

Abstract
Recent studies have shown that insulin growth factor‐1 (IGF‐1) and either erythropoietin (EPO) or the long‐acting EPO analog Darbepoetin alfa (DA) protect the heart against ischemia/reperfusion (I/R) and myocardial infarction (MI). The present study examined the cardioprotective effect of simultaneous treatments with IGF‐1 and DA in these models of cardiac injury. Rats were subjected to I/R or MI and were treated with IGF‐1, DA, and a combination of IGF‐1 and DA, or vehicle treatment. IGF‐1 and DA treatments imparted similar protective effect by reducing infarct size. Moreover, these treatments led to improvement of cardiac function after I/R or MI compared to vehicle. In the reperfused heart, apoptosis was reduced with either or both IGF‐1 and DA treatments as measured by reduced TUNEL staining and caspase‐3 activity. In addition, after MI, treatment with IGF‐1 or DA significantly induced angiogenesis. This angiogenic effect was enhanced significantly when IGF‐1 and DA were given simultaneously compared to vehicle or either agents alone. These data indicate simultaneous pharmacological treatments with IGF‐1 and DA protect the heart against I/R and MI injuries. This protection results in reduced infarct size and improved cardiac function. Moreover, this treatment reduces apoptosis and enhances angiogenesis in the ischemic heart.
Keywords: myocardial ischemia, ischemia/reperfusion injury, cardioprotection, insulin‐like growth factor‐1, darbepoetin alfa
Introduction
Cardiovascular disease represents the leading cause of mortality in developed countries. Ischemic heart disease contributes to this health concern and outcomes in congestive heart failure have not improved. 1 Cardioprotection during ischemia and reperfusion is a target of investigation for the development of innovative protective therapies to limit the death of heart cells. 2 An approach that could be used clinically is the protection of ischemic and/or reperfused heart cells against death. 2 Of importance, limiting infarct size after myocardial infarction may prevent or delay ventricular dysfunction and thus decrease the future occurrence of chronic heart failure. Therapy before, during, or after ischemia, and during reperfusion with pharmacological compounds has been shown to be beneficial. 2 Two of these compounds, erythropoietin (EPO) and insulin‐like growth factor 1 (IGF‐1) are growth factors that, when administered during the course of myocardial infarction and ischemia‐reperfusion (I/R), result in such protection. 2 , 3
EPO, a hematopoietic cytokine primarily synthesized in kidney peritubular cells, has physiologic effects mediated through its interaction with its specific cellular receptor (EPO‐R). 4 EPO is synthesized in a variety of other tissues, including liver, peripheral endothelial cells, vascular smooth muscle cells, and cardiomyocytes, 5 and its receptor has also been located in those cells. 6 , 7 , 8 , 9 Recombinant human EPO (rhEPO) administration is protective in various organs under ischemia or other types of injuries. 10 , 11 , 12 , 13 , 14 , 15 In the cardiovascular system, EPO (through the binding to the EPO‐R) is reported to induce endothelial cell proliferation and angiogenesis. 16 , 17 Moreover, rhEPO has been shown to exert marked myocardial protective effects against I/R injury in rats, rabbits, and dogs when administrated at early time points. 18 , 19 , 20
Darbepoetin alfa (DA), a unique analog of EPO with 3‐fold longer plasma half‐life that binds to the same receptor, 21 has also been shown to be cardioprotective in various models of myocardial infarction and I/R. 22 , 23 , 24 , 25 Studies from our lab as well as others have shown reduced left ventricular (LV) infarct size, enhanced recovery of LV function, reduced apoptotic cell death, and activated the salvage proteins phosphoinositide‐3 kinase (PI3K) and Akt. 19 , 20 , 22 , 26 , 27 These protective effects of EPO and DA have all been demonstrated to be independent of a rise in hematocrit.
Insulin‐like growth factor‐1 (IGF‐1) is a single‐chain polypeptide that is structurally related to proinsulin (reviewed in Ref. 28). Its effects are mediated mainly by binding the IGF‐1 receptor located on cell surfaces. 29 This receptor is also widely expressed in various tissues, including the heart. 30 IGF‐1 represents another molecule that has been shown to be cardioprotective in vitro 31 , 32 , 33 and in vivo. 34 , 35 , 36 , 37 These studies have demonstrated reduction in infarct size as well as myocardial apoptosis mediated by the PI3K‐Akt intracellular signaling pathway.
Conversely, limited information is available regarding simultaneous pharmacological therapy with these two growth factors in settings of I/R and myocardial infarction. Concurrent administration of two different pharmacological compounds could impart synergistic cardioprotection and lead to further additive beneficial effects compared to either therapy alone. Thus, we examined the cardioprotective effects of DA and IGF‐1, either as therapy alone or in combination, on the rat heart in (1) an I/R model and (2) a permanent coronary occlusion (ischemia) model. Our results show that either therapy alone displays robust myocardial protection after ischemic injury.
Methods
Experimental groups and protocol
Experiments were carried out according to National Institutes of Health Guidelines on the Use of Laboratory Animals and all procedures were approved by the Thomas Jefferson University Committee on Animal Care. Male Sprague–Dawley rats (200–250 g) were randomly assigned to the following groups: sham, vehicle (saline), insulin‐like growth factor‐1 at 1 μg/kg (IGF‐1; Sigma), darbepoetin alfa at 30 μg/kg (DA; Amgen, Thousand Oaks, CA, USA) or a combination of IGF‐1 and DA at the initiation of ischemia intravenously (i.v.; via tail vein). Pharmacological dosages were chosen in accordance with pilot study data and those obtained in a previously described study. 22 For the permanent coronary occlusion (MI) protocol, rats were randomly assigned to the following groups: sham, vehicle (saline), IGF‐1 at 1 μg/kg, DA at 20 μg/kg, or a combination of IGF‐1 and DA at the initiation of ischemia (i.v.), and a weekly intraperitoneal (i.p.) treatment was given to rats with the following doses: IGF‐1 at 1 μg/kg and DA at 10 μg/kg. For the I/R protocol, rats were reperfused for 72 hours. For the MI protocol, rats were kept for 4 weeks.
In vivo ischemia‐reperfusion (I/R) and permanent coronary occlusion (MI)
For the I/R model, surgical procedures were performed as previously described. 22 Animals were anesthetized with 2% isoflurane inhalation. A skin incision was made over the left thorax and the pectoral muscles were retracted to expose the ribs. The heart was exposed and exteriorized through a left thoracotomy at the level of the fifth intercostal space. A slipknot was made around the left anterior descending coronary artery (LAD) 2–3 mm from its origin with a 6–0 silk suture. After the slip knot was tied, the heart was immediately placed back into the intrathoracic space followed by manual evacuation of pneumothoraces and closure of muscle and the skin suture by means of the previously placed purse‐string suture. Sham‐operated animals were subjected to the same surgical procedures except that the suture was passed under the LAD but was not tied. Animals recovered from anesthesia within 5 minutes after the completion of surgery and received appropriate postsurgery analgesia. Following 30 minutes of ischemia, the slipknot was released and the myocardium was reperfused. For the MI model, rats were subjected to the same surgical procedure but the suture was tied permanently and no reperfusion was induced.
