

Abstract
Spinocerebellar ataxias 6 and 7 (SCA6 and SCA7) are neurodegenerative disorders caused by expansion of CAG repeats encoding polyglutamine (polyQ) tracts in CACNA1A, the alpha1A subunit of the P/Q-type calcium channel, and ataxin-7 (ATXN7), a component of a chromatin-remodeling complex, respectively. We hypothesized that finding new protein partners for ATXN7 and CACNA1A would provide insight into the biology of their respective diseases and their relationship to other ataxia-causing proteins. We identified 118 protein interactions for CACNA1A and ATXN7 linking them to other ataxia-causing proteins and the ataxia network. To begin to understand the biological relevance of these protein interactions within the ataxia network, we used OMIM to identify diseases associated with the expanded ataxia network. We then used Medicare patient records to determine if any of these diseases co-occur with hereditary ataxia. We found that patients with ataxia are at 3.03-fold greater risk of these diseases than Medicare patients overall. One of the diseases comorbid with ataxia is macular degeneration (MD). The ataxia network is significantly (P= 7.37 × 10−5) enriched for proteins that interact with known MD-causing proteins, forming a MD subnetwork. We found that at least two of the proteins in the MD subnetwork have altered expression in the retina of Ataxin-7266Q/+ mice suggesting an in vivo functional relationship with ATXN7. Together these data reveal novel protein interactions and suggest potential pathways that can contribute to the pathophysiology of ataxia, MD, and diseases comorbid with ataxia.
INTRODUCTION
Spinocerebellar ataxias (SCAs) are a class of neurodegenerative disorders, characterized by the loss of balance, progressive motor dysfunction and degeneration of the cerebellar Purkinje cells (PC). Several SCAs are caused by the expansion of unstable CAG repeats that encode a polyglutamine (polyQ) tract in the respective protein (1). Outside of the CAG tract, the proteins do not share homology. In all of the proteins, the length of the polyQ tract correlates inversely with the age of disease onset (reviewed in 2). The cellular and molecular pathophysiology underlying most of the SCAs is not understood and many of the implicated proteins appear to be functionally distinct (3–14). The common pathology in these diseases, however, raised the possibility that common molecular mechanisms might underlie the pathogenesis of SCAs. To gain insight into the functions of the ataxia-causing proteins and to explore potential common molecular pathways in the SCAs, our laboratory generated a protein interaction network for 23 ataxia-causing proteins and their known interacting partners using a high-throughput yeast two-hybrid (Y2H) screen (15). The ataxia network (interactome) successfully led to the identification of shared pathways and key interactions relevant to the pathophysiology of ataxia (16–26). One of the proteins identified in the original screen and investigated further by our lab was RNA binding motif protein 17 (RBM17). RBM17 is an RNA binding protein that functions in a spliceosome complex (27,28). In vivo studies demonstrated a molecular role for RBM17 in SCA1 gain of function (18). Together these findings highlight the utility and quality of the screen and the ataxia network.
However, a few of the bait proteins failed to identify many interacting partners (15). The proteins that failed had, on average, fewer different bait fragments, and many were only full-length baits. Two of the proteins that failed to identify interactors in the original screen were CACNA1A (zero interactors) and ataxin-7 (three interactors), the disease causing proteins for SCA6 and SCA7, respectively.
Patients with SCA6 have dysarthria, oculomotor abnormalities and an ataxic gait. SCA6 is caused by an expansion of a CAG repeat in CACNA1A, a pore-forming subunit of the P/Q-type calcium channels. There are three reported isoforms of CACNA1A that result from alternative splicing at the C-terminus; however, two of these, MPI and MPc, make up 99% of the transcripts (8,29). These two splice variants differ in the splice acceptor site for the final exon such that MPI includes exon 47 and the CAG coding repeat is translated into a polyglutamine (polyQ) tract in the cytoplasmic tail of the protein, whereas MPc splices to an immediate stop codon, resulting in a shorter cytoplasmic tail lacking the polyQ tract. Since the polyQ tract is in the cytoplasmic tail of the MPI isoform, it is readily available for protein–protein interactions. Furthermore, data from Kordasiewicz et al. (30) suggest that the cytoplasmic tail of CACNA1A can be cleaved and translocated to the nucleus, possibly indicating a novel function for this protein distinct from its role as a channel subunit.
Patients with SCA7 have macular degeneration (MD) in addition to ataxia (31,32). SCA7 is caused by an expansion of a coding CAG repeat near the N-terminus of ataxin-7. Ataxin-7 is evolutionarily conserved with the yeast SAGA complex component, Sgf73, particularly in two blocks within the N-terminal portion of the respective proteins (9,33–35). Ataxin-7 is a component of a STAGA/TFTC histone acetyl transferase complex (9). In mammals, glutamine-expanded ataxin-7 interferes with the proper function of the complex and leads to misregulation of rod photoreceptor gene expression (9,36,37). Interestingly, in mouse and human tissues, ataxin-7 is localized in both the nucleus and cytoplasm and it can shuttle between the nucleus and the cytoplasm in live cells (38–47). There is currently no known functional role for ataxin-7 in the cytoplasm.
We hypothesized that identifying protein partners of CACNA1A and ataxin-7 will provide insight into SCA6 and SCA7 pathogenesis. Boxem et al. (48) demonstrated that using fragments in an Y2H screen increases the sensitivity without reducing the specificity. Therefore, we hypothesized that using partial segments of the coding sequences of both wild-type and polyQ expanded CACNA1A and ataxin-7 would increase the likelihood of identifying interacting partners.
Additionally, we hypothesized that the expanded ataxia interactome could provide insight into ataxia pathogenesis. We present data here integrating the ataxia interactome with patient medical records to uncover molecular associations for diseases comorbid with ataxia. Furthermore, we show that some of the proteins predicted to be relevant for one comorbid disease, MD, are altered in a mouse model of SCA7 exhibiting MD.
RESULTS
Identification of novel ataxin-7 and CACNA1A interacting partners
To identify interacting partners for CACNA1A and ataxin-7, we generated 28 baits representing overlapping fragments of each protein (Fig. 1 and Supplementary Material, Table S1). The fragments of ataxin-7 were chosen based on known evolutionarily conserved or functional domains (9,38,49). CACNA1A baits were generated from the cytoplasmic tail of both isoforms. For the MPI isoform, we used multiple polyQ lengths; 11Q is common in wild-type alleles (one bait was 15Q for the wild-type length due to expansion during cloning), 23Q is disease-causing and 72Q is hyper-expanded (8,29). For clarity, throughout this manuscript, we refer to these as MPI-11Q, MPI-23Q and MPI-72Q, respectively, and MPI refers to this isoform regardless of polyQ length. All of the baits were fused to the DNA binding domain of Gal4 using the Invitrogen Gateway system and were used in a high-stringency Y2H screen against an adult human brain cDNA library. We identified 152 unique interactions with CACNA1A_MPI, CACNA1A_MPc and ataxin-7 (Supplementary Material, Table S2). For CACNA1A, we identified 59 interactors with the MPI isoform and 44 with the MPc isoform; for ataxin-7, we identified 49 interactors. Seventeen prey proteins were shared by both CACNA1A and ataxin-7, and 17 prey proteins interacted with both CACNA1A isoforms; in many instances, we identified multiple independent clones of the same protein. Thus, a total of 118 different prey proteins were identified.
Figure 1.

