterone concentrations were not associated with alterations in plasma corticosterone levels, which although significantly increased with age (F (1,39) = 15.7, p < 0.001), did not vary with genotype nor show genotype × age interactions (Fig. 3).
Figure 2.
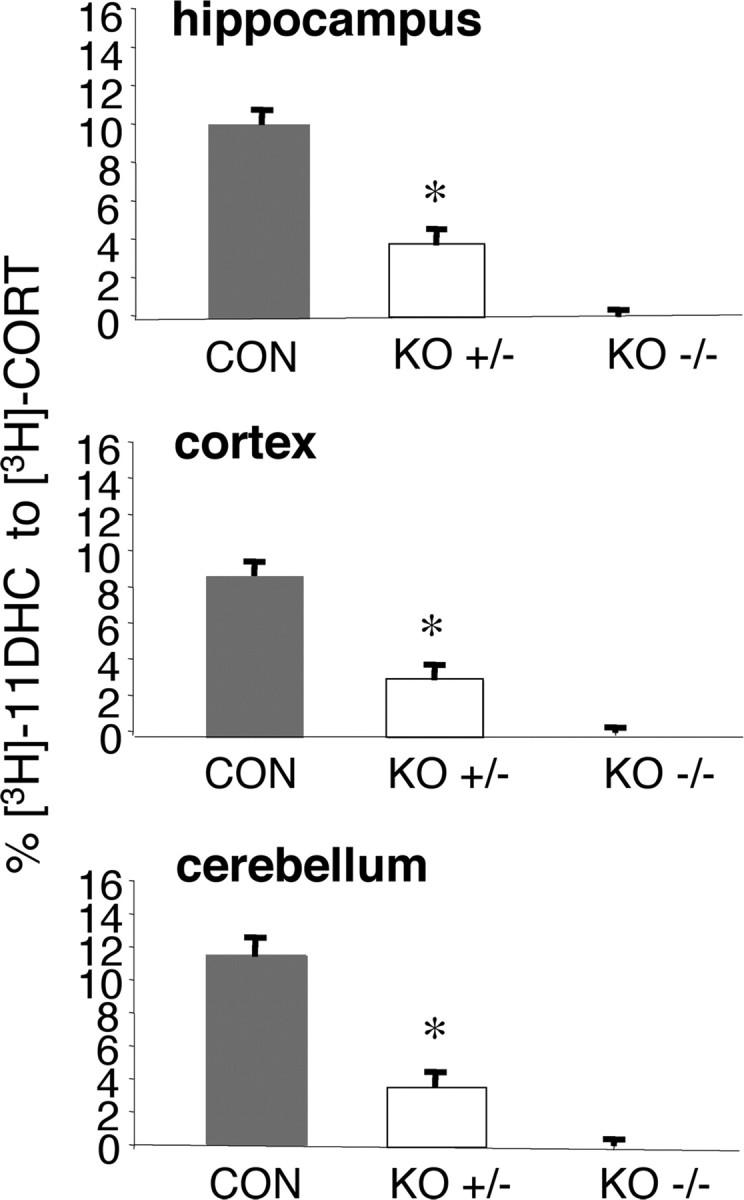
11β-Reductase activity is reduced in brain of aged 11β-HSD1+/− mice. The 11β-reductase activity was measured in tissue homogenates of the aged mice [C57BL/6J (CON) controls, 11β-HSD1+/− (KO+/−), and 11β-HSD1−/− (KO−/−), n = 7–8/genotype] incubated with [3H]-11-dehydrocorticosterone (11-DHC), and the percentage conversion to active [3H]-corticosterone (CORT) was measured. Enzyme activity levels in the tissues of aged 11β-HSD1−/− mice were not detectable. Values are means ± SEM. *p < 0.001 compared to C57BL/6J controls.
Figure 3.