In vivo echocardiographic measurements
We assessed in vivo cardiac function following 4 weeks of permanent coronary occlusion via echocardiographic measurements (Vevo 770, VisualSonics, Toronto, Canada). Rats were anesthetized with 2% isoflurane and the fractional shortening and ejection fraction were measured as previously described. 38
In vivo hemodynamic measurements
We assessed in vivo cardiac hemodynamic function following either 72 hours of reperfusion (I/R protocol) or 4 weeks of ischemia (MI protocol). Rats were anesthetized with 2% isoflurane and the right common carotid artery was cannulated and advanced retrograde into the left ventricule (LV) with a 2.0 French micromanometer (Millar Instruments, Houston, TX, USA). 22 , 38 , 39 LV pressure, LV end‐diastolic pressure (LVEDP), and heart rate (HR) were measured by this catheter advanced into the LV cavity, and data were recorded and analyzed on a PowerLab System (AD Instruments Pty Ltd., Mountain View, CA, USA) as previously described. 22 , 38 , 39 These parameters as well as LV + dP/dt max, as a measure of cardiac contractility and LV − dP/dt min, as a measure of relaxation, were recorded at baseline and after administration of the β‐adrenergic receptor (βAR) agonist, isoproterenol (0.1–1,000 ng) as described. 22 , 38 , 39
Determination of LV area at risk and infarct size
For the I/R model, infarct size was determined as previously described. 22 Briefly, at the end of a 72‐hour reperfusion period, the ligature around the LAD was re‐tied through the previous ligation site and 2% Evans blue dye was injected. The dye was circulated uniformly and distributed in the heart to areas perfused by the open coronary arteries. The heart was quickly excised, sliced, and the sections were then incubated in a 1% 2,3,5‐triphenyltetrazolium chloride (TTC, Sigma) solution and then digitally photographed. The area not at risk (ANAR, Evan's blue‐stained area), and the area at risk (AR, including both TTC‐stained and TTC‐negative staining area (infarcted tissue, I)) were measured using the computer‐based image analyzer SigmaScan Pro 5.0 (SPSS Science, Chicago, IL, USA). Myocardial infarct size was expressed as a percentage of the AR (I/AR) and the AR was expressed as the percentage of total LV (AR/AR + ANAR).
For the MI model, at 4 weeks post‐MI, hearts were quickly excised, and then sliced into 6 pieces. The sections were incubated in a 1% TTC solution and then digitally photographed. The length of infarction scar (TTC‐negative stained section) and noninfarcted LV (TTC‐stained) were analyzed using the same software. Myocardial infarct size was expressed as a percentage of the length of scar to the circumference of the LV.
Determination of myocardial apoptosis (I/R protocol)
Myocardial apoptosis was determined as previously described with minor modifications. 22 Terminal deoxynucleotidyl‐transferase mediated dUTP nick‐end labeling (TUNEL) staining and caspase‐3 activity assay were performed. For TUNEL staining, at the end of 3 hours of reperfusion, rats (n= 12 for the sham group, n= 15 for the vehicle group, n= 10 for the DA group, n= 7 for the IGF‐1 group, and n= 7 for the IGF‐1 + DA group) were anesthetized. Hearts were removed, perfused, and fixed with 4% paraformaldehyde and then stored at 4°C. The hearts were then embedded in paraffin and cut into 6‐μm thickness sections. The sections were treated as instructed with an in situ Cell Death Detection kit (Roche Applied Science, 11 684 795 910, Indianapolis, IN, USA). Sections were also stained for troponin I (TnI) for assessing myocytes. Slides were covered with a glass cover slide applied with mounting media containing DAPI. The entire population was visualized under a fluorescence microscope with the DAPI filter (330–380 nm); the same population was also examined with an FITC filter (465–495 nm) and apoptotic cells with green fluorescence were counted under a high‐power field; furthermore, myocytes were examined with a TRITC filter (545–565 nm). More than 5 fields in >3 different sections/animals in the infarction border zone were examined.
Cardiac caspase‐3 activity was measured as previously described 22 by using a caspase colorimetric assay kit (Chemicon International, Inc., Temecula, CA, USA) following the manufacturer's instructions. Briefly, at the end of the 3‐hour reperfusion period, rats (n= 7 for the sham group, n= 5 for the vehicle group, n= 6 for DA group, n= 6 for the IGF‐1 group, and n= 5 for the IGF‐1 + DA group) were anesthetized, hearts were removed, and stored at −80°C until analysis. The myocardial tissue was homogenized in ice‐cold lysis buffer for 30 seconds using a tissue homogenizer. The homogenates were centrifuged for 5 minutes at 10,000 ×g at 4°C, supernatants were collected, and protein concentrations were measured by the bicinchoninic acid method (Pierce Chemical, Rockford, IL, USA). To each well of a 96‐well plate, supernatant containing 200 μg of protein was loaded and incubated with 25 μg Ac‐DEVD‐pNA as colorimetric‐specific substrate at 37°C for 1.5 hours. The pNA was cleaved from DEVD by the caspase‐3 and this free pNA was quantified using a SpectraMax‐Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) at 405 nm. Changes in caspase activity in the I/R tissue samples were calculated against the mean value of caspase activity in the sham tissue and expressed as pmol pNA/mg protein.
Determination of vessel density
Following 4 weeks of ischemia, rats (n= 6 for the sham group, n= 8 for the vehicle group, n= 7 for DA group, n= 7 for the IGF‐1 group, and n= 5 for the IGF‐1 + DA group) were anesthetized. Hearts were removed, perfused and fixed with 4% paraformaldehyde, and then stored at 4°C. The hearts were then embedded in paraffin and cut into 6‐μm‐thickness sections. Immunohistochemistry for von Willebrand factor (vWF; Dako Cytomation, A‐0082) and hematoxylin counterstaining were performed. The number of vWF‐positive vessels was then counted either at the border zone or at a remote area.
Measurement of hematocrit
Hematocrit was measured at sacrifice as described previously. 19 , 20 , 22
Statistical analysis
All values in the text and figures are presented as mean ± SEM of independent experiments from given n‐sizes. Statistical significance of multiple treatments was determined by one‐way or two‐way analysis of variance (ANOVA) followed by the Bonferroni post hoc test when appropriate. p values of <0.05 were considered significant.
Results
Ischemia‐reperfusion model
1. Cardiac function is enhanced by the different pharmacological treatments
Cardiac function was measured at 72 hours of reperfusion via cardiac catheterization. As shown in Figure 1, no differences at baseline (Basal) were observed in either contractility (+dP/dt) and relaxation (−dP/dt) or in heart rate. Also, even if the LVEDP seemed higher in the vehicle group, no statistical difference was observed (p > 0.05). On the other hand, after β‐adrenergic stimulation (isoproterenol administration), statistical differences in contractility and relaxation were observed. Following progressive doses of isoproterenol (10–1,000 ng), sham, IGF‐1, DA, and IGF‐1 + DA groups had significantly enhanced cardiac contractility (Figure 1A) compared to vehicle (p < 0.01) but were not different from each other, indicating a better contraction subsequent to the I/R injury. As for the relaxation (Figure 1B), the same differences between sham, IGF‐1, DA, and IGF‐1 + DA groups were observed. Indeed, relaxation was significantly enhanced compared to vehicle under isoproterenol challenge. Similarly, all treated groups were similar to sham. Importantly, no differences were noted in HR (Figure 1C) either at baseline or under sympathetic stimulation. Taken together, these results demonstrate significant cardiac dysfunction at 72 hours of reperfusion in the vehicle group as assessed by hemodynamic measurements.