Baits used to identify proteins that interact with ataxin-7 and CACNA1A. (A) Ataxin-7 baits. Conserved and functional domains are indicated with boxes and an inverted triangle indicates the polyQ tract (10 and 52Q were used). Amino acid range, transformation efficiency and the number of identified proteins are given for all the constructs in Supplementary Material, Table S1. Though all had greater than 7.56 × 105 transformation efficiency, not all baits found interacting partners. (B) CACNA1A baits. The full-length protein is shown with the transmembrane domains and polyQ tract as indicated. The C-terminus of the protein (cytoplasmic tail) is shown in greater detail with the amino acid indicated for the beginning of each fragment. In the MPI isoform, the inverted triangle indicates the polyQ tract (11, 23 and 72Q were used for each construct).
We next sought to recapitulate the interactions in mammalian cells. We therefore tagged the bait and prey proteins with GST and myc, respectively, and tested 47 of 152 of the Y2H-based interactions using glutathione-sepharose affinity co-purifications (GST-APs) in HEK293T cells (15,50,51). Specifically, we were able to recapitulate the interactions for 78.7% of the protein pairs tested (Fig. 2 and Supplementary Material, Table S2). This rate of reproducibility of Y2H interactions in mammalian cells is similar to that seen in other studies and confirms the high quality of the screen (15,50,51).
Figure 2.

Representative western blots of GST-APs recapitulating the interactions identified for various myc-tagged prey proteins. Blotting antibodies are anti-myc and anti-GST as indicated. (A) Ataxin-7 interactors. (B) CACNA1A_MPc interactors.
CACNA1A interacts with several proteins in an isoform-specific manner
Given that only one of the two predominant isoforms of CACNA1A contains the disease-causing CAG tract, we hypothesized that interactions specific to the disease-causing isoform, MPI, may be particularly relevant to pathogenesis. Therefore, we further explored the CACNA1A interactions, specifically searching for proteins that interact in a polyQ length-dependent and/or isoform specific manner. In the initial Y2H screen, we identified 44 different protein interactions with MPc, the short isoform, and 59 interactions with MPI, the polyQ containing isoform. Of these, 17 were identified by both classes of baits. Since the screen was not saturated, some of the interactions may have been found only with one isoform by chance. Cross-testing all of the interactions that were unique to each isoform by a yeast mating assay against the other isoform revealed that 71 of 86 proteins interacted with both isoforms, whereas 15 of the interactions were exclusive to the MPI (glutamine-containing) isoform in yeast. None of the proteins identified was unique to MPc, consistent with MPc containing domains that are also present in MPI (Fig. 1B).
We then specifically tested the 15 MPI partners by GST-AP to explore these interactions in a mammalian system (Table 1, Fig. 3). Of the 15, 5 are not expressed at detectable levels in this system and therefore could not be evaluated. Of the remaining 10 that interact exclusively with MPI in the Y2H system, 6 proved to interact with both CACNA1A isoforms in mammalian cells (Fig. 3A and B). Three of these, RBM12B, LPHN1 and SIAHBP1, preferentially interact with the pathogenic MPI-23Q and hyper-expanded MPI-72Q GST fusion and also with MPc, but not with the wild-type length MPI-11Q construct. This suggests that the polyglutamine expansion within the MPI isoform may cause aberrant protein–protein interactions with MPc interacting partners. Another 3, of the 10 proteins tested (ABI1, BZRAP1, YLPM1), recapitulate the Y2H interactions and interact only with the MPI isoforms (Fig. 3C and D). Furthermore, ABI1 and YLPM1 preferentially interact with the disease-causing MPI-23Q and hyper-expanded MPI-72Q polyQ form and not with the wild-type length MPI-11Q.
Table 1.
CACNA1A isoform specific interactions identified by Y2H and recapitulated by GST-AP in mammalian cells
| Entrez ID | HGNC Symbol | MPc | Wild-type MPI-11Q, or MPI-15Q | Pathogenic MPI-23Q or MPI-72Q |
|---|---|---|---|---|
| Y2H | ||||
| 10006 | ABI1 | No | + | + |
| 9256 | BZRAP1 | No | + | + |
| 114897 | C1QTNF1 | No | + | + |
| 9793 | CKAP5 | No | + | No |
| 51232 | CRIM1 | No | + | No |
| 25822 | DNAJB5 | No | + | No |
| 8997 | KALRN | No | + | + |
| 22859 | LPHN1 | No | + | No |
| 389677 | RBM12B | No | + | + |
| 23504 | RIMBP2 | No | + | + |
| 22827 | SIAHBP1 | No | + | No |
| 8848 | TSC22D1 | No | + | No |
| 7407 | VARS | No | + | No |
| 56252 | YLPM1 | No | + | + |
| 353355 | ZNF233 | No | + | No |
| GST-AP | ||||
| 10006 | ABI1 | No | No | + |
| 9256 | BZRAP1 | No | + | + |
| 51232 | CRIM1 | + | + | + |
| 25822 | DNAJB5 | + | No | No |
| 22859 | LPHN1 | + | No | + |
| 389677 | RBM12B | + | No | + |
| 22827 | SIAHBP1 | + | No | + |
| 8848 | TSC22D1 | + | + | + |
| 7407 | VARS | + | + | + |
| 56252 | YLPM1 | No | No | + |
Figure 3.

Representative western blots of GST-APs for proteins that interacted with the CACNA1A_MPI but not CACNA1A_MPc isoform by Y2H mating assay (Table 1). The myc-tagged prey and GST-tagged baits are indicated (empty vector, MPc or MPI polyQ length). Blotting antibodies are anti-myc and anti-GST as indicated. (A) The myc-TSC22D1 interaction is neither isoform nor polyQ length dependant in mammalian cells. (B) The myc-RBM12B interaction with CACNA1A is not isoform specific, but for MPI is polyQ length dependant, interacting with the disease-causing 23Q and hyper-expanded 72Q but not the wild-type 11Q baits. (C) The myc-BZRAP1 interaction with CACNA1A is isoform, but not polyQ length dependant. (D) The myc-YLPM1 interaction with CACNA1A is both isoform and polyQ length dependant.
Analysis of the ataxia network including CACNA1A and ataxin-7 partners
To learn about the biological properties of the new ataxia network, it is necessary to analyze its topological properties as they typically bring useful insight into organizational principles of biological networks (52,53). We explored the network properties of the ataxia interactome, including its degree distribution, which is the distribution of the number of proteins each protein interacts with, the identification of proteins with high betweenness centrality (a measure of the importance of a node to the overall network) and enrichment of ataxia subnetworks (54). Our previously published network looked at the degree distribution of an expanded network which included the literature-curated interactions to allow comparison of the degree distribution to that of other interactome studies of model organisms (15). Here we generated the ataxia interactome solely based on data generated from the Y2H screen we performed (Fig. 4) and did not include literature-curated interactions to avoid biases emerging from well-studied proteins (55). This ataxia interactome has a noisy degree distribution even with the logarithmic binning due to less rich statistics compared with the literature-expanded network. Yet, the distribution is still consistent with a power law, in agreement with previously found scaling exponent −2.2 (Supplementary Material, Fig. S1) indicating the importance of hubs in the ataxia network.
Figure 4.

Ataxia interaction network. Red nodes are ataxia-causing proteins. CACNA1A and ataxin-7, screened in this study, are outlined in orange. Newly identified interactors are light blue and previously reported interactors are dark blue. The properties of this network are described in Supplementary Material, Figure S1 and Table S3.
