

Specific modification of functional groups in aminoglycosides poses a significant synthetic challenge. In this report, a chemoenzymatic route for modification of aminoglycosides is disclosed. The critical feature of this approach is the discovery that the aminoglycoside 3-N-acetyltransferase AAC(3)-IV from Escherichia coli [1] accepts azido acetyl coenzyme A (AzAcCoA) as a substrate similarly as the natural substrate, acetyl coenzyme A (AcCoA). After enzymatic delivery of an azido acetyl group, it can be chemically modified via a Huisgen dipolar cycloaddition reaction (HDCR)[2] enabling further diversification. Thus, this method accelerates access to modified compounds with diversity beyond that which can be installed directly via AAC(3) and a modified CoA thioester. The approach was further developed to study modification of aminoglycosides by AAC(3), which causes broad-scale aminoglycoside inactivation, using a fluorescence-based microarray platform. This platform is a useful analytical tool for the facile identification of both protein and carbohydrate substrates for acetyltransferases, which play critical roles in a multitude of cellular processes.[3]
Aminoglycosides represent one of the largest classes of antibacterials with activity against both gram negative and gram positive bacteria. Many aminoglycosides exert their antibacterial activity by binding to the decoding site (A-site) in 16S rRNA.[4] Binding to this site affects recognition of cognate and non-cognate tRNAs by the ribosome. As is common with all antibiotics, resistance to aminoglycosides has been emerging since their initial introduction as therapeutic agents.[5] One of the most important resistance-causing mechanisms towards aminoglycosides is enzymatic modification, for example by acetyltransferases (AAC’s). Aminoglycosides have also been used to facilitate translational read-through in diseases caused by non-sense mutations including Duchenne’s muscular dystrophy, cystic fibrosis, and hemophilia.[6] To better exploit these targets, simple access to modified aminoglycosides with improved biological activity, such as reduced susceptibility to modification by resistance-causing enzymes or more specific recognition of target RNAs, is needed. Synthesis of designer aminoglycosides, however, is difficult because they contain a variety of amino and hydroxyl groups with similar reactivities.
In an effort to develop expedited protocols leading to site-specifically derivatized aminoglycosides, we studied the ability of AAC(3) to accept modified acetyl coenzyme A substrates to install functional groups that can be further diversified chemically. Towards this end, AzAcCoA was synthesized and its ability to acylate the aminoglycoside apramycin was studied (Figure 1A). AzAcCoA was used because the azido-group can be diversified by reaction with alkynes using a Cu(I)-catalyzed HDCR. Modification of apramycin by AzAcCoA was monitored by UV-vis[1] and mass spectrometry, and each study unambiguously showed that AzAcCoA is accepted as a substrate (Figure 1, B and C). In fact, AzAcCoA is accepted in a similar manner as the natural substrate. For example, AzAcCoA has a KM and a kcat that are 4-fold and 3-fold lower, respectively, than AcCoA (Figure 1B). The specificity constants (kcat/KM) are similar; 5. 1 × 105 M−1s−1 for AcCoA and 6. 3 ×105 M−1s−1 for AzAcCoA. In contrast, when propionyl CoA, malonyl CoA, and butyryl CoA substrates were studied, they had kcat/KM’s that are 7-, 23-, and 1,730-fold reduced relative to AcCoA, respectively.[1] Another recent and extensive study has shown that a variety of aminoglycosides can be modified by CoA derivatives to provide new aminoglycosides. The kcat/KM of the modified CoA substrates in that study are also lower by about 10-fold compared to AcCoA.[7] Thus, the similarity in specificity constants of AzAcCoA and AcCoA as substrates for AAC(3) points to unique features for AzAcCoA interacting with the enzyme.
Figure 1.