Figure 1.
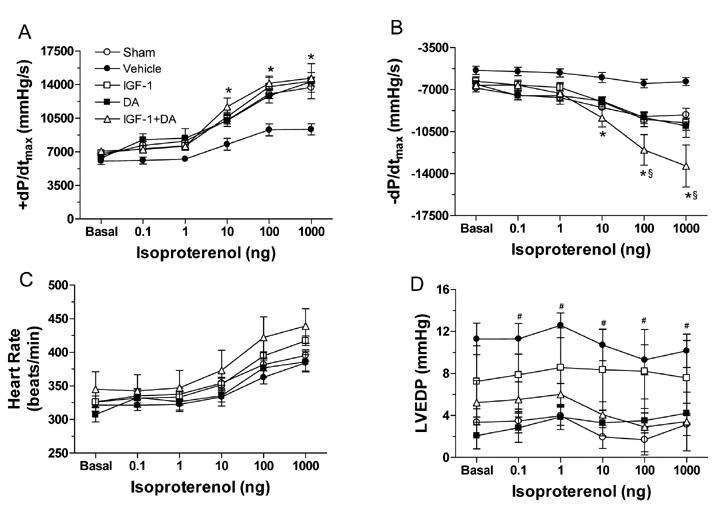
Cardiac hemodynamics function at 72 hours of reperfusion. Rats were subjected to sham operation (sham) or 30 minutes ischemia followed by 72 hours of reperfusion and treated with vehicle, IGF‐1, DA, or IGF 1 + DA at the onset of ischemia. (A) LV + dP/dtmax, (B) LV − dP/dtmin, (C) heart rate, and (D) LVEDP. Cardiac function was recorded at baseline (B) and upon isoproterenol (0.1–1,000 ng) administration. (n= 8–10/group; *p < 0.05 vs. vehicle, # p < 0.05 vs. sham, § p < 0.05 vs. DA and IGF‐1 alone.)
2. Infarction size is reduced by the different pharmacological treatments
To evaluate the effect of IGF, DA, or a combination of these two drugs in the I/R model, we measured LV infarction sizes after 72 hours of reperfusion. As shown in Figure 2A, extensive damages (white region in the AR) are present in the vehicle group compared to sham or IGF‐1, DA, and IGF‐1 + DA groups. Infarction size was 30.2 ± 3.0% in the vehicle group and significantly reduced in all drug‐treated groups to 15.8 ± 2.6% for the IGF‐1 group, 11.2 ± 3.0% for the DA group, and 8.5 ± 2.6% for the IGF + DA group (Figure 2B). As expected, the sham group did not have any damages in the AR. Moreover, no difference was noted in the size of the AR, which represented approximately 40% of the LV among groups (Figure 2C). Taken together, these data indicate that the three drug regimens provide similar cardioprotection.
Figure 2.
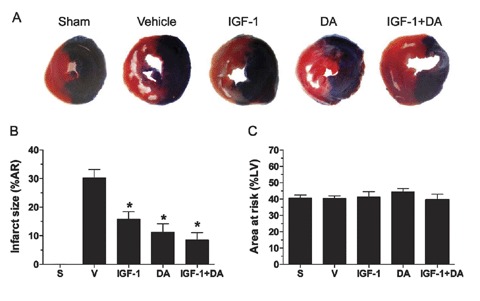
Post‐I/R infarction size. (A) Representative photographs of TTC‐stained rat heart sections from the different treatment groups. (B) LV infarct size expressed as percentage of the area at risk (AR; ischemic) in each group. (C) LV AR expressed as percentage of the total LV. (n= 5–10/group; *p < 0.05 vs. vehicle or sham; S, sham, V, vehicle.)
3. Apoptosis is reduced by the different pharmacological treatments
Two different methods were used to measure apoptosis. TUNEL staining was one of these and the infarct‐border zone was studied. As shown in Figure 3A, no staining was observed in the sham group. In contrast, the vehicle group demonstrated extensive TUNEL staining. All pharmacological regimens led to a drastic and significant reduction in TUNEL staining, which is quantified in Figure 3. Either IGF‐1 and DA alone or the combination of IGF‐1 and DA led to a significant reduction in the number of apoptotic nuclei (TUNEL positive cells) compared to the vehicle group. Moreover, no differences were observed between IGF‐1, DA, and IGF‐1 + DA groups. Consistent with TUNEL data, caspase‐3 activity was significantly reduced in all drug‐treated groups compared to vehicle.
Figure 3.
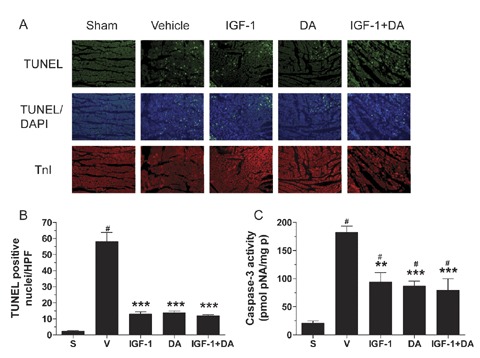
Post‐I/R apoptosis. (A) Representative photographs of TUNEL‐stained tissue sections from the different treatment groups. TUNEL (green), apoptotic nuclei, DAPI (blue), total nuclei, and TnI (red), myocytes. (B) TUNEL‐positive nuclei quantification represented as number per high‐power field (HPF). (C) Caspase‐3 activity represented as pmol of substrate (pNA) per mg of protein (mg p). (n= 5–15/group; **p < 0.01 or ***p < 0.001 vs. vehicle, # p < 0.05 vs. sham; S, Sham, V, vehicle.)
Permanent occlusion model
1. Cardiac function is enhanced by the different pharmacological treatments
Echocardiography was used as a noninvasive method to measure cardiac function 4 weeks after permanent coronary artery occlusion. As shown in Figure 4A, the vehicle group (b) exhibits severely impaired and akinetic anterior wall when compared to sham hearts (a). IGF‐1 (c), DA (d), and IGF‐1 + DA (e) groups demonstrate better wall movement in contrast to vehicle. Figure 4B and 4C shows LV ejection fraction (LVEF) and fractional shortening (LVFS), respectively. Vehicle‐treated infarcted rats have significantly impaired cardiac function compared to sham. On the other hand, all drug treatments led to an improvement of both LVEF and LVFS compared to vehicle. These drug‐treated groups are still showing significantly lower LVEF and LVFS compared to sham.
Figure 4.
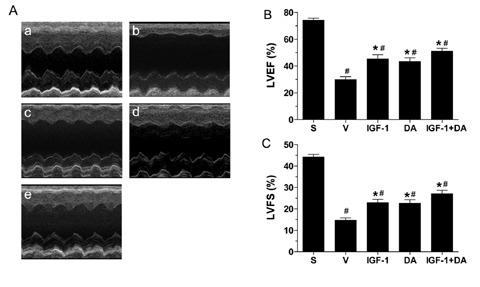
Cardiac echocardiographic function at 4 weeks of coronary occlusion. Rats were subjected to sham operation (sham) or 4 weeks of myocardial ischemia and treated with vehicle, IGF‐1, DA, or IGF‐1 + DA at the onset of ischemia. (A) Representative echocardiogram for sham (a), vehicle (b), IGF‐1 (c), DA (d), and IGF‐1 + DA (e) groups. (B) LV ejection fraction (LVEF). (C) LV fractional shortening (LVFS). Cardiac function was recorded at 4 weeks of myocardial ischemia. (n= 6–13/group; *p < 0.01 vs. vehicle, # p < 0.01 vs. sham.)