The proteins with the highest betweenness centrality in the interactome tend to act as bridges connecting functional modules (56). In this network, CACNA1A emerges as one of these proteins. The difference between the previously published ataxia interactome and the new updated network is due to the large number of newly identified interactors for CACNA1A. This result emphasizes the importance of multiple, overlapping protein fragments as baits for identifying interacting preys. Overall, the expanded network properties are statistically unchanged from the earlier network by the addition of two new hubs (compared in Supplementary Material, Table S3) (15).
One of the most useful analyses in terms of understanding the biology of the network is the identification of disease pathways by locating subnetworks or modules within the overall network (57–61). We searched for proteins linking pairs of ataxia-causing proteins that we call ‘ataxia triples.’ A protein in a triple directly interacts with two different ataxia-causing proteins in the form: Ataxia-protein1–Interactor–Ataxia-protein2. All of the new triples that did not appear in the previous network included CACNA1A and/or ataxin-7 as one of the ataxia proteins, suggesting that CACNA1A and ataxin-7 are linked to other ataxia-causing proteins through multiple interacting partners. There are 116 ataxia triples in our network compared with 53 and 63 in the previous Y2H- and literature-expanded ataxia interactomes, respectively (15). We calculated the significance of the observed number of triples, by randomizing the ataxia interaction network and preserving its degree distribution. With 1000 random realizations, on average, we observed 18 (± 9) triples, which renders the number of triples, 116, in our network highly significant (P< 0.001). The large number of links from ataxia-causing proteins most likely explains the large number of triples compared with the randomized networks. It is also useful to compare the number of triples in networks constructed from proteins associated with a phenotypically diverse group of disorders. This control, while focusing on disease proteins, removes biases for a particular disease phenotype and permitted us to analyze the extent to which the phenotype-based ataxia network has unusual topological properties. The number of triples in the ataxia interactome was significantly higher (P< 0.005) than phenotypically diverse networks with a mean of 31 triples, suggesting the importance of these triples in ataxia pathology (15).
Gene-disease associations and comorbidity analysis
The interaction network we mapped experimentally is based on screening with proteins involved in one common phenotype, ataxia, and contains many proteins of diverse functions. We hypothesized that if the protein interactions in the ataxia network are biologically relevant and relevant to the pathogenesis of ataxia, then other diseases associated with proteins in the network are more likely to occur in patients with ataxia. Therefore, using the network data together with Medicare patient records, we asked three questions. First, we asked whether patients with ataxia are at an increased risk of having any other diseases compared with other Medicare patients; secondly, if proteins implicated in these other diseases are found in the ataxia interactome; and finally, what relationships exist between proteins implicated in the same diseases?
We first performed comorbidity analysis in the large set of Medicare patient medical history data in the form of ICD-9 codes from 13 million patients. We aimed to make the analysis as specific to the hereditary ataxias as possible. Therefore, we removed records from both our patient and control group with ICD codes corresponding to toxic effects from non-medical substances (codes 980–989). We then defined the ataxia patient population by only including ICD-9 codes corresponding to specific ataxia diagnoses therefore excluding most non-degenerative ataxias (see Materials and Methods for ICD-9 codes corresponding to ataxia). We also removed ataxia patients with documented alcoholism since chronic alcohol abuse can cause ataxia. In our medical history data set, we identified 11 265 patients who have a diagnosis of ataxia meeting our criteria and 13 022 828 controls. By looking for diseases that appear more often in ataxia patients than in the patient population as a whole, we identified diseases that are comorbid with ataxia. We measured relative risk based on the number of occurrences of any one diagnosis with ataxia, and its prevalence in the ataxia and general patient populations (see Materials and Methods). We found over 500 ICD-9 codes with a relative risk of greater than 1.00 (Supplementary Material, Table S4A). Overall, within the 99% confidence interval, the average relative risk among the ataxia patients is 1.63, meaning that patients with ataxia are 1.63 times more likely to also have any other diagnosis (a comorbid condition) than Medicare patients without an ataxia diagnosis.
There are several built-in positive controls as well as limitations of using ICD-9 codes. For example, nystagmus (code 379.50), a known symptom of cerebellar dysfunction, has a relative risk of 25.1, meaning this diagnosis is 25.1 times more likely in a patient also diagnosed with ataxia. Conversely, despite our efforts to use strict criteria to classify patients, a limitation of this method is the inclusion of non-genetically caused ataxia due to the diagnosis/billing codes used. For example, although we removed patients with documented toxic substance exposure and alcoholism, some cases of alcoholic ataxia are possibly included and thus there is high relative risk of alcohol-induced persisting amnestic disorder (code 291.1) with a relative risk of 5.3, which is not due to a common molecular pathway, but rather to prolonged alcohol exposure. However, assessing the presumed false-positive comorbidities on a case-by-case basis introduces investigator bias. Ataxia is generally an outpatient condition and patients may have been seeking medical care for some other diagnosis, which may or may not be related to their ataxia; however, we cannot infer a priori which diagnoses are directly related.
We therefore used gene-disease associations and comorbidity analysis to relate proteins in the ataxia interaction network to disease phenotypes and to analyze whether molecular level gene-disease relationships are observed in the population (62–64). To relate the ataxia protein–protein interaction network to population disease patterns, we interrogated the ataxia network for proteins with links to disease phenotypes by searching the Online Inheritance in Man (OMIM) database (http://www.ncbi.nlm.nih.gov/omim/) for known gene-disease associations and we generated an overlap list of diseases that are represented in the ataxia network and appear in ataxia patients. We used this overlap list to assess the relative risk for patients diagnosed with ataxia to also have a disease whose associated protein is present in the ataxia network (Supplementary Material, Table S5). Accounting for a molecular relationship this risk is 3.03, which is significantly higher than the general comorbidity risk of 1.63 (Supplementary Material, Table S4B). This enrichment indicates that detailed protein interaction maps can shed light into population disease patterns. Furthermore, we can use the list of diseases that occur more often in ataxia patients and have known molecular causes to investigate the biological origin of the comorbidity relationships between diseases.
Macular degeneration associated proteins are tightly linked to one another and form a subnetwork
Interestingly, although not entirely unexpected, MD (code 362.50) was 2.7 times more likely to be diagnosed in ataxia patients than controls. Additionally, other similar visual diagnoses were also overrepresented in patients (Supplementary Material, Table S6). We were particularly intrigued that MD was comorbid with ataxia since it is considered a unique feature of SCA7 and we wanted to further understand the molecular relationship between ataxin-7 and other MD-causing proteins and to verify the comorbidity analysis. We hypothesized that our network was enriched for proteins associated with retinal degeneration. We therefore used the OMIM database to identify proteins associated with macular or retinal degeneration or dystrophy (see Materials and Methods for search terms). Surprisingly, of the 33 proteins that met these criteria, only fibulin 5 (FBLN5) and EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1) appeared in the ataxia network. Ataxin-7 is not included in the OMIM database as an MD-associated gene, despite the fact that glutamine-expanded ataxin-7 causes MD (31,32,39,43,45).