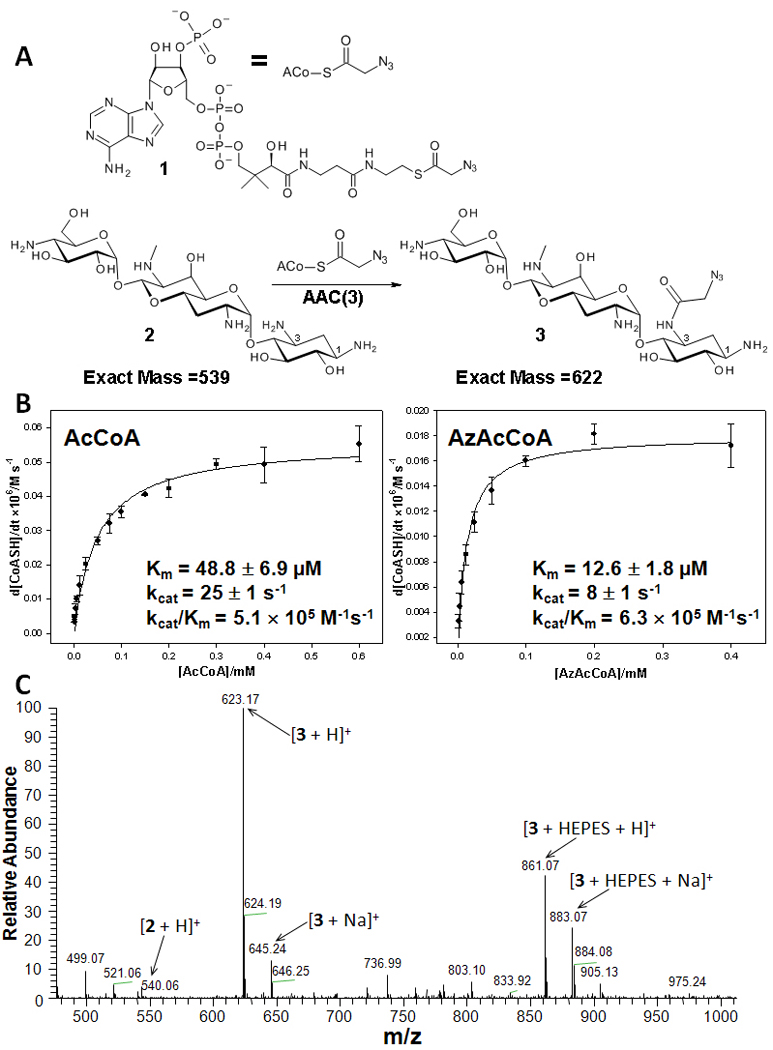
Studying the ability of AAC(3) to accept AzAcCoA as a substrate. A, Scheme for reaction of AzAcCoA with apramycin. B, Michaelis-Menten plot for modification of apramycin by AcCoA and AzAcCoA. C, Mass spectral analysis of reaction of apramycin with AzAcCoA.
Encouraged by the results of enzymatic azidoacetyl transfer, the scope of the 3-N-azidoacetyl-apramycin (3) modification via HDCR was further investigated. In initial studies, dansyl alkyne (4) was used to modify the azidoacetyl aminoglycosides (Figure 2). 3-N-azidoacetyl-apramycin (3) was partially purified by passing the reaction mixture over Dowex ion exchange resin (HO- form) to capture any CoA-containing materials. This material was then subjected to HDCR modification with 4. Mass spectral analysis indicated complete conversion to 5. The reaction was purified by HPLC and 5 was obtained in 44% isolated yield from the two steps.