To further confirm the cardiac functional changes observed via echocardiogram, we measured hemodynamics at 4 weeks following coronary occlusion via cardiac catheterization. As shown in Figure 5, no differences at baseline (Basal) were observed in contractility, relaxation, or heart rate. On the other hand, under sympathetic stimulation, statistical differences in contractility and relaxation were observed. Following isoproterenol challenge (all doses), sham animals demonstrated significantly enhanced cardiac contractility (Figure 5A) compared to vehicle (p < 0.05). Interestingly, IGF‐1 did not lead to better contractility (p > 0.05) and was between the sham and vehicle values. On the other hand, DA and IGF‐1 + DA treatments enhanced the +dP/dt significantly compared to vehicle. These two groups were also not different from sham. Cardiac relaxation, as measured by LV − dP/dt was severely impaired at baseline (Basal) and under β‐adrenergic stimulation (Figure 5B). Similar to contractility, the IGF‐1 group was not statistically different from the vehicle group and values were between vehicle and sham. Baseline values were similar to sham but different when stimulated with isoproterenol. However, DA and IGF‐1 + DA treatments led to significant improvement of cardiac relaxation after isoproterenol stimulation.
Figure 5.
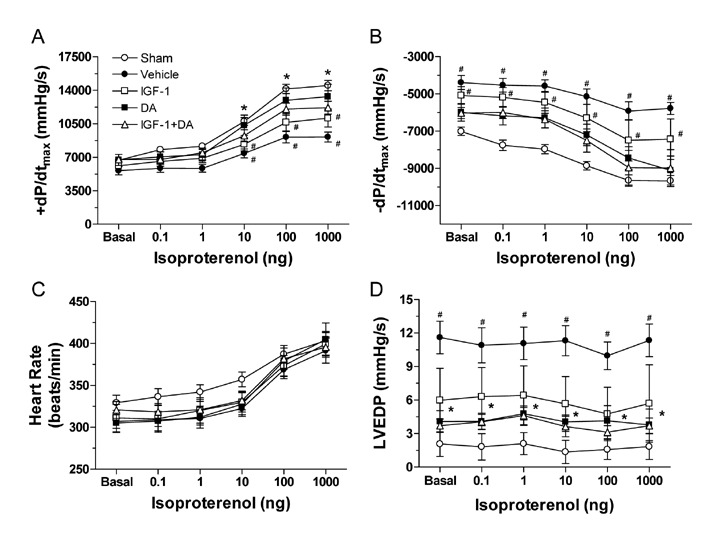
Cardiac hemodynamics function at 4 weeks of coronary occlusion. Rats were subjected to sham operation (sham) or 4 weeks of myocardial ischemia and treated with vehicle, IGF‐1, DA, or IGF‐1 + DA at the onset of ischemia. (A) LV + dP/dt max, (B) LV − dP/dt min, (C) heart rate, and (D) LVEDP. Cardiac function was recorded at baseline (B) and upon isoproterenol (0.1–1,000 ng) administration. (n= 8–10/group; *p < 0.05 vs. vehicle, # p < 0.05 vs. sham.)
Conversely, LVEDP was significantly higher in the vehicle group compared to all other groups (p > 0.05; Figure 5D). Similarly, all treated groups were similar to sham. Importantly, no differences were noted in HR (Figure 4C) either at baseline or under sympathetic stimulation. Of note, all treatments were similar between each other (no statistical difference), IGF‐1 being the treatment that led to the smallest (not significant) enhancement and DA and IGF‐1 + DA leading to the strongest (significant) improvement of cardiac function.
2. Infarction size and physiological parameters are reduced by the different pharmacological treatments
Infarction sizes were measured following 4 weeks of ischemia via TTC staining. Figure 6A shows extensive damage (white, thin region) in the vehicle group compared to sham (no damage) or IGF‐1, DA, and IGF‐1 + DA groups. Infarction size was 40.7 ± 1.7% in the vehicle group and significantly reduced in all drug‐treated groups to 30.3 ± 2.4% for the IGF‐1 group, 28.2 ± 2.1% for the DA group, and 25.7 ± 3.3% for the IGF + DA group (Figure 6B). However, no differences were observed between IGF‐1, DA, and IGF‐1 + DA. Again, as expected, the sham group did not have any damages.
Figure 6.
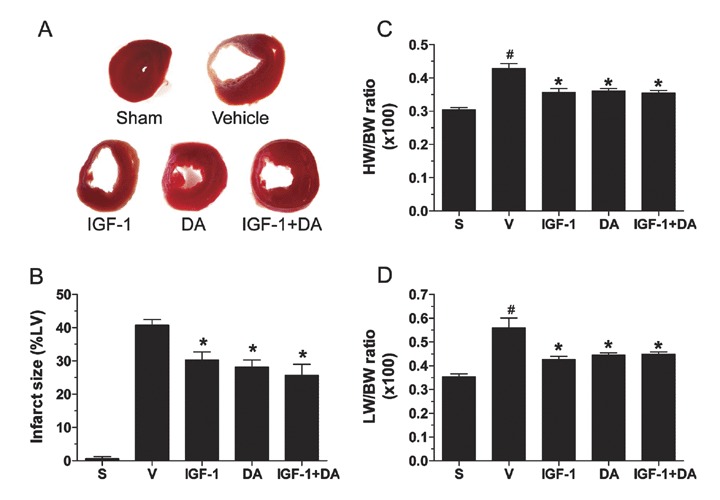
Postischemia infarction size. (A) Representative photographs of TTC‐stained rat heart sections from the different treatment groups. (B) LV infarct size expressed as percentage of the LV in each group. (C) Heart weight (HW)‐to‐body weight (BW) ratio. (D) Lung weight (LW)‐to‐body weight (BW) ratio. (n= 4–22/group; *p < 0.05 vs. vehicle, # p < 0.05 vs. sham; S, sham, V, vehicle.)
Heart weight‐to‐body weight (HW//BW) and lung weight‐to‐body weight (LW/BW) ratios were significantly enhanced in the vehicle compared to sham group. IGF‐1, DA, and a combination of IGF‐1 and DA administration led to a significant reduction of cardiac and lung mass compared to vehicle (p < 0.001). Moreover, it is interesting to note that these same ratios for the drug‐treated groups were not different from the sham group. Thus, it is clear that IGF‐1, DA, or a combination of IGF‐1 and DA leads to cardioprotection and altered remodeling.