When we did not find enrichment for retinal degeneration-associated proteins as we expected, we investigated the proteomic relationship of MD-associated proteins (FBLN5, EFEMP1 and ATXN7) within the ataxia interactome, hypothesizing that the relationships between these proteins may provide insights into retinal degeneration. We looked for MD subnetworks, finding two MD triples in the ataxia network where an MD triple is defined as three interacting proteins in the form: MD protein1–Interactor–MD protein2 and where the three MD proteins are FBLN5, EFEMP1 and/or ATXN7. Interestingly, we identified 80 MD quadruples (MD protein1–Interactor–Interactor–MD protein2) (Fig. 5A). Ataxin-7, owing to being a hub, is central to the MD network and removing it results in a collapse of the MD subnetwork (Fig. 5B).
Figure 5.

(A) MD subnetwork endpoints are ATXN7, EFEMP1 and FBLN5, indicated in orange. Ataxia-causing proteins are red. New interactors are in light blue and interactors also present in the earlier ataxia network are in dark blue. There are 80 quadruples involving 32 proteins (Table 2). All binary interactions recapitulated by GST-APs (after testing 20 pairs) are indicated with red lines. (B) MD subnetwork is highly dependent on ATXN7, when the ATXN7 node is removed the subnetwork collapses.
In order to determine the significance of the number of observed quadruples, we measured the number of quadruples between any three proteins in the ataxia network. On average, we found 0.48 quadruples rendering the 80 observed MD quadruples in the ataxia interaction network highly significant (P= 7.37 × 10−5). Since ataxin-7 was a bait protein with a large number of interacting partners, a more strict control would be to find the quadruples between prey proteins and ataxia (bait) proteins. In this case, we found 2.56 such quadruples on average, again significantly less than the 80 we observed based on the three MD-causing proteins (P= 0.000596). Thus, this high number of observed quadruples indicates that the three proteins known to be involved in MD are highly connected. Since the proteins in the MD subnetwork are quite diverse, we examined the protein pairs in the network and found that of the 83 interacting pairs 17 had been validated by GST-APs in the course of our validation of this Y2H screen and the previously published data in Lim et al. (15).
Given the significant enrichment of MD quadruples within the ataxia network, we hypothesized that these interactors are possible candidates for MD-causing or modifying loci or otherwise important in establishing or maintaining retinal health.
All MD subnetwork proteins are expressed in retina
Since our screening used an adult brain cDNA library, we first tested the hypothesis that the proteins included in the MD subnetwork are also expressed in the retina. Literature searches revealed that 31 of the 32 MD quadruple proteins (or their paralogues) demonstrate retinal expression by either RNA or protein detection methods (Table 2) (65–72). The remaining protein, GFI1B, a transcriptional repressor, was used as a bait protein in the earlier interactome studies and thus is a hub within the MD subnetwork; therefore, it was essential that we validate its retinal expression. We first used commercial human retina polyA RNA for quantitative real-time PCR and detected GFI1B transcript (data not shown). We also sought evidence of GFI1B protein expression using immunofluorescence in wild-type mouse retina. We found that GFI1B is expressed most prominently in the outer nuclear layer (ONL) (Fig. 6A). Taken together, these results demonstrate that all of the proteins in the MD subnetwork are expressed in the retina.
Table 2.
Proteins in the MD subnetwork and other ataxin-7 interactors
| Human symbol | Entrez ID | Mouse symbol | Entrez ID | Expression studies | Human mapped loc. | Overlapping RetNet unmapped? |
|---|---|---|---|---|---|---|
| ACTN1a | 87 | Actn1 | 109711 | Blackshaw et al. (65) | 14q22–q24 | No |
| ACTN2 | 88 | Actn2 | 11472 | Schulz et al. (72) | 1q42–q43 | No |
| ACTN4 | 81 | Actn4 | 60595 | Schulz et al. (72) | 19q13 | MCDR5 |
| ADAM33 | 80332 | Adam33 | 110751 | UniGene Hs.173716 | 20p13 | No |
| AGRNa | 375790 | Agrn | 11603 | Blackshaw et al. (65) | 1p36.33 | LCA9 |
| ATN1a | 1822 | Atn1 | 13498 | Blackshaw et al. (65), Zhang et al. (71) | 12p13.31 | No |
| ATXN1La | 342371 | Atxn1L | 52335 | Blackshaw et al. (65) | 16q22.2 | No-FHASD close |
| ATXN7a | 6314 | Atxn7 | 246103 | Ström et al. (70) and this study | 3p21.1–p12 | Yes-itself |
| CACNA1Aa | 773 | Cacna1a | 12286 | Xu et al. (87) and Blackshaw et al. (65) | 19p13.2–p13.1 | No |
| CARD10 | 29775 | Card10 | 105844 | Schulz et al. (72) | 22q13.1 | OPA5 and VRD1 |
| CEP70 | 80321 | Cep70 | 68121 | Blackshaw et al. (65) | 3q22–q23 | No |
| CEP72 | 55722 | Cep72 | 74470 | Blackshaw et al. (65) | 5p15.33 | MCDR3 |
| COG6 | 57511 | Cog6 | 67542 | Blackshaw et al. (65) | 13q13.3 | No |
| CRIM1a | 51232 | Crim1 | 50766 | Lovicu et al. (78), Kinna et al. (98), Pennisi et al. (81) | 2p21 | No |
| EFEMP1a | 2202 | Efemp1 | 216616 | Stone et al. (84) | 2p16 | No |
| EFEMP2a | 30008 | Efemp2 | 58859 | Blackshaw et al. (65) | 11q13 | VRNI |
| FBLN5a | 10516 | Fbln5 | 23876 | Blackshaw et al. (65) | 14q32.1 | ACHM1 and itself |
| FBN1 | 2200 | Fbn1 | 14118 | Blackshaw et al. (65) | 15q21.1 | No |
| FN1 | 2335 | Fn1 | 14268 | Blackshaw et al. (65), Siegert et al. (83) | 2q34 | No |
| GFI1Ba | 8328 | Gfi1b | 14582 | This study | 9q34.13 | No |
| GOLGA2 | 2801 | Golga2 | 99412 | Blackshaw et al. (65) | 9q34.11 | No |
| GOLGA3 | 2802 | Golga3 | 269682 | Blackshaw et al. (65) | 12q24.33 | No |
| GPRASP1 | 9737 | Gprasp1 | 67298 | Blackshaw et al. (65) | Xq22.1 | No |
| GRNa | 2896 | Grn | 14824 | Blackshaw et al. (65) | 17q21.32 | CORD4 |
| JAG2a | 3714 | Jag2 | 16450 | Blackshaw et al. (65) | 14q32 | ACHM1 |
| KALRNa | 8997 | Kalrn | 545156 | Unigene Hs.8004 | 3q21.1–q21.2 | No |
| LAMB1a | 3912 | Lamb1-1 | 16777 | Sarthy and Fu (68) | 7q22 | No |
| LAMB2 | 3913 | Lamb2 | 16779 | Blackshaw et al. (65) | 3p21 | No |
| LRSAM1 | 90678 | Lrsam1 | 227738 | Blackshaw et al. (65) | 9q33.3–q34.11 | No |
| LTBP4a | 8425 | Ltbp4 | 108075 | Blackshaw et al. (65) | 19q13.1–q13.2 | No-MCDR5 close |
| MATN2a | 4147 | Matn2 | 17181 | Fuchshofer et al. (75) | 8q22 | OPA6 |
| MEGF6a | 1953 | Megf6 | 230971 | Blackshaw et al. (65) | 1p36.3 | LCA9 |
| MEGF8a | 1954 | Megf8 | 269878 | Blackshaw et al. (65) | 19q12 | No |