Plasma corticosterone levels are increased with age but are not affected by lifelong 11β-HSD1 deficiency. Basal corticosterone (CORT) levels were measured from tail venesection blood samples taken in the morning 1 h after lights on from young (6 months old; n = 6–8/genotype) and aged (24 months old; n = 7–8/genotype) C57BL/6J (CON) controls, 11β-HSD1+/− (KO+/−), and 11β-HSD1−/− (KO−/−) mice. Values are means ± SEM. *p < 0.05 compared to corresponding young mice of same genotype.
Compound UE1961 is a selective inhibitor of 11β-HSD1 and improves spatial memory in aged C57BL/6J mice
Compound UE1961 (Fig. 4 A) is a 11β-HSD1 inhibitor displaying nanomolar potency against both human and rodent enzymes in intact cellular assays (IC50 values: 479 nm, 528 nm, and 287 nm for human, mouse, and rat 11β-HSD1, respectively). In such assays the nonselective licorice-derived 11β-HSD inhibitor carbenoxolone has an IC50 for human 11β-HSD1 of 2 μm. UE1961 is selective for 11β-HSD1 (IC50 for human 11β-HSD2 >10,000 nm; for human GR 7% binding at 10 μm). When dosed twice daily intraperitoneally for 3 d in 12-month-old C57BL/6J mice, UE1961 produced 27.1 ± 4.6% and 20.4 ± 3.2% reduction in 11β-HSD1 activity in the hippocampus and cortex ex vivo, respectively (F (1,15) = 6.1, p < 0.05 and F (1,16) = 15.9, p < 0.01) (Fig. 4 B).
Figure 4.

Treatment of aged C57BL/6J mice with a selective 11β-HSD1 inhibitor UE1961 decreases 11β-reductase activity in the hippocampus and improves spatial memory retention in the Y-maze. A, Chemical structure of UE1961. B, 11β-Reductase activity was measured in tissue homogenates of the aged mice following administration of 10 mg/kg UE1961 (inhibitor) incubated with [3H]-11-dehydrocorticosterone (11-DHC) and the percentage conversion to active [3H]-corticosterone (CORT) was measured. C, Spatial memory retention as measured in the Y-maze with a 2 h ITI was significantly better in the UE1961-treated mice than vehicle-treated controls (n = 9–10/group). Values are means ± SEM. *p < 0.05 compared to corresponding novel arm. § p < 0.001 compared to novel arm of vehicle-treated controls.
Treatment of the aged (24 months old) mice with either vehicle or UE1961 by intraperitoneal injections twice daily for 10 d had no effect on body weight, and the injections were tolerated well. Consistent with untreated aged mice, aged C57BL/6J mice given intraperitoneal vehicle could not distinguish the novel arm of the Y-maze after a 2 h ITI (Fig. 4 C). In contrast, after 10 d of UE1961 treatment, aged mice spent more time in the novel arm compared to the other two arms (p < 0.05) and also compared to vehicle-treated controls (F (1,18) = 19.1, p < 0.001) (Fig. 4 C).
This improvement in glucocorticoid-associated spatial memory performance with UE1961 was not due to lowered circulating corticosterone, indeed basal plasma corticosterone levels were higher in UE1961-treated aged C57BL/6J mice (8.58 ± 1.44 μg/dl) than in vehicle-treated controls (3.39 ± 0.53 μg/dl; F (1,17) = 12.5, p < 0.005). Adrenal weights of the aged mice were unaltered by UE1961 treatment [vehicle: 0.129 ± 0.018 mg/g body weight (bw); UE1961: 0.114 ± 0.013 mg/g bw].
Discussion
Mice heterozygous for a 11β-HSD1 null allele, with ∼60% enzyme deficiency, resisted spatial memory deficits with aging. Importantly, even short-term administration of a selective 11β-HSD1 inhibitor to achieve ∼27% reduction in hippocampal enzyme activity improved spatial memory in already aged mice. The data indicate that short-term reductions in intracellular glucocorticoid levels have cognitive benefits with aging and support the utility of CNS-active selective inhibitors of 11β-HSD1.