3. Vessel density is enhanced by the different pharmacological treatments
Vessel density was assessed via von Willebrand factor (vWF) immunohistochemical stainings. This protein was chosen since small (new or old) capillaries and microcapillaries bear this protein 40 , 41 whereas smooth‐muscle actin does not on small vessels. Stainings were done following 4 weeks of coronary occlusion. Representative photographs of stained sections are shown in Figure 7A (infarct border‐zone, anterior wall). As seen, vehicle (b), IGF‐1 (c), DA (d), and a combination of both drugs (e) reveal more vessels than sham (a). Additionally, quantitative analysis has demonstrated that vehicle had more vessels in the anterior wall than sham, this being the result of the ischemic insult to the ventricle. Moreover, pharmacological treatments had a profound effect on the number of vessels in the infarct border zone (Figure 7B). Indeed, both drugs alone led to a significant enhancement of the number of vessels in that region. Also, DA led to even more vessels than IGF‐1 alone. Furthermore, the combination of both drugs gave rise to a significantly higher number of vessels compared to either IGF‐1 or DA alone, demonstrating the powerful synergistic effect of the combination of both drugs following 4 weeks of ischemia.
Figure 7.
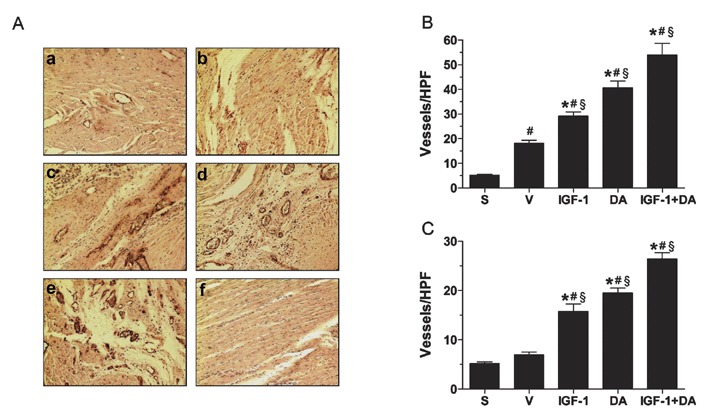
Vessel density quantification. (A) Representative photographs of von Willbrand factor (vWF)‐stained tissue sections from sham (a), vehicle (b), IGF‐1 (c), DA (d), and IGF‐1 + DA (e) groups as well as a negative control (f). The vWF is stained in brown with a counterstaining with hematoxylin in purple. (B) Quantification of vessels in the infarct border zone. (C) Quantification of vessels in the remote area (septal wall). (n= 5–8/group; *p < 0.05 vs. vehicle, # p < 0.05 vs. sham, § p < 0.05 vs. other pharmacological treatments; S, sham, V, vehicle, HPF, high power field.)
The nonischemic, remote septal wall was also analyzed. As seen in Figure 7C, sham and vehicle had similar vessel density. However, IGF‐1 or DA treatment alone led to a significant increase of vessel number in the septal wall compared to vehicle or sham but being similar between each other. Most amazingly, simultaneous administration of IGF‐1 and DA resulted in a higher number of vessels. Again, this effect is synergistic when compared to either drug alone. Taken together, these results suggest that either treatment with IGF‐1 or DA leads to enhanced angiogenesis and vessel density in the infarcted myocardium, both around the infarct itself and in the nonischemic, remote area. Besides, simultaneous therapy with IGF‐1 and DA enhances these processes synergistically.
Hematocrit (both models)
Hematocrit in both the I/R (Figure 8A) and permanent coronary occlusion (Figure 8B) models were significantly enhanced in the DA alone and IGF‐1 + DA groups. On the other hand, hematrocrit levels were not changed in the sham, vehicle, or in the IGF‐1 alone groups. DA being an hematopoietic drug, 21 higher hematrocrit levels are the result of the drug administration. It is important to note that this increase in hematocrit does not take place until 3 days after treatment with DA (data not shown). Thus, the cardioprotection seen is independent of a rise in hemoglobin since a reduction of infarct size is seen before this time period.
Figure 8.
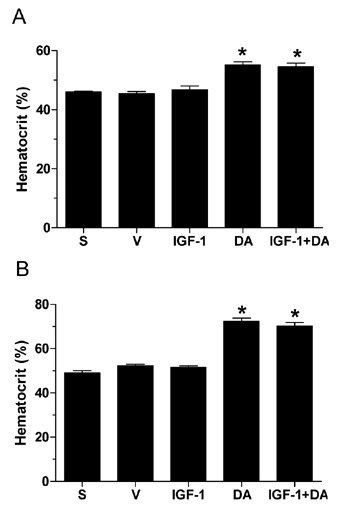
Hematocrit content. (A) Whole blood hematocrit content measured at 72 hours of reperfusion. (B) Whole blood hematocrit content measured at 4 weeks of myocardial ischemia. (n= 4–20/group; *p < 0.05 vs. vehicle, sham, and IGF‐1; S, sham, V, vehicle.)
Discussion
In this study, we have demonstrated the cardioprotective properties of IGF‐1 and DA, as well as the combination of both drugs. Several studies have investigated this cardioprotection by IGF‐1 34 , 36 or DA. 22 , 23 , 24 , 25 , 42 , 43 , 44 However, limited information has been provided regarding post‐I/R or permanent coronary occlusion cardiac function, apoptosis, and angiogenesis, as well as the effect of the combination of these two drugs. We report that either IGF‐1 or DA alone, as well as a combination of IGF‐1 and DA, lead to cardioprotection, reducing infarct size and myocardial apoptosis, and enhancing cardiac function. A novel finding is that the combination of IGF‐1 and DA leads to a synergistic effect on angiogenesis.
Like other growth factors, IGF‐1 has been previously shown to be protective in I/R settings when administered before the ischemia or in early reperfusion with a dose as low as 1 μg per animal. 34 , 36 We therefore based our experiment on these previous studies but modified the regimen to 1 μg/kg (or approximately 0.2–0.4 μg per animal). In both MI and I/R injury, infarct size was significantly reduced by the IGF‐1 therapy and, consequently, we demonstrated that an even lower dose of IGF‐1 as previously used could lead to cardioprotection. Similar to IGF‐1, administration of the erythropoiesis‐stimulating protein DA also imparts cardioprotection. Our group has already demonstrated the potent cardiac protection afforded by DA when given before the ischemia, at the ischemia, at the reperfusion, in early reperfusion, and even 24 hours after induction of reperfusion. 22 Similar to these results, LV infarct size was significantly reduced by administration of DA. Simultaneous administration of IGF‐1 and DA also led to a significant lowering of the infarct size compared to vehicle; however, it was not different from the reduction in LV infarct size observed with either agent alone. This could be explained by the fact that both drugs lead to maximal infarct size reduction and cardioprotection with the dosage used. Perhaps lower doses could have been employed to generate intermediate infarction sizes by the drugs alone and IGF‐1 and DA treatment may have produced a synergistic effect.
The smaller infarcts with IGF‐1 and DA led to enhanced cardiac function. This was seen both after 3 days of reperfusion (I/R model) and more importantly after 4 weeks of coronary occlusion (MI model). Interestingly, cardiac contractility is only enhanced after isoproterenol administration and was similar to sham values when treated with cardioprotective regimens. This is explained by the fact that contractile reserve in response to β‐adrenergic stimulation is preserved after IGF‐1 and or DA treatments.