Figure 2.
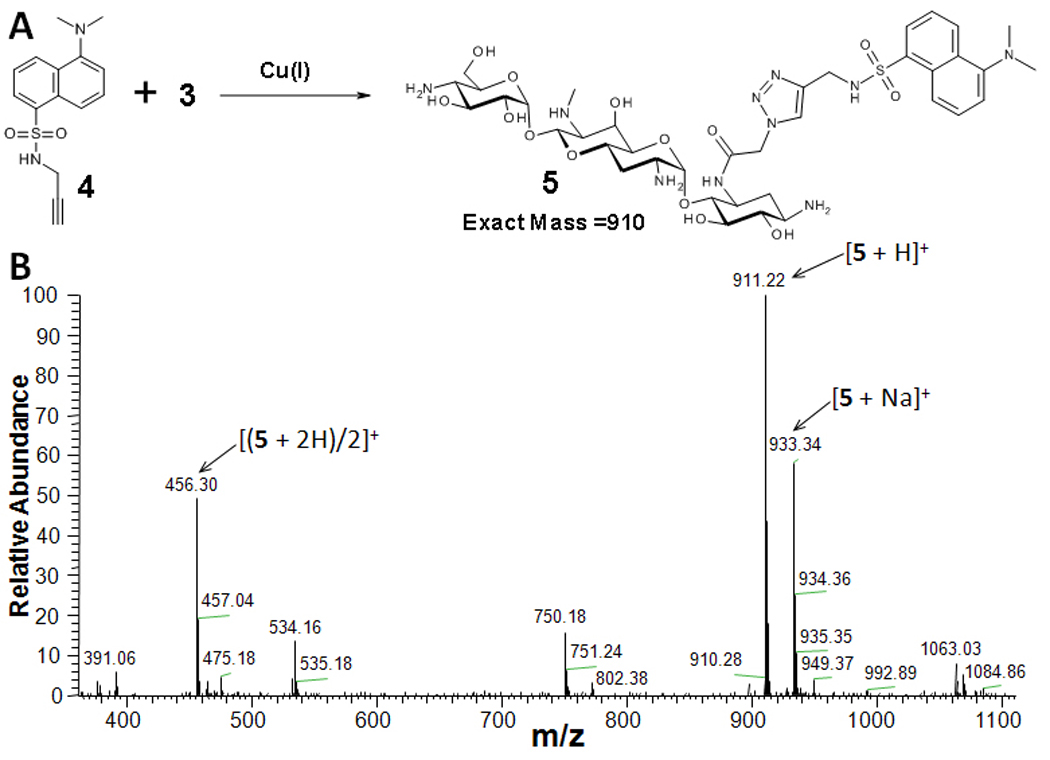
HDCR modification of 3-N-azidoacetyl-apramycin, 3. A, Reaction between 3 and 4. B, Mass spectral analysis of the reaction to form 5.
Armed with these results, a microarray-based method to study the modification of 2-deoxystreptamine aminoglycosides by AAC(3) was developed. In these experiments, the extent of AAC(3) modification on the array surface was monitored by reaction of the generated azidoacetyl aminoglycosides with an alkyne dye via HDCR (Figures 3 and 4).
Figure 3.
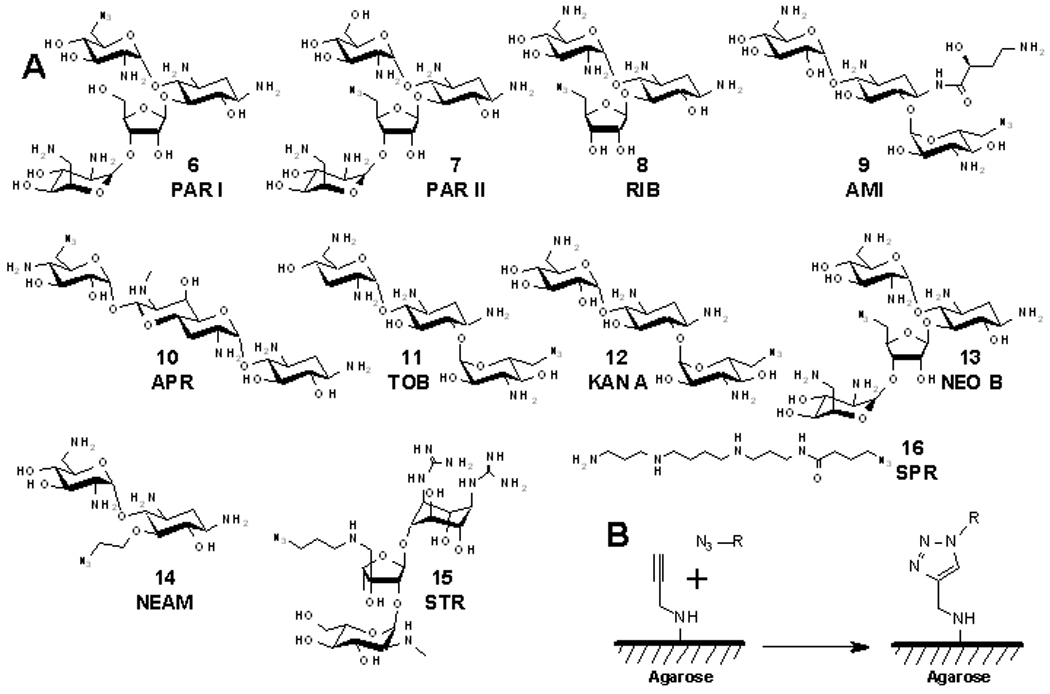
The compounds used to construct microarrays and the microarray immobilization chemistry used in this study. A, Structures of azide-functionalized aminoglycosides 6–15 and spermine 16. B, The HDCR used to anchor azide-containing small molecules onto alkyne-functionalized agarose microarray surfaces. (N3–R represents azide-functionalized compounds 6–16.)