| MPRIP | 23164 | Mprip | 26936 | Blackshaw et al. (65) | 17p11.2 | No |
| NELL1a | 4745 | Nell1 | 338352 | Piri et al. (67) | 11p15.2–p15.1 | No |
| NELL2a | 4753 | Nell2 | 54003 | Blackshaw et al. (65) | 12q13.11–q13.12 | No-CODA1 close |
| NOC2La | 26155 | Noc2l | 57741 | Hublitz et al. (66) | 1p36.33 | LCA9 |
| NUP62 | 23636 | Nup62 | 18226 | Blackshaw et al. (65) | 19q13.33 | No-MCDR5 close |
| PICK1a | 9463 | Pick1 | 18693 | Blackshaw et al. (65) | 22q13.1 | OPA5 and VRD1 |
| PNMA1 | 9240 | Pnma1 | 70481 | Schulz et al. (72) | 14q24.3 | ACHM1 |
| PPP1R12Ca | 54776 | Ppp1r12c | 232807 | Schulz et al. (72) | 19q13.42 | No |
| RABEP1 | 9135 | Rabep1 | 54189 | Blackshaw et al. (65) | 17p13.2 | CACD |
| RAD23A | 5886 | Rad23a | 19358 | Blackshaw et al. (65) | 19p13.2 | No |
| REREa | 473 | Rere | 68703 | Blackshaw et al. (65) | 1p36 | LCA9 |
| TRIM27a | 5987 | Trim27 | 19720 | Blackshaw et al. (65) and this study | 6p22 | No |
| RNF31a | 55072 | Rnf31 | 268749 | Schulz et al. (72) | 14q11.2 | MCDR4 |
| SIAH1a | 6477 | Siah1a | 20437 | Schulz et al. (72), Yego and Mohr (89) | 16q12 | No |
| Siah1b | 20438 | |||||
| SNX1 | 6642 | Snx1 | 56440 | Blackshaw et al. (65) | 15q22.31 | No |
| SPRY1a | 10252 | Spry1 | 24063 | Blackshaw et al. (65) | 4q28.1 | No |
| SSFA2 | 6744 | Ssfa2 | 70599 | Blackshaw et al. (65) | 2q31.3 | No |
| THY1 | 7070 | Thy1 | 21838 | Blackshaw et al. (65) | 11q22.3–q23 | No |
| TMEM25a | 84866 | Tmem25 | 71687 | Blackshaw et al. (65) | 11q23.3 | No |
| TP53BP2 | 7159 | Trp53bp2 | 209456 | Blackshaw et al. (65) | 1q42.1 | No |
| TRAF2 | 7186 | Traf2 | 22030 | Blackshaw et al. (65) | 9q34 | No |
| TRIM23 | 373 | Trim23 | 81003 | Blackshaw et al. (65) | 5q12.3 | No |
| TRIM54 | 57159 | Trim54 | 58522 | Blackshaw et al. (65) | 2p23.3 | No |
| TXNDC11a | 51061 | Txndc11 | 106200 | Schulz et al. (72) | 16p13.13 | No |
| UBQLN2 | 29978 | Ubqln2 | 54609 | Blackshaw et al. (65) | Xp11.23–p11.1 | OPA2 |
| VWFa | 7450 | Vwf | 22371 | Blackshaw et al. (65) | 12p13.3 | No |
Entrez IDs and symbols are shown for both human and mice. Expression in the retina is based on the noted reference(s). RetNet (http://www.sph.uth.tmc.edu/retnet/) loci that are overlapping or near (in case of unmatched markers) the chromosomal location of the indicated protein.
aProtein is in the MD subnetwork. The remainder interact with ataxin-7 but are not part of the MD subnetwork.
Figure 6.

(A) GFI1B immunostaining in wild-type mouse retina. (B) RNA in situ hybridization for Ataxin-7 and sense control as labeled.
Given that our screen was not performed using a retina cDNA library, nor was the screen saturated and given central importance of ataxin-7 in the MD subnetwork, we considered that other ataxin-7 interactors might also be important in retinal health. We thus expanded our literature review and found that the 27 additional ataxin-7 interactors are all expressed in the eye or retina. Overall, we show that all 59 ataxin-7 interactors or MD quadruple components are expressed in the mammalian retina; interestingly, some of the genes map to loci previously associated with familial retinal degeneration (Table 2), suggesting they may be candidates for disease genes, although overall we would predict the MD-associated loci would be genetic modifiers of retinal health (31,32,47,65–99).
Ataxin-7 knock-in mice display altered expression of some interacting partners in retina
We next hypothesized that some of the ataxin-7 interactors and/or MD quadruple proteins would be altered in the retina of a previously characterized SCA7 knock-in mouse model, Ataxin-7266Q/+ (39). In a mixed genetic background, visual dysfunction begins around 5 weeks of age and ataxin-7 micro-aggregates are seen beginning at 8–10 weeks of age with more and larger inclusions by 15 weeks (39). However, as the mice have been backcrossed to a pure C57BL6/J background, the Ataxin-7266Q/+ mice die at 10–11 weeks of age and inclusions remain very small.
To validate the functional relevance of the MD subnetwork in vivo, we examined the expression of some MD subnetwork components by immunofluorescence in 5-, 8- and 10-week-old Ataxin-7266Q/+ retina and wild-type littermate controls. Unfortunately, in vivo mouse ataxin-7 protein studies are limited by a lack of an antibody that recognizes normal expression of either the wild-type mouse or knock-in protein, precluding co-localization studies until after protein inclusions have formed.
We therefore used RNA in situ hybridization (ISH) to uncover Atxn7 expression in the ONL, inner nuclear (INL) and ganglion cell layers (GCL) of wild-type C57BL6/J mouse retinal sections (Fig. 6B). These data support previous data from other groups using immunostaining in SCA7 patient and control retina tissue that find ataxin-7 expression in these same retinal layers (40,46,70,100). Furthermore, Cancel et al. (40) and Einum et al. (100) demonstrate ataxin-7 staining in control patient retina in the inner plexiform layer (IPL), specifically in both cytoplasm and processes of the ganglion cells, the outer plexiform layer (OPL) and also in the inner and outer segments of the rods and cones. Thus, our ISH data and patient protein data taken together demonstrate that ataxin-7 is normally expressed throughout the retina.
We next randomly selected 7 of the 59 MD subnetwork and/or ataxin-7 interacting proteins for immunofluorescence studies. We used retina from wild-type and Ataxin-7266Q/+ littermates collected at 5, 8 and 10 weeks of age and immunostained for TRIM27, TRIM23, GRN, RAD23A, TRIM54, SIAH1 and CARD10 protein expression.
TRIM27 is a zinc finger protein that is reported to be a transcriptional repressor (101,102); however, similarly to ataxin-7, in some cell types TRIM27 is reportedly cytoplasmic (103). In mouse retinal sections, TRIM27 is strongly expressed in the ONL (Fig. 7A). Additionally, it is expressed in the outer segments of the photoreceptors, INL and GCL (data not shown). At 5 weeks, staining levels in wild-type and Ataxin-7266Q/+ retina are approximately equivalent. However, in both genotypes, staining increases with age in the nuclear layers. The increase is more obvious in knock-in retina such that at 10 weeks the staining is many times brighter in the ONL than either earlier Ataxin-7266Q/+ or age-matched wild-type retina. Importantly, the alteration in TRIM27 expression occurs at a protein level as Trim27 RNA is not altered in Ataxin-7266Q/+ retina by qRT–PCR (data not shown).