Apoptosis, a key modulator of reperfusion injury, 45 , 46 was also reduced in our model of pharmacological cardioprotection. For IGF‐1 and DA alone as well as with the combination of both agents, this was observed with both TUNEL staining and caspase‐3 activity assay. IGF‐1 has been shown previously to reduce apoptosis in I/R. 34 , 36 Deficiency in that growth factor promotes apoptosis following myocardial infarction, 47 demonstrating the powerful effect of IGF‐1 in modulating myocardial apoptosis. As well, DA has been shown to reduce apoptosis following I/R. 22 , 23 , 42 , 43 Again, similar to the infarction limitation, the anti‐apoptotic effect seems to be maximal with either drug alone. Indeed, even if the combination of IGF‐1 and DA did reduce apoptosis significantly compared to vehicle, this decrease was not different from the drugs alone.
Previous reports have demonstrated that IGF‐1 is protective in the setting of permanent coronary occlusion. Li et al. 48 showed that overexpression of IGF‐1 can prevent the deleterious remodeling following coronary occlusion, thus reducing ventricular dilation, loading, and hypertrophy. Matthews and collaborators 49 have also reported a beneficial effect of IGF‐1 in an ovine model of heart failure where IGF‐1 increased cardiac function.
DA has been shown to be protective in a similar model of permanent coronary occlusion. 24 This group reported that an early DA administration (just after the occlusion with and without weekly administration) leads to a reduced infarction. This infarction reduction was to the same extent as that seen in our study (approximately 25%). However, this group used a higher DA dose compared to our study (initially 40 μg/kg and 40 μg/kg every 3 weeks in Ref. 24 vs. 20μg/kg and 10 μg/kg every week in our study). On the other hand, the same group showed that late administration of DA (3 weeks after occlusion) did not alter infarction size, 25 therefore demonstrating that timing of administration is critical for infarct size modulation.
Of interest, physiological parameters of heart failure (HW/BW and LW/BW ratios) were normalized by the IGF‐1 and DA therapies. Indeed, the vehicle group had significant hypertrophy (higher HW/BW ratio) and index of failure (higher LW/BW ratio) compared to sham group. Drug treatments led to significant reduction of these two ratios, compared to vehicle, to values not different from sham. Consequently, these data indicate that pharmacological drug administration normalizes cardiac hypertrophy and heart failure and appears to protect against deleterious remodeling. This is probably due to powerful and acute cardioprotection as reduced infarcts do allow for a more viable myocardium, which can no doubt lead to this chronic improvement in cardiac function. An interesting novel finding of this study that could also lead to improved cardiac function and less LV remodeling was that both drugs enhanced apparent neoangiogenesis as measured by vessel number. Interestingly, the vehicle group also had a small but significant increase in vessel number in that area, probably due to infarct scarring, shrinking, and repair. In the remote area (septal wall), the drug therapies also led to increased vessel number with similar enhancement as observed in the infarct border‐zone. Of note, a study by Duerr et al. 50 failed to show an increase of vessel density in the infarcted heart following coronary occlusion and 2 weeks of continuous IGF‐1 administration via mini‐pump implantation. This group used a dose higher than that used in our study (3 mg/kg/day) and they started the administration of the compound ≥24 hours after coronary occlusion, a time point where IGF‐1 cannot impart myocardial protection (at least on infarct size). On the other hand, DA has been reported to enhance vessel density. 24 , 25 Interestingly, they reported that this density was only “normalized” to sham values. In contrast, we observed enhanced vessel density in the DA‐treated group compared to sham animals. Differences in drug regimen, timing of administration, and protocol could at least in part explain the discrepancies between our group and others. More studies would be needed to resolve these issues. However, our results clearly show that IGF‐1, DA, and the combination of both drugs can lead to enhanced vessel density. One could expect these results because of the growth factor nature of both pharmacological compounds, therefore having angiogenic properties and enhancing vessel density following myocardial ischemia. Interestingly, the combination of both molecules induced a greater angiogenic response than either treatment alone and results with concurrent therapy were clearly synergistic. More vessels were present both in the infarct border zone and in the remote septal wall after dual therapy. These results could be explained by the fact that two known growth factors are being applied concurrently. Because they act on different receptor targets but activate similar downstream signaling (reviewed in Refs. 51, 52, 53, 54), it would be possible that synergistic angiogenic effects and enhanced vessel density are observed compared to either drug alone.
One potential mechanism for enhanced cardiac function that may go beyond limitation of infarct size is the stimulation of cardiac regeneration via resident or circulation and homing of stem cells. We did not test this possibility in our models and this represents one limitation of our study. IGF‐1 has been reported to activate DNA synthesis in vitro. 55 Moreover, IGF‐1 overexpression in mice increases cardiac stem cell division, delays senescence, and preserves the functional reservoir of these cells. 56 As for DA, it has been shown to promote circulation of progenitor cells. 25 Thus, we cannot exclude the fact that the endothelium and myocardium could have been regenerated and that enhanced vessel density is a direct or indirect result of stem cell stimulation and differentiation.
Conclusion
Results indicate that simultaneous pharmacological treatments with IGF‐1 and DA protect the heart in models of I/R and permanent coronary occlusion. This protection results in reduced infarct size and improved cardiac function via reduced apoptosis and a more viable myocardium. Of interest is the finding that IGF‐1 and DA together lead to a significant effect on angiogenesis in the ischemic heart.
Clinical perspective
In the present study, we have shown that pharmacological therapies using IGF‐1 and DA are beneficial for the reperfused myocardium and the failing heart. Previous studies have shown that these compounds alone can protect the myocardium but limited information is available concerning simultaneous administration of these (or any other) compounds. As reported, concurrent administration of IGF‐1 and DA led to synergistic effects regarding angiogenesis. Of note, IGF‐1 effect on the ischemic heart was more modest compared to DA regarding the improvement in cardiac function. However, a combination of these two peptides was beneficial and improved the function of the reperfused and ischemic hearts.
One should consider the combination of cardioprotective drugs for future studies. Indeed, limited protection might be provided with one drug, but the combination of two different compounds aiming at similar or completely different targets might lead to significantly enhanced heart function improvement in comparison with only one compound. Combinatory therapy is therefore of great clinical interest to ultimately improve the morbidity and mortality of patients suffering from ischemic heart disease.
Overall, this study provides us with clear evidence that pharmacological compounds concurrent to either reperfusion therapy or classic heart failure treatment could be used, leading to beneficial outcomes in patients with myocardial infarction.
Acknowledgments
This study was supported in part by a National Institutes of Health (NIH) grant R01 HL56205 (to W. J. K.). M. B. is supported by an American Heart Association postdoctoral fellow award and a Fonds de la Recherche en Santé du Québec postdoctoral scholarship. In addition, DA was provided by Amgen Corporation (Thousand Oaks, CA, USA) as part of a Sponsored Research Agreement (to W. J. K.). W. J. K. is the W.W. Smith Professor of Medicine at Thomas Jefferson University.