Figure 4.
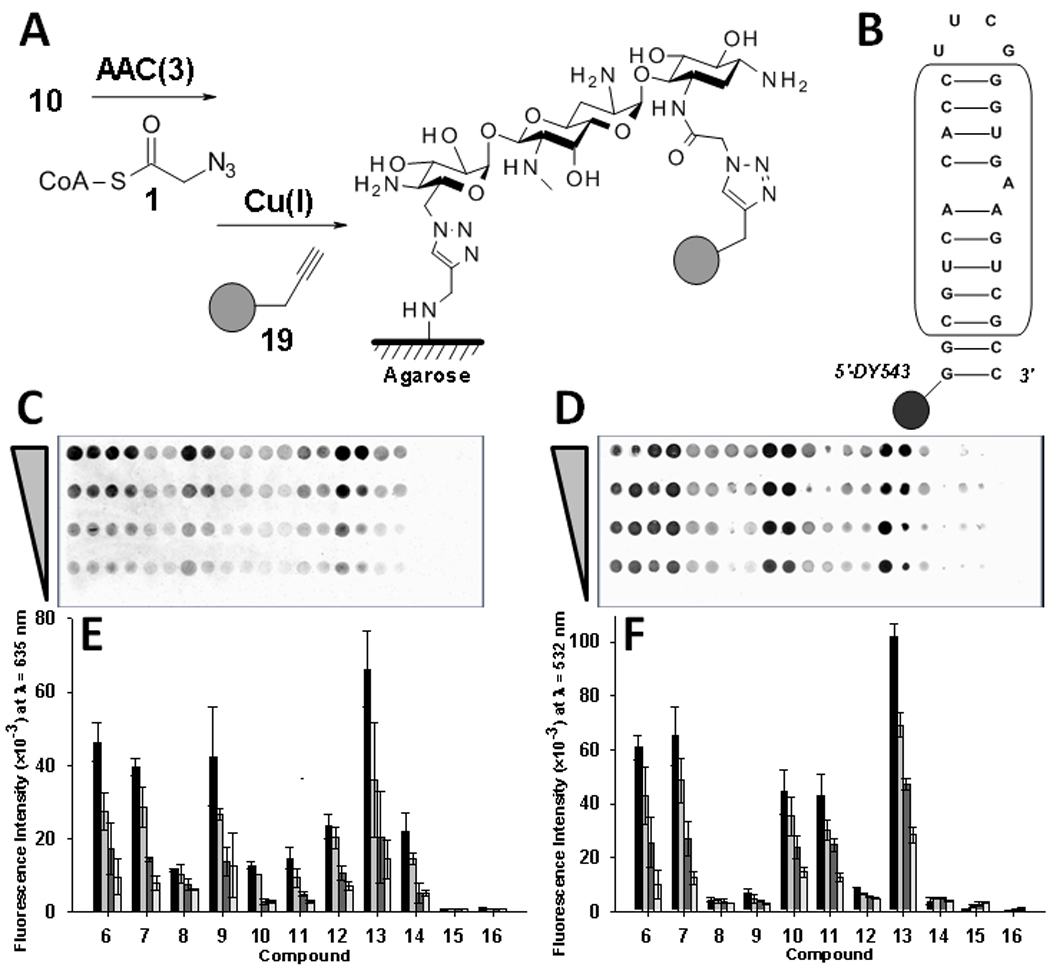
Probing aminoglycoside recognition via microarray. A, Scheme for the labeling reaction. B, Secondary structure of the bacterial rRNA A-site mimic (boxed nucleotides represent those derived from the native sequence). C, Image of a microarray to study AAC(3) modification of aminoglycosides. D, Image of a microarray to study binding of an oligonucleotide mimic of the bacterial rRNA A-site. E, Representative plot of data for AAC(3) modification. F, Representative plot of data for binding of the rRNA A-site mimic. Each group of bars in the plots from left to right corresponds to delivery of 500, 250, 125, 62, and 0 picomoles of material to an array surface.