Recent data show that hippocampal 11β-HSD1 expression increases (∼30%) with aging in C57BL/6J mice and correlates with spatial memory deficits (Holmes et al., 2010). Modeling this by transgenic forebrain-specific overexpression (50% in hippocampus) of 11β-HSD1 accelerates cognitive decline with aging (Holmes et al., 2010). Clearly modest changes in brain 11β-HSD1 may impact on cognition with aging. 11β-HSD1 heterozygous knock-out mice showed ∼60% decrease in 11β-reductase activity in the brain. This was greater than the anticipated 50% change but may reflect reduced expression of 11β-HSD1 from the unaffected 11β-HSD1 allele since GCs themselves induce 11β-HSD1 gene expression (Sai et al., 2008). Decreased 11β-reductase activity was reflected in lower hippocampal tissue corticosterone levels in aged 11β-HSD1+/− mice. This was not a consequence of genotype-specific alterations in circulating corticosterone levels, emphasizing the importance of intracellular metabolism in determining GC signaling, especially as plasma corticosterone levels rise with age. Thus modest decreases in 11β-HSD1 across the lifespan exert cognitive protection with aging. Since no apparent cognitive benefits are seen in young animals even with complete 11β-HSD1 deficiency, the implication is that the cognitive effects of deficiency of the enzyme only becomes manifest with age, plausibly in association with chronic elevation of GC levels and perhaps also reduced hippocampal glucocorticoid receptors (Murphy et al., 2002) and/or the accumulation of allostatic load (McEwen, 2002).
To address any short-term effects of reduced 11β-HSD1 activity on cognitive function, we developed UE1961, a highly selective 11β-HSD1 inhibitor that penetrates the CNS. Peripheral administration of UE1961 caused ∼27% reduction in 11β-HSD1 activity in hippocampal homogenates. This is likely an underestimate of inhibition in vivo since the effectiveness of inhibitors declines rapidly ex vivo (Hermanowski-Vosatka et al., 2005). Nonetheless, UE1961 improved spatial memory performance of aged mice in the Y-maze after only 10 d treatment. This occurred despite increased plasma corticosterone levels, an effect unexpected from 11β-HSD1 inhibition in C57BL/6J mice (Carter et al., 2009). The implication is that intracellular corticosterone levels in brain (part of which comes from free corticosterone from the periphery) is still sufficiently lower than in aged wild-type mice, because of the reduced 11β-HSD1 activity, to improve spatial memory performance.
Inhibition of 11β-HSD1 activity may lead to beneficial metabolic effects such as decreased fasting glucose and improved insulin sensitivity (Hermanowski-Vosatka et al., 2005), and 11β-HSD1-deficient mice show improved glucose tolerance on a high-fat diet (Morton et al., 2001). Hyperglycemia associates with cognitive decline with aging in humans and some animal models (Convit et al., 2003; McCall, 2005) and hippocampal memory processes in rats are modulated by insulin and high-fat-induced insulin resistance (McNay et al., 2010). Whether the UE1961 effects on cognition in aged mice is mediated centrally or via such peripheral effects is unclear. However, our recent unpublished findings in aged 11β-HSD1−/− mice with transgenic “rescue” of the enzyme in the forebrain show spatial memory impairments compared to 11β-HSD1−/− mice (J. L. W. Yau and J. R. Seckl, unpublished results). This suggests that inhibition of 11β-HSD1 activity in brain by UE1961 is pivotal in the improved cognitive function.