Figure 7.

(A) TRIM27 immunostaining in the retina ONL of wild-type and Ataxin-7266Q/+ mouse retina at 5, 8 and 10 weeks of age, as indicated. The knock-in mice have increased expression of TRIM27, which is particularly evident at the later disease point. (B) TRIM23 immunostaining in wild-type and Ataxin-7266Q/+ mouse retina at 5, 8 and 10 weeks of age, as indicated. The staining is more pronounced in the WT mice as they age compared with knock-in mice. (C) Calbindin (green) and TRIM23 (red) are shown for wild-type and knock-in samples at 10 weeks.
TRIM23 staining is in the OPL and IPL (Fig. 7B) in both knock-in and wild-type retina. In contrast to TRIM27, which is increased, TRIM23 immunostaining is reduced in knock-in tissue, particularly in the OPL. Given that the Ataxin-7266Q/+ mice have progressive retinal degeneration we co-labeled with anti-calbindin, a marker of horizontal and amacrine cells, to better visualize the OPL (Fig. 7C). Fluorescence of the TRIM23 in Ataxin-7266Q/+ tissue was lower than the dynamic range in wild-type tissue under the same sample preparation and imaging conditions. Thus, in order to confirm that the reduced fluorescence was due to decreased protein levels rather than cell loss, we scaled the intensity of both the wild-type and Ataxin-7266Q/+ images by a factor of 3× (Supplementary Material, Fig. S2). Scaling reveals that the TRIM23 pattern is indeed preserved in both Ataxin-7266Q/+ and wild-type tissue, confirming reduced TRIM23 expression in the absence of cell loss. Additionally, by qRT–PCR, Trim23 RNA levels are equivalent in wild-type and Ataxin-7266Q/+ retina thus demonstrating the reduction is at the protein level (data not shown).
We found that both GRN and RAD23A are expressed in the GCL and there are no differences between wild-type and Ataxin-7266Q/+ retina at the time points tested (Supplementary Material, Fig. S3). Similarly, there are no differences in TRIM54, expressed in the INL and ganglion cell nuclei. SIAH1 appears similar between wild-type and Ataxin-7266Q/+ retina, although it may be slightly brighter in wild-type tissue. Finally, CARD10, a caspase recruitment domain protein that is reportedly involved in GPCR signaling (104), exhibits strong immunostaining in the outer segments in wild-type mice but is not present in the Ataxin-7266Q/+ mice at any age. There is also weaker CARD10 staining of the ONL, in a pattern similar to both GFI1B and TRIM27 staining. As previously reported, Ataxin-7266Q/+ mice exhibit early photoreceptor degeneration. We therefore collected light microscope images at the same time as CARD10 imaging; the light images indicate that the difference in outer segment staining is likely attributable to photoreceptor degeneration (data not shown) (39).
Thus, the proteins we tested from the MD quadruples and other ataxin-7 interactors are all expressed in the retina, confirming previous data from other labs. Additionally, at least two, TRIM27 and TRIM23, demonstrate altered expression in Ataxin-7266Q/+ mouse retina compared with wild-type littermates. Taken together the GST-AP validations, literature support and in vivo expression data provide strong support for the biological relevance and importance of the MD quadruples identified in this study.
DISCUSSION
The earlier ataxia interactome provided new insight into the pathogenesis of inherited ataxias (16–26), but it lacked interactors for two SCA-causing proteins, CACNA1A and ataxin-7. We hypothesized that using many fragments of the two proteins rather than full-length proteins to rescreen an adult human brain library in a highly stringent Y2H screen would generate better coverage and yield more interactors. By using 28 bait fragments, we identified 118 interactors for CACNA1A and ataxin-7 and were able to include these interactions in the ataxia interactome leading to a more expanded ataxia-protein network. The fact that we were more successful in finding binding partners using portions of the coding region in comparison with the full-length proteins raises the possibility that fragments might expose interaction domains that are otherwise impossible to expose in a yeast system, especially if certain cell-specific modifications are required to uncover such surfaces.
CACNA1A has two predominant splice forms that are expressed at roughly equal levels in wild-type mice. However, in knock-in mice carrying a polyQ expansion of either 14, 30 or 84 glutamines, there is a shift in the ratio of expression such that the MPI isoform is expressed at up to 80% of the transcripts (29). In post-mortem SCA6 patient brain samples, there was also an increased amount of the MPI isoform compared with control brains (105). It is unclear whether the increased prevalence of MPI is caused directly by the expansion and how the shift in isoform stoichiometry may alter disease pathogenesis. In this study, we used the C-termini of two predominant CACNA1A isoforms, MPI and MPc, as bait in an Y2H screen. Additionally, for the MPI bait fragments, we used wild-type (MPI-11Q), disease-causing (MPI-23Q) and hyper-expanded (MPI-72Q) polyQ alleles. We identified interactors that are isoform specific and also interactions that seem to make the MPI isoform with an expanded polyQ tract act more similarly to the alternative, MPc splice form. Interactors that are exclusive to MPI may mediate gain of function effects upon the expansion of the polyQ tract, such that more available MPI results in more association of these proteins. Given the altered MPc to MPI ratio seen in both patients and mouse models of SCA6 and that in a SCA1 mouse model a change in stoichiometry between two endogenous ataxin-1 containing complexes contributes to neuropathology, we propose that differences in the affinity of interactors for either MPI or MPc could modify the SCA6 disease phenotype (18). Overall, we suggest that the CACNA1A interactions that are isoform specific may be important in the SCA6 disease process and that the newly identified CACNA1A interacting partners may provide insight into the molecular understanding of SCA6. Additionally, the identification of isoform-specific interactions for CACNA1A raises the possibility that future protein-interaction studies could provide additional insights when the individual isoforms are considered as nodes instead of including them together as a single protein, particularly when only one isoform is associated with a disease.
Ataxin-7 is highly evolutionarily conserved. In fact, yeast lacking the ataxin-7 homolog, Sgf73, can be rescued by wild-type ataxin-7 expression (106). Our bait constructs took into account the regions of highest homology across species and paralogues. Interestingly, baits containing only the first half of the protein or the middle section were unable to identify interacting partners. This is likely a result of two related factors; first, the Y2H system that we used is highly stringent and does not result in the high levels of overexpression seen in some other screens. Second, the two baits that succeeded in identifying interacting partners are the C-terminal fragments that contain mostly regions not conserved in yeast, whereas those fragments that failed predominantly contained the polyglutamine tract, block I and/or block II, conserved regions that are known to be required for recruitment into yeast SAGA and mammalian STAGA/TFTC complexes (9,33–35). We suspect that in yeast, the interaction of ataxin-7 with SAGA components might compromise the Y2H reporter assay. Interestingly, block III is not found in Sgf73 and may have a specific function aside from SAGA complex formation (9). Furthermore, the successful baits begin near the reported caspase-7 cleavage sites, suggesting a possible role for this C-terminal protein fragment in vivo, although to date most studies and antibodies have targeted the N-terminal cleavage product (38,43,49). Despite being limited to only two successful baits, we identified many novel ataxin-7 interactors and revealed potentially interesting candidate partners.