In order to site-specifically immobilize the aminoglycosides onto a microarray, a small library of azide-modified aminoglycosides was synthesized (Figure 3). These compounds can be anchored onto alkyne-functionalized agarose microarrays.[8] Syntheses of compounds 6–14 were completed according to modification of previously published procedures in which a primary hydroxyl group is activated by reaction with 2,4,6-triisopropylbenzenesulfonyl chloride. The resulting adduct is displaced with sodium azide. [8b, 9] Compound 15 was synthesized via reductive amination of streptomycin with 3-azidopropylamine. Compound 16 was synthesized by acylation of spermine with 4-azidobutyric acid. Structures 6–14 are known to bind to the bacterial rRNA A-site,[10] and most have been studied as AAC(3) substrates.[1] Structures 15 (streptomycin derivative) and 16 (spermine derivative) are not known to be specific bacterial rRNA A-site binders. Compounds 15 and 16 are not expected to be substrates for AAC(3) because previous studies have shown that guanidiniylated aminoglycosides are not modified by acetyltransferases.[11]
The azido-aminoglycosides and spermine derivatives were arrayed onto alkyne-agarose microarrays in the presence of Cu(I). Serial dilutions from 500 to 62 picomoles of compound were delivered to the surface via a pin transfer replicator. After reaction to conjugate the ligands onto an array, the arrays were quenched [12] and then modified with AzAcCoA via AAC(3). After washing the arrays, they were reacted with an alkyne-functionalized BODIPY dye via HDCR (Figure 4A). Scans of modified arrays showed clear spots of modification for 6–14 whereas 15 and 16 were not labeled, as expected (Figure 4C and 4E). Array data for modification by AAC(3) were then compared to previous solution-based studies of modification.[1] In general, the two studies are in agreement. In solution-based studies neomycin B has the highest kcat/KM value followed by tobramycin, paromomycin, apramycin, ribostamycin, kanamycin and amikacin. The order of the extent of modification on the arrays at the highest spot loading is, neomycin B (13) > paromomycin (6 and 7) = amikacin (9) > kanamycin A (12) = neamine (14). Other aminoglycosides are modified to a lesser extent.
Arrays were also probed for binding to a fluorescently labeled mimic of the bacterial rRNA A-site (Figure 4D). The data are in good agreement with a study of aminoglycoside binding to the A-site by SPR.[10] For example, neomycin B (13-like) gives the highest signal for binding to the A-site and is the highest affinity binder in the series (Figure 4F). The second and the third highest binding signals are observed for the paromomycin derivatives (7 and 6), and the apramycin (10) and tobramycin (11) derivatives, respectively. The other aminoglycosides that were expected to bind the A-site gave lower, but measureable signals. The lowest binding signals are observed for the non-specific binders, streptomycin (15) and spermine (16) derivatives, as expected.
Our fluorescence-based approach is advantageous over other microarray approaches that have been developed to probe aminoglycoside modification by resistance-causing enzymes that use radioisotopes. For example, detection of 14C acetylated aminoglycosides requires several days of exposure in a phosphorimager cassette.[13] Furthermore, the resolution of a phosphorimager is significantly diminished relative to that of a fluorescent microarray scanner. Enhanced resolution allows for a significant increase in the number of features (spots) that can be analyzed on a microarray.
During the course of this work, a series of modified aminoglycosides were synthesized using modified coenzyme A derivatives.[7] Interestingly, these modified antibiotics displayed antibacterial activity despite the fact that they were modified by resistance-causing enzymes.[7] The results herein expand that work, demonstrating that aminoglycosides can be modified by coenzyme A’s that install reactive groups that can be easily diversified chemically. Thus, the diversity of modifications introduced into aminoglycosides is not solely limited by the ability of an AAC to accept a modified coenzyme A as a substrate. Rather, reactive tags can be installed on an aminoglycoside via a CoA derivative followed by subsequent chemical modification to provide access to novel compounds.