Ten days of treatment with UE1961 in already aged (24 months old) mice had similar effects on spatial memory performance to lifelong partial deficiency of 11β-HSD1. This suggests that the memory deficit in the aged mice was not linked to an irreversible molecular or structural impairment. Moreover, the inhibitor effects are unlikely a consequence of hippocampal structural changes but more likely due to the shorter-term effects of reduced intracellular corticosterone exposure at the time of memory testing. In support, spatial memory impairments as a consequence of hippocampal lesions or chronic restraint stress-induced hippocampal dendritic atrophy are prevented by a single pretesting injection of the corticosterone synthesis blocker metyrapone (Roozendaal et al., 2001; Wright et al., 2006). Thus, spatial memory deficits may arise from hippocampal damage-induced corticosterone hypersecretion rather than as a direct effect of hippocampal structural changes per se. The data presented here suggest that short-term 11β-HSD1 inhibition or lifelong partial 11β-HSD1 deficiency decrease intracellular corticosterone levels sufficiently to ameliorate GC impairing effects on memory in the aged mice. This parallels two randomized, double-blind, placebo-controlled, cross-over studies in which short-term treatment with carbenoxolone, a nonselective 11β-HSD inhibitor, improved verbal fluency in healthy elderly men and improved verbal memory in patients with type 2 diabetes (Sandeep et al., 2004). While one potential side effect of 11β-HSD1 inhibition is the possibility of HPA axis stimulation to compensate for the reduced tissue regeneration of active GCs, the limited data in humans treated with 11β-HSD1 inhibitors report no change in cortisol (Sandeep et al., 2004; Rosenstock et al., 2009).
The findings raise the issue of the normal role of 11β-HSD1 in the forebrain and whether there are negative consequences of enzyme deficiency. While chronically elevated GCs have adverse impacts on cognition, dynamic increases in GCs facilitate the consolidation of emotionally arousing experiences (Abrari et al., 2009; Burman et al., 2010). Any role of 11β-HSD1 on such effects is unknown but is currently under investigation. Whatever the outcome, our data suggest that brain-active selective 11β-HSD1 inhibitors may have cognitive benefits in the already aged.
Footnotes
This work was supported by a Medical Research Council (MRC) Project Grant G0501596 (J.L.W.Y., J.R.S.) and Wellcome Trust Seeding Drug Discovery Award 078269 (B.R.W., S.P.W., J.R.S.). J.L.W.Y. holds a Research Councils UK Academic Fellowship. J.L.W.Y. and J.R.S. are members of The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology, part of the cross-council Lifelong Health and Wellbeing Initiative (G0700704/84698). Funding from the Biotechnology and Biological Sciences Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council, and MRC and the support of the British Heart Foundation Centre of Research Excellence are gratefully acknowledged. We thank Drs. David Pallin and Hazel Hunt at Argenta and Dr. Peter Ward for their expertise in medicinal chemistry.
References
- Abrari K, Rashidy-Pour A, Semnanian S, Fathollahi Y, Jadid M. Post-training administration of corticosterone enhances consolidation of contextual fear memory and hippocampal long-term potentiation in rats. Neurobiol Learn Mem. 2009;91:260–265. doi: 10.1016/j.nlm.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Al-Dujaili EAS, Williams BC, Edwards CRW. The development and application of a direct radioimmunoassay for corticosterone. Steroids. 1981;37:157–176. doi: 10.1016/s0039-128x(81)80015-3. [DOI] [PubMed] [Google Scholar]
- Brown RW, Chapman KE, Edwards CRW, Seckl JR. Human placental 11β-hydroxysteroid dehydrogenase: partial purification of and evidence for a distinct NAD-dependent isoform. Endocrinology. 1993;132:2614–2621. doi: 10.1210/endo.132.6.8504762. [DOI] [PubMed] [Google Scholar]