References
- 1. Rich MW. Epidemiology, pathophysiology, and etiology of congestive heart failure in older adults. J Am Geriatr Soc. 1994; 45: 968–974. [DOI] [PubMed] [Google Scholar]
- 2. Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovasc Res. 2006; 70: 212–221. [DOI] [PubMed] [Google Scholar]
- 3. Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia–reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)‐pathway. Cardiovas Res. 2004; 61: 448–460. [DOI] [PubMed] [Google Scholar]
- 4. Constantinescu SN, Ghaffari S, Lodish HF. The erythropoietin receptor: structure, activation and intracellular signal transduction. Trends Endocrinol Metab. 1999; 10: 18–23. [DOI] [PubMed] [Google Scholar]
- 5. Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005; 293: 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anagnostou A, Lee ES, Kessimian N, Levinson R, Steiner M. Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proc NatI Acad Sci USA. 1990; 87: 5978–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anagnostou A, Liu Z, Steiner M, et al Erythropoietin receptor mRNA expression in human endothelial cells. Proc NatI Acad Sci USA. 1994; 91: 3974–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R, Xie QW, Smart J, Su‐Rick CJ, Pobre E, Diaz D, Gomez D, Hand C, Coleman T, Cerami A. Erythropoietin mediates tissue protection through an erythropoietin and common β‐subunit heteroreceptor. Proc NatI Acad Sci USA. 2004; 101: 14907–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wright GL, Hanlon P, Amin K, Steenbergen C, Murphy E, Arcasoy MO. Erythropoietin receptor expression in adult rat cardiomyocytes is associated with an acute cardioprotective effect for recombinant erythropoietin during ischemia‐reperfusion injury. FASEB J. 2004; 18: 1031–1033. [DOI] [PubMed] [Google Scholar]
- 10. Erbayraktar S, Yilmaz O, GÃkmen N, Brines M. Erythropoietin is a multifunctional tissue‐protective cytokine. Curr Hematol Rep. 2004; 2: 465–470. [PubMed] [Google Scholar]
- 11. Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM. Erythropoietin administration protects retinal neurons from acute ischemia‐reperfusion injury. Proc NatI Acad Sci USA. 2002; 99: 10659–10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sakanaka M, Wen TC, Matsuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc NatI Acad Sci USA. 1998; 95: 4635–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi M, Nielsen J, Gerwien J, Kallunki P, Larsen AK, Helboe L, Christensen S, Pedersen LO, Nielsen M, Torup L, Sager T, Sfacteria A, Erbayraktar S, Erbayraktar S, Gokmen N, Yilmaz O, Cerami‐Hand C, Xie QW, Coleman T, Cerami A, Brines M. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004; 305: 239–242. [DOI] [PubMed] [Google Scholar]
- 14. Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S, Bigini P, Barbera S, Fumagalli E, Mennini T, Vezzani A, Rizzi M, Coleman T, Cerami A, Brines M, Ghezzi P, Bianchi R. Delayed administration of erythropoietin and its non‐erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol. 2006; 172: 27–37. [DOI] [PubMed] [Google Scholar]
- 15. Wang L, Zhang Z, Wang Y, Zhang R, Chopp M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004; 35: 1732–1737. [DOI] [PubMed] [Google Scholar]
- 16. Yasuda Y, Masuda S, Chikuma M, Inoue K, Nagao M, Sasaki R. Estrogen‐dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J Biol Chem. 1998; 273: 25381–25387. [DOI] [PubMed] [Google Scholar]
- 17. Ribatti D, Presta M, Vacca A, Ria R, Giuliani R, Dell‐Era P, Nico B, Roncali L, Dammacco F. Human erythropoietin induces a pro‐angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Blood. 1999; 93: 2627–2636. [PubMed] [Google Scholar]
- 18. Hirata A, Minamino T, Asanuma H, Sanada S, Fujita M, Tsukamoto O, Wakeno M, Myoishi M, Okada K, Koyama H, Komamura K, Takashima S, Shinozaki Y, Mori H, Tomoike H, Hori M, Kitakaze M. Erythropoietin just before reperfusion reduces both lethal arrhythmias and infarct size via the phosphatidylinositol‐3 kinase‐dependent pathway in canine hearts. Cardiovasc Drugs Ther. 2005; 19: 33–40. [DOI] [PubMed] [Google Scholar]
- 19. Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, Thompson RB, Petrofski JA Annex BH, Stamler JS, Koch WJ. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003; 112: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parsa CJ, Kim J, Riel RU, Pascal LS, Thompson RB, Petrofski JA, Matsumoto A, Stamler JS, Koch WJ. Cardioprotective effects of erythropoietin in the reperfused ischemic heart: a potential role for cardiac fibroblasts. J Biol Chem. 2004; 279: 20655–20662. [DOI] [PubMed] [Google Scholar]
- 21. Macdougall IC, Gray SJ, Elston O, Breen C, Jenkins B, Browne J, Egrie J. Pharmacokinetics of novel erythropoiesis stimulating protein compared with epoetin alfa in dialysis patients. J Am Soc Nephrol. 1999; 10: 2392–2395. [DOI] [PubMed] [Google Scholar]
- 22. Gao E, Boucher M, Chuprun JK, Zhou RH, Eckhart AD, Koch WJ. Darbepoetin alfa, a long‐acting erythropoietin analog, offers novel and delayed cardioprotection for the ischemic heart. Am J Physiol. 2007; 293: H60–H68. [DOI] [PubMed] [Google Scholar]
- 23. Prunier F, Pfister O, Hadri L, Liang L, Del Monte F, Liao R, Hajjar RJ. Delayed erythropoietin therapy reduces post‐MI cardiac remodeling only at a dose that mobilizes endothelial progenitor cells. Am J Physiol. 2007; 292: H522–H529. [DOI] [PubMed] [Google Scholar]
- 24. Van Der Meer P, Lipsic E, Henning RH, Boddeus K, Van Der Velden J, Voors AA, Van Veldhuisen DJ, Van Gilst WH, Schoemaker RG. Erythropoietin induces neovascularization and improves cardiac function in rats with heart failure after myocardial infarction. J Am Coll Cardiol. 2005; 46: 125–133. [DOI] [PubMed] [Google Scholar]
- 25. Westenbrink BD, Lipsic E, Van Der Meer P, Van Der Harst P, Oeseburg H, Du Marchie Sarvaas GJ, Koster J, Voors AA, Van Gilst WH, Schoemaker RG. Erythropoietin improves cardiac function through endothelial progenitor cell and vascular endothelial growth factor mediated neovascularization. Eur Heart J. 2007; 28: 2018–2027. [DOI] [PubMed] [Google Scholar]
- 26. Rafiee P, Shi Y, Su J, Pritchard Jr. KA , Tweddell JS, Baker JE. Erythropoietin protects the infant heart against ischemia‐reperfusion injury by triggering multiple signaling pathways. Basic Res Cardiol. 2005; 100: 187–197. [DOI] [PubMed] [Google Scholar]
- 27. Verdier F, Chretien S, Billat C, Gisselbrecht S, Lacombe C, Mayeux P. Erythropoietin induces the tyrosine phosphorylation of insulin receptor substrate‐2. An alternate pathway for erythropoietin‐induced phosphatidylinositol 3‐kinase activation. J Biol Chem. 1997; 272: 26173–26178. [DOI] [PubMed] [Google Scholar]