Since the known role for ataxin-7 is as a STAGA/TFTC component, we expected to find ataxin-7 interactors related to histone modifications and transcriptional regulation. However, since the baits which successfully identified partners do not contain the regions responsible for STAGA/TFTC complex inclusion, it is not surprising that the proteins we identified have other functions. Although ataxin-7 cytoplasmic localization and nuclear-cytoplasmic shuttling have been reported, most studies to date have focused on the role of ataxin-7 in the nucleus. The ataxin-7 interactors we identify include nuclear proteins with known roles in transcriptional regulation and other proteins that are predicted to be cytoplasmic or secreted. Wild-type ataxin-7 was also found to colocalize with BiP, a marker for the endoplasmic reticulum in neurons, strengthening the likelihood that ataxin-7 has other functions besides its role in the STAGA/TFTC complexes (40). Thus, our studies provide a unique foundation for future studies to uncover novel roles of ataxin-7.
Our studies of CACNA1A and ataxin-7 interacting partners lead to an expansion of the ataxia interactome (15). We then used the updated ataxia interactome as a tool for exploring diseases comorbid with ataxia, hypothesizing that comorbid conditions would be associated with proteins found in the ataxia network. This study is the first that we are aware of in which patient medical records and a phenotype-based interactome are used together to explore the molecular basis of comorbid conditions. By interrogating 32 million patient records, we found that ataxia patients are indeed at increased risk of any comorbid diagnosis compared with the control population. Significantly, the relative risk of comorbidity increased from 1.63 to 3.03 when we considered the diseases represented in the ataxia interactome.
Importantly, the relative risk of 3.03 is likely an underrepresentation of the true comorbidity rate for ataxia and other diseases represented by the ataxia network. These calculations are based on currently available data sets. For instance, the ataxia network is not saturated and thus some interacting proteins are missing by chance. Second, the Medicare patient records are in the form of ICD-9 codes. These codes do not correspond in a one-to-one relationship for each disease, but can correspond to specific features and symptoms of diseases. For example, some ICD-9 codes are specific—such as MD (code 326.50), whereas some MD cases could instead be coded as low-vision (codes 369.60, 369.4, 369.3, 369.61 and others). These potentially overlapping ICD-9 codes can decrease the power (number of patients) one may see if some of the overlapping codes are considered together, as we did with some of the ataxia codes. Conversely, inclusion of misdiagnosed or miscoded patients can cause false-positive associations, such as alcohol-induced persisting amnestic disorder (code 291.1). Finally, when we account for the liabilities in our patient records by overlapping comorbid diseases with the ataxia interactome, we were limited to gene-disease relationships recorded in OMIM. Unfortunately, OMIM records are incomplete, even for the associations already reported in the literature, as exemplified by searching for MD associations and not finding ataxin-7 among the results. Thus, there is a disconnect between a patient having a molecular diagnosis of, for example, SCA7 and then having medical records including some form of ataxia and some form of vision loss. Thus, there are many nuances in patient records and disease–gene relationship databases that decrease the power of using a combined molecular and bioinformatics approach to uncover molecular relationships between comorbid diseases. Despite these limitations, our results show that this approach is fruitful for identifying these relationships and such analysis will only improve as electronic medical records are improved, and more disease-causing genes are identified and included into public databases. Furthermore, the approach and data in this study will serve as a framework for future studies that will benefit from better electronic medical data and accurate genetic diagnostic codes.
Overall, our comorbidity analysis and OMIM-based searches were limited to our ataxia interactome and 13 million patients and yet provided candidate proteins for a comorbid disease, MD. From these criteria, we found a highly significant subnetwork anchored by three MD causing proteins. The MD subnetwork contains 32 proteins that may have a role in MD or macular health. It should be noted that ataxin-7 is central in this subnetwork. Initially, we were surprised by the fact that many of these MD subnetwork proteins are predicted to be cytoplasmic or extracellular, however further literature searches and studies provide strong support for the biological relevance of this network. Specifically, 31 of the 32 proteins were previously reported to be expressed in the retina (65–72,75,78,81,85,87,89,98). EFEMP1, one of the MD proteins that anchor the network, is a secreted protein; however, the disease-causing mutation R345W results in the retention of the protein within the cells (107,108). In the mammalian retina, EFEMP1 and ataxin-7 are expressed in the same cell types (40,46,70,100,107,109,110). Additionally, MEGF8, another component of the subnetwork, was predicted to be in the cytoplasm or extracellular space, but recent studies have demonstrated that it co-localizes with GFI1B in the nucleus (111). Given the lack of saturation of this screen and also the lack of other MD proteins as baits in the screen, it seemed likely that some or many of the other ataxin-7 interactors that are not connected to the subnetwork are also involved in macular health, thus we also included some of these proteins for in vivo studies. Our own studies demonstrate that randomly selected proteins from within the MD quadruples and other ataxin-7 interactors are expressed in the mouse retina and some show altered expression in a mouse model of SCA7. Thus, we suggest that further investigation of these proteins could provide valuable insight into age-related MD.
More than 35 transgenic mouse models are available for a variety of the genes in the MD quadruples. We suggest that these mice should be further investigated given that mice are able to freely move, obtain chow and mate without vision, thus home cage behavior would not be changed even if these mice have retinal defects. Therefore, it would be interesting to look at the retina and its integrity from these mice at different ages as well as potential genetic interactions between seemingly benign mutants and existing models of retinal degeneration.
In addition to SCA7, there have been several reports of human diseases and mouse models that involve both cerebellar or PC and retinal degeneration, including a possible variant of Boucher–Neuhauser syndrome and PC degeneration (pcd) mice (112–117), and these cell types express a common subset of genes (118). Our study shows a systematic underlying relationship between ataxia and MD.
Overall, our studies demonstrate that taken together disease-based interaction networks and patient medical records can reveal molecular associations underlying comorbidities in the patient population. Our data provide candidate proteins for further investigation into the molecular pathogenesis of SCA6, SCA7 and MD. Additionally, researchers investigating other diseases represented in the network and comorbid with ataxia will be able to use the data to interrogate possible molecular relationships. As researchers continue to develop interaction networks based on a common phenotype and medical records are increasingly made electronic, the power of comorbidity analysis to identify common molecular pathways in the pathogenesis of many diseases will increase too.
MATERIALS AND METHODS
Yeast two-hybrid
Y2H baits were designed based on known homology domains between species for ataxin-7 and for CACNA1A based on reports of translocation to the nucleus and splice isoforms (9,30,38,49). The baits were cloned using Invitrogen Gateway technology as previously described (15).
Glutathione-sepharose affinity co-purifications (GST-APs)
Baits were tagged with GST using the Invitrogen Gateway system as previously described (15,51). Preys were tagged with myc in the same manner. Prey proteins were ORFeome clones from the Human ORFeome collection [http://horfdb.dfci.harvard.edu/ and (119)] or were rescued from the yeast colonies. Briefly, for yeast rescue, the colony was grown in liquid culture and lysed. The purified plasmid was then transformed into E. coli. Since there is no bacterial selection marker that differs for the bait and prey plasmids in this system, three to five colonies were selected and sequenced to identify the prey colonies. After identification, we used the LR reaction from the Invitrogen Gateway system to transfer the prey coding sequence from the yeast-destination vector to the entry vector. Using a BP reaction, the coding sequence was subsequently transferred to the myc-vector as previously described (15,51).
GST-APs were performed in HEK293T cells. Eight-hundred nanograms of the GST-bait and myc-prey vectors were transfected into the cells using Lipofectamine 2000 and affinity purifications were performed as previously described (15). Western blots were performed as described previously (15).