- Brown RW, Chapman KE, Kotelevtsev Y, Yau JL, Lindsay RS, Brett L, Leckie C, Murad P, Lyons V, Mullins JJ, Edwards CR, Seckl JR. Cloning and production of antisera to human placental 11 beta-hydroxysteroid dehydrogenase type 2. Biochem J. 1996;313:1007–1017. doi: 10.1042/bj3131007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman MA, Hamilton KL, Gewirtz JC. Role of corticosterone in trace and delay conditioned fear-potentiated startle in rats. Behav Neurosci. 2010;124:294–299. doi: 10.1037/a0018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RN, Paterson JM, Tworowska U, Stenvers DJ, Mullins JJ, Seckl JR, Holmes MC. Hypothalamic-pituitary-adrenal axis abnormalities in response to deletion of 11beta-HSD1 is strain-dependent. J Neuroendocrinol. 2009;21:879–887. doi: 10.1111/j.1365-2826.2009.01899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JL, Mason PJ. Murine hexose-6-phosphate dehydrogenase: a bifunctional enzyme with broad substrate specificity and 6-phosphogluconolactonase activity. Arch Biochem Biophys. 2003;415:229–234. doi: 10.1016/s0003-9861(03)00229-7. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Lupien SJ, McEwen BS. Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol Learn Mem. 1999;72:39–46. doi: 10.1006/nlme.1998.3898. [DOI] [PubMed] [Google Scholar]
- Convit A, Wolf OT, Tarshish C, de Leon MJ. Reduced glucose tolerance is associated with poor memory performance and hippocampal atrophy among normal elderly. Proc Natl Acad Sci U S A. 2003;100:2019–2022. doi: 10.1073/pnas.0336073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellu F, Mayo W, Cherkaoui J, Le Moal M, Simon H. A two-trial memory task with automated recording: study in young and aged rats. Brain Res. 1992;588:132–139. doi: 10.1016/0006-8993(92)91352-f. [DOI] [PubMed] [Google Scholar]
- Dhingra D, Parle M, Kulkarni SK. Memory enhancing activity of Glycyrrhiza glabra in mice. J Ethnopharmacol. 2004;91:361–365. doi: 10.1016/j.jep.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Diaz R, Brown RW, Seckl JR. Ontogeny of mRNAs encoding glucocorticoid and mineralocorticoid receptors and 11β-hydroxysteroid dehydrogenases in prenatal rat brain development reveal complex control of glucocorticoid action. J Neurosci. 1998;18:2570–2580. doi: 10.1523/JNEUROSCI.18-07-02570.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner MJ, Corol DI, Havlíková H, Honour JW, Fry JP. Identification of neuroactive steroids and their precursors and metabolites in adult male rat brain. Endocrinology. 2006;147:179–190. doi: 10.1210/en.2005-1065. [DOI] [PubMed] [Google Scholar]
- Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, et al. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MC, Yau JL, Kotelevtsev Y, Mullins JJ, Seckl JR. 11β-Hydroxysteroid dehydrogenases in the brain: two enzymes two roles. Ann N Y Acad Sci. 2003;1007:357–366. doi: 10.1196/annals.1286.035. [DOI] [PubMed] [Google Scholar]
- Holmes MC, Carter RN, Noble J, Chitnis S, Dutia A, Paterson JM, Mullins JJ, Seckl JR, Yau JL. 11β-Hydroxysteroid dehydrogenase type 1 expression is increased in the aged mouse hippocampus and parietal cortex and causes memory impairments. J Neurosci. 2010;30:6916–6920. doi: 10.1523/JNEUROSCI.0731-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa AM, Rowe W, Gauthier S, Meaney MJ. Hypothalamic-pituitary-adrenal activity in aged, cognitively impaired and cognitively unimpaired rats. J Neurosci. 1990;10:3247–3254. doi: 10.1523/JNEUROSCI.10-10-03247.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CRW, Seckl JR, Mullins JJ. 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science. 1981;214:581–584. doi: 10.1126/science.6270791. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NPV, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- McCall AL. Altered glycemia and brain-update and potential relevance to the aging brain. Neurobiol Aging. 2005;26(Suppl 1):70–75. doi: 10.1016/j.neurobiolaging.2005.08.009. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging. 2002;23:921–939. doi: 10.1016/s0197-4580(02)00027-1. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Cameron H, Chao HM, Gould E, Magarinos AM, Watanabe Y, Woolley CS. Adrenal steroids and plasticity of hippocampal neurons: toward an understanding of the underlying cellular and molecular mechanisms. Cell Mol Neurobiol. 1993;13:457–482. doi: 10.1007/BF00711583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem. 2010;93:546–553. doi: 10.1016/j.nlm.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH, van Berkel C, Bhatnagar S, Sapolsky RM. Effect of neonatal handling on age-related impairments associated with the hippocampus. Science. 1988;239:766–768. doi: 10.1126/science.3340858. [DOI] [PubMed] [Google Scholar]