- 28. Ren J, Samson WK, Sowers JR. Insulin‐like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol. 1999; 31: 2049–2061. [DOI] [PubMed] [Google Scholar]
- 29. LeRoith D. Insulin‐like growth factors. New Engl J Med. 1997; 336: 633–640. [DOI] [PubMed] [Google Scholar]
- 30. Wang PH. Insulin‐like growth factor 1 and the heart In: Levin ER, Nadler JL, eds. Endocrinology of Cardiovascular Function. Boston : Kluwer Academic Publishers; 1998: 225–234. [Google Scholar]
- 31. Wang L, Ma W, Markovitch R, Chen JW, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin‐like growth factor I. Circ Res. 1998; 83: 516–522. [DOI] [PubMed] [Google Scholar]
- 32. Hong F, Kwon SJ, Jhun BS, Kim SS, Ha J, Kim SJ, Sohn NW. Kang C, Kang I. Insulin‐like growth factor‐1 protects H9c2 cardiac myoblasts from oxidative stress‐induced apoptosis via phosphatidylinositol 3‐kinase and extracellular signal‐regulated kinase pathways. Life Sci. 2001; 68: 1095–1105. [DOI] [PubMed] [Google Scholar]
- 33. Mehrhof FB, MÃller FU, Bergmann MW, Li P, Wang Y, Schmitz W, Dietz R, Von Harsdorf R. In cardiomyocyte hypoxia, insulin‐like growth factor‐I‐induced antiapoptotic signaling requires phosphatidylinositol‐3‐OH‐kinase‐dependent and mitogen‐activated protein kinase‐dependent activation of the transcription factor cAMP response element‐binding protein. Circulation. 2001; 104: 2088–2094. [DOI] [PubMed] [Google Scholar]
- 34. Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K, Lefer AM. Cardioprotective effect of insulin‐like growth factor I in myocardial ischemia followed by reperfusion. Proc NatI Acad Sci USA. 1995; 92: 8031–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamamura T, Otani H, Nakao Y, Hattori R, Osako M, Imamura H. IGF‐I differentially regulates Bcl‐xL and Bax and confers myocardial protection in the rat heart. Am J Physiol. 2001; 280: H1191–H1200. [DOI] [PubMed] [Google Scholar]
- 36. Yamashita K, Kajtsura J, Discher DJ, Wasserlauf BJ, Bisphoric NH, Anversa P, Webster KA. Reperfusion‐activated Akt kinase prevents apoptosis in transgenic mouse overexpressin insulin‐like growth factor‐1. Circ Res. 2001; 88: 609–614. [DOI] [PubMed] [Google Scholar]
- 37. Davani E, Brumme Z, Singhera G, Cote H, Harrigan PR, Dorscheid D. Insulin‐like growth factor‐1 protects ischemic murine myocardium from ischemia/reperfusion associated injury. Crit Care. 2003; 7: R176–R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pleger ST, Remppis A, Heidt B, Volkers M, Chuprun JK, Kuhn M, Zhou RH, Gao E, Szabo G, Weichenhan D, Muller OJ, Eckhart AD, Katus HA, Koch WJ, Most P. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Ther. 2005; 12: 1120–1129. [DOI] [PubMed] [Google Scholar]
- 39. Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PML, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004; 114: 1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith JM, Meinkoth JH, Hochstatter T, Meyers KM. Differential distribution of von Willebrand factor in canine vascular endothelium. Am J Vet Res. 1996; 57: 750–755. [PubMed] [Google Scholar]
- 41. Page C, Rose M, Yacoub M, Pigott R. Antigenic heterogeneity of vascular endothelium. Am J Pathol. 1992; 141: 673–683. [PMC free article] [PubMed] [Google Scholar]
- 42. Van Der Meer P, Lipsic E, Henning RH, De Boer RA, Suurmeijer AJH, van, Veldhuisen DJ, Van Gilst WH. Erythropoietin improves left ventricular function and coronary flow in an experimental model of ischemia‐reperfusion injury. Eur J Heart Fail. 2004; 6: 853–859. [DOI] [PubMed] [Google Scholar]
- 43. LipÅic E, Van Der Meer P, Henning RH, Suurmeijer AJH, Boddeus KM, Van Veldhuisen DJ, Van Gilst WH, Schoemaker RG. Timing of erythropoietin treatment for cardioprotection in ischemia/reperfusion. J Cardiovasc Pharmacol. 2004; 44: 473–479. [DOI] [PubMed] [Google Scholar]
- 44. Baker JE. Erythropoietin mimics ischemic preconditioning. Vascul Pharmacol. 2005; 42: 233–241. [DOI] [PubMed] [Google Scholar]
- 45. Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994; 94: 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996; 79: 949–956. [DOI] [PubMed] [Google Scholar]
- 47. Palmen M, Daemen MJ, Bronsaer R, Dassen WRM, Zandbergen HR, Kockx M, Smits JFM, Van Der Zee R, Doevendans PA. Cardiac remodeling after myocardial infarction is impaired in IGF‐1 deficient mice. Cardiovasc Res. 2001; 50: 516–524. [DOI] [PubMed] [Google Scholar]
- 48. Li B, Setoguchi M, Wang X, Andreoli AM, Leri A, Malhotra A, Kajstura P, Anversa P. Insulin‐like growth factor‐1 attenuates the detrimental impact of nonocclusive coronary artery constriction on the heart. Circ Res. 1999; 84: 1007–1019. [DOI] [PubMed] [Google Scholar]
- 49. Matthews KG, Devlin GP, Stuart SP, Jensen JA, Doughty RN, Conaglen JV, Bass JJ. Intrapericardial IGF‐1 improves cardiac function in an ovine model of heart failure. Heart Lung Circ. 2005; 14: 98–103. [DOI] [PubMed] [Google Scholar]
- 50. Duerr RL, Huang S, Miraliakbar HR, Clark R, Chien KR, Ross Jr. J . Insulin‐like growth factor‐1 enhances ventricular hypertrophy and function during the onset of experimental cardiac failure. J Clin Invest. 1995; 95: 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fisher JW. Erythropoietin: physiology and pharmacology update. Exp Biol Med. 2003; 228: 1–14. [DOI] [PubMed] [Google Scholar]
- 52. Yoshimura A, Misawa H. Physiology and function of the erythropoietin receptor. Curr Opin Hematol. 1998; 5: 171–176. [DOI] [PubMed] [Google Scholar]
- 53. Hoover HE, Thuerauf DJ, Martindale JJ, Glembotski CC. β‐crystallin gene induction and phosphorylation by MKK6‐activated p38. J Biol Chem. 2000; 275: 23825–23833. [DOI] [PubMed] [Google Scholar]
- 54. Suleiman MS, Singh RJR, Stewart CEH. Apoptosis and the cardiac action of insulin‐like growth factor I. Pharmacol Ther. 2007; 114: 278–294. [DOI] [PubMed] [Google Scholar]
- 55. Reiss K, Cheng W, Pierzchalski P, Kodali S, Li B, Wang S, Liu Y, Anversa P. Insulin‐like growth factor‐1 receptor and its ligand regulate the reentry of adult ventricular myocytes into the cell cycle. Exp Cell Res. 1997; 235: 198–209. [DOI] [PubMed] [Google Scholar]
- 56. Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, Urbanek K, Nadal‐Ginard B, Kajstura J, Anversa P, Leri A. Cardiac stem cell and myocyte aging, heart failure, and insulin‐like growth factor‐1 overexpression. Circ Res. 2004; 94: 514–524. [DOI] [PubMed] [Google Scholar]