In summary, a chemoenzymatic route toward the synthesis of aminoglycosides is reported. New compounds synthesized by this method can be used to identify new RNA-ligand interactions via Two-Dimensional Combinatorial Screening (2DCS).[8b, 8c] Our chemo-enzymatic approach has a wide scope as any amine, branched aliphatic group, heterocycle, etc. of any size, bulkiness, and/or formal charge could be conjugated with aminoglycosides via Huisgen cycloaddition reaction. Thus, derivatization of aminoglycosides is independent of CoA-thioester substrate specificity. Therefore, acylation of 3-amine followed by “click” chemistry can be used for preparation of compounds with a triazole-containing linker and a new functionality at the periphery of aminoglycosides. Introduction of additional peripheral amino-group could lead to a new family of compounds similar to amicacin, which posesses an acylated 1-amino group of 2-deoxystreptamine ring with a terminal amine. For example, such modification could be accomplished using propargylamine as an alkyne reagent. That is to say, since amicacin exhibits a diminished modification by resistance enzymes, these 3-amine acylated compounds are of a special importance. Moreover, our chemo-enzymatic approach is synthetically faster and gives greater possibilities for diversification of aminoglycosides in a high-throughput manner.
In addition, this method was used to develop a high throughput and sensitive microarray-based method to study the modification of aminoglycosides by acetyltransferases that confer resistance. These developments have broad applications in both the synthesis of diversified aminoglycosides and in developing microarray-based platforms to probe modification of biomolecules by acetyltransferases, including proteins and carbohydrates whose activities are modulated by acetyltransferases.[3]
Supplementary Material
{kind=link}
Acknowledgements
We thank Prof. Jessica Childs-Disney for critical review of the manuscript and Prof. John Blanchard for the gift of the plasmid encoding AAC(3). This work was supported by the National Institutes of Health (RO1-GM079235). MDD is a Cottrell Scholar from the Research Corporation, a J. D. Watson Young Investigator from NYSTAR, and a Camille and Henry Dreyfus New Faculty Awardee.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
Chemoenzymatic Synthesis
A chemoenzymatic route for modification of aminoglycosides is disclosed. The critical feature of this approach is the discovery that an aminoglycoside 3-N-acetyltransferase AAC(3)-IV from Escherichia coli accepts azido acetyl coenzyme A (AzAcCoA) as a substrate that can be further chemically diversified. This approach was utilized to develop a carbohydrate microarray method to study modification of aminoglycosides.
References
- 1.Magalhaes ML, Blanchard JS. Biochemistry. 2005;44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 3.a) Mellert HS, McMahon SB. Trends Biochem. Sci. 2009;34:571–578. doi: 10.1016/j.tibs.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schauer R. Curr. Opin. Struct. Biol. 2009;19:507–514. doi: 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Moazed D, Noller HF. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]; b) Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]; c) Yoshizawa S, Fourmy D, Puglisi JD. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]; d) Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. Nat. Struct. Mol. Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]; e) Yonath A. ChemBioChem. 2003;4:1008–1017. doi: 10.1002/cbic.200300683. [DOI] [PubMed] [Google Scholar]
- 5.Magnet S, Blanchard JS. Chem. Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 6.Kellermayer R. Eur. J. Med. Genet. 2006;49:445–450. doi: 10.1016/j.ejmg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Green KD, Chen W, Houghton JL, Fridman M, Garneau-Tsodikova S. ChemBioChem. 2010;11:119–126. doi: 10.1002/cbic.200900584. [DOI] [PubMed] [Google Scholar]
- 8.a) Disney MD, Childs-Disney JL. ChemBioChem. 2007;8:649–656. doi: 10.1002/cbic.200600569. [DOI] [PubMed] [Google Scholar]; b) Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. ACS Chem. Biol. 2007;2:745–754. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]; c) Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, Tran T, Velagapudi SP, Wu M, Childs-Disney JL. J. Am. Chem. Soc. 2008;130:11185–11194. doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Tor Y. Angew. Chem. Int. Ed. Engl. 1998;37:109–111. [Google Scholar]
- 10.Wong CH, Hendrix M, Priestley ES, Greenberg WA. Chem. Biol. 1998;5:397–406. doi: 10.1016/s1074-5521(98)90073-4. [DOI] [PubMed] [Google Scholar]
- 11.Disney MD, Magnet S, Blanchard JS, Seeberger PH. Angew. Chem. Int. Ed. Eng. 2004;43:1591–1594. doi: 10.1002/anie.200353236. [DOI] [PubMed] [Google Scholar]
- 12.Arrays were quenched to remove any reactive azide that could have been left on the array surface.Furthemore, control experiments in the presence of AAC(3) that excluded AzAcCoA did not deposit dye onto the surface.
- 13.Barrett OJ, Pushechnikov A, Wu M, Disney MD. Carbohydr. Res. 2008;343:2924–2931. doi: 10.1016/j.carres.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.