- Morton NM, Holmes MC, Fiévet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. Improved lipid and lipoprotein profile, hepatic insulin sensitivity and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- Murphy EK, Spencer RL, Sipe KJ, Herman JP. Decrements in nuclear glucocorticoid receptor (GR) protein levels and DNA binding in aged rat hippocampus. Endocrinology. 2002;143:1362–1370. doi: 10.1210/endo.143.4.8740. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM, McGaugh JL. Memory retrieval impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat Neurosci. 2001;4:1169–1171. doi: 10.1038/nn766. [DOI] [PubMed] [Google Scholar]
- Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, Hollis G, Flores R, Levy R, Williams WV, Seckl JR, Huber R INCB13739-202 Principal Investigators. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33:1516–1522. doi: 10.2337/dc09-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar GD, Larson DM. Carbenoxolone inhibits junctional transfer and upregulates Connexin43 expression by a protein kinase A-dependent pathway. J Cell Biochem. 2006;98:1543–1551. doi: 10.1002/jcb.20870. [DOI] [PubMed] [Google Scholar]
- Sai S, Esteves CL, Kelly V, Michailidou Z, Anderson K, Coll AP, Nakagawa Y, Ohzeki T, Seckl JR, Chapman KE. Glucocorticoid regulation of the promoter of 11beta-hydroxysteroid dehydrogenase type 1 is indirect and requires CCAAT/enhancer-binding protein-beta. Mol Endocrinol. 2008;22:2049–2060. doi: 10.1210/me.2007-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandeep TC, Yau JL, MacLullich AM, Noble J, Deary IJ, Walker BR, Seckl JR. 11Beta-hydroxysteroid dehydrogenase inhibition improves cognitive function in healthy elderly men and type 2 diabetics. Proc Natl Acad Sci U S A. 2004;101:6734–6739. doi: 10.1073/pnas.0306996101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seckl JR, Yau J, Holmes M. 11Beta-hydroxysteroid dehydrogenases: a novel control of glucocorticoid action in the brain. Endocr Res. 2002;28:701–707. doi: 10.1081/erc-120016990. [DOI] [PubMed] [Google Scholar]
- Webster SP, Ward P, Binnie M, Craigie E, McConnell KM, Sooy K, Vinter A, Seckl JR, Walker BR. Discovery and biological evaluation of adamantyl amide 11beta-HSD1 inhibitors. Bioorg Med Chem Lett. 2007;17:2838–2843. doi: 10.1016/j.bmcl.2007.02.057. [DOI] [PubMed] [Google Scholar]
- Webster SP, Seckl JR, Walker BR, Ward P, Pallin TD, Dyke HJ, Perrior TR. Amido thiophene compounds and their use as 11-beta-HSD1 inhibitors. PCT Intl WO/2009/074789. 2009 [Google Scholar]
- Wright RL, Lightner EN, Harman JS, Meijer OC, Conrad CD. Attenuating corticosterone levels on the day of memory assessment prevents chronic stress-induced impairments in spatial memory. Eur J Neurosci. 2006;24:595–605. doi: 10.1111/j.1460-9568.2006.04948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau JL, Noble J, Kenyon CJ, Hibberd C, Kotelevtsev Y, Mullins JJ, Seckl JR. Lack of tissue glucocorticoid reactivation in 11beta -hydroxysteroid dehydrogenase type 1 knockout mice ameliorates age-related learning impairments. Proc Natl Acad Sci U S A. 2001;98:4716–4721. doi: 10.1073/pnas.071562698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau JL, McNair KM, Noble J, Brownstein D, Hibberd C, Morton N, Mullins JJ, Morris RG, Cobb S, Seckl JR. Enhanced hippocampal long-term potentiation and spatial learning in aged 11β-hydroxysteroid dehydrogenase type 1 knock-out mice. J Neurosci. 2007;27:10487–10496. doi: 10.1523/JNEUROSCI.2190-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]