Bioinformatic analyses
The sequence data were processed as previously described (15). Briefly, the sequencing results obtained for the prey clones from the Y2H screen were submitted to BlastN and tBlastX searches to identify the protein-coding genes in NCBI database. The sequence databases used for alignment analysis were obtained from NCBI. The protein sequence data for alignments were obtained from ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/protein/protein.fa.gz, while nucleotide data were obtained from ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/RNA/rna.fa.gz. The cross-tabulated protein and nucleotide alignment results were compared (filtered) for high quality based on E-value as well as for agreement between the nucleotide and protein result pairs. Discordant pairs and low-quality results were manually checked and repaired. Entrez and symbol identifiers were determined for each prey protein passing the alignment filter. Sequences that had poor results or questionable BLAST were discarded. The resulting data were compiled by both bait protein and bait fragment. A network (undirected graph) was constructed using the unique bait–prey protein pairs, determined by the analysis (see below).
Network generation
The network was generated from the published ataxia network (15) and our screen data. We also included unpublished data from further rescreening with three of the baits previously published (15). The ataxia network was drawn using Cytoscape (120).
Comorbidity analysis
Medicare patient ICD-9 codes were used to analyze whether the molecular level gene–disease relationships are also present in the patient population. The Medicare data set consists of over 32 million hospitalization records of 13 039 018 patients. We first removed all patients with a diagnosis of ‘toxic effects of substances chiefly nonmedical as to source’ (ICD-9 codes 980–989) from our data set. We then defined patients as ataxia patients if they had one or more of the following ataxia-related ICD-9 codes in their history: primary cerebellar degeneration (code 334.2), spinocerebellar disease, unspecified (code 334.9), cerebellar ataxia NOS (code 334.3), Friedreich's ataxia (code 334.0), cerebral ataxia (code 331.89), ataxia-telangiectasia (code 334.8) and cerebellar ataxia in diseases classified elsewhere (code 334.4), but not including patients with alcoholism (codes 303.0–303.9). This left 13 022 828 controls and 11 265 patients with some form of ataxia. These ataxia patients’ records were searched for other disease diagnoses (comorbidities). For a given disease, the relative risk, RR, is measured by
where Cxa is the number of patients that have the disease x and ataxia (a), and 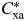 is the expected number of patients that have disease x and ataxia. The expected comorbidity is
is the expected number of patients that have disease x and ataxia. The expected comorbidity is 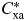 =Ix /N * Ia /N where Ix and Ia are the prevalence of disease x and ataxia, respectively, and N is the total number of patients. The confidence intervals were measured by the Katz method (121).
=Ix /N * Ia /N where Ix and Ia are the prevalence of disease x and ataxia, respectively, and N is the total number of patients. The confidence intervals were measured by the Katz method (121).
Macular degeneration search terms in OMIM
Search terms were: macular corneal dystrophy, MD, macular dystrophy, maculopathy, retinal cone dystrophy, retinal degeneration, retinal dystrophy and degeneration of the retina.
In situ hybridization
Probes were designed based on Allen Brain Atlas probes that were successful in the brain. ISH experiments were performed as previously described (122–125). Primers used included a T7 promoter for the sense probe and SP6 promoter for the anti-sense. Gene-specific primers are as follows: Atxn7-for 5′-CACAGCTATGGAAGAAAATCCC-3′; Atxn7-rev 5′-AGGCTCACCGAGTGTGTTTTAT-3′.
Immunofluorescence in wild-type and Ataxin-7266Q/+ mice
Ataxin-7266Q/+ and wild-type littermates were sacrificed at 5, 8 or 10 weeks in accordance with our approved animal protocol. Eyes were immediately removed and placed in 4% paraformaldehyde in 1xPBS for 1–2 days on a rotator in a cold room. Tissue was then cryoprotected in a sucrose gradient of 5, 10 and 30 sucrose in 1xPBS overnight at 4°. The tissues were embedded in Tissue-Tek O.C.T Compound (Sakura Finetek U.S.A., Inc. Torrance, CA 90501, USA) and stored at −80° until sectioning. Tissue was sectioned on a cryostat at 12 μm, mounted on slides and dried overnight at room temperature. Slides were stored at −80° until immunostaining.
Slides were brought to room temperature, washed in PBS and permeablized with 0.3% Triton X-100 in PBS and blocked for 2 h at RT with 1% serum, 1% BSA, 0.05% Triton X-100 in 1xPBS. The slides were washed in PBS and incubated with the primary antibody (1:200 unless otherwise noted) without Triton X-100 for 3 days in a humid box at 4°. Primary antibodies are: TRIM27 (gtx102048), TRIM23 (gtx100057), CARD10 (gtx111222), SIAH1 (gtx104715), GRN (gtx100803), RAD23A (gtx100425), Genetex Inc., Irvine, CA 92606, USA; GFI1B, D-19 (sc-8559) used at (1:50), Santa Cruz Biotechnology, Inc., Santa Cruz, CA 95060 USA; and calbindin, D-28K, clone CB-955, (c-9849) used at (1:500), Sigma-Aldrich, St Louis, MO 63103 USA. After washing, secondary antibody [goat anti-rabbit Alexa Fluor 647 (A21245), donkey anti-goat Alexa Fluor 647 (A21447) and goat anti-mouse Alexa Fluor 488 (A11001) Invitrogen Corporation, Carlsbad, CA 92008 USA] was added at (1:250) to incubate for 2 days. The slides were then washed and incubated for 5 min with DAPI (2.5 μg/ml) in PBS, washed again and mounted with Prolong Gold antifade reagent (P36934), Invitrogen Corporation. After curing, the slides were stored at −20° until imaging. Images were taken on a Leica confocal microscope and processed using ImageJ software.
For each time point, two to three pairs of mice were collected. Each antibody was used to stain one pair of 5-, 8- and 10-week-old knock-in and wild-type mice during at least two independent experiments. Representative images are selected.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by Award Number P30HD024064 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development (to H.Y.Z.) and by the Ellison Foundation (to DFCI CCSB, M Vidal, Director). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health. J.J.K. was supported by a training grant from the National Eye Institute (T32 EY07102), and the research on SCA6 was supported by a generous gift from a foundation that asked that its identity not be disclosed (to H.Y.Z.). H.Y.Z. is an investigator with the Howard Hughes Medical Institute. Funding to pay the Open Access publication charges for this article was provided by Howard Hughes Medical Institute.
ACKNOWLEDGEMENTS
We thank Agnes Liang, Dr Christina Thaller and Dr Richard Atkinson for technical assistance; Dr Nicholas A. Christakis for sharing the Medicare phenotypic network and patient medical history data; and Dr Melissa Ramocki, Dr Hsiao-Tuan Chao and Dr John Fryer for input on the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Gatchel J.R., Zoghbi H.Y. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 2005;6:743–755. doi: 10.1038/nrg1691. doi:10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 2.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. doi:10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 3.Black B.E., Paschal B.M. Intranuclear organization and function of the androgen receptor. Trends Endocrinol Metab. 2004;15:411–417. doi: 10.1016/j.tem.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Kao C.C., Lieberman P.M., Schmidt M.C., Zhou Q., Pei R., Berk A.J. Cloning of a transcriptionally active human TATA binding factor. Science. 1990;248:1646–1650. doi: 10.1126/science.2194289. doi:10.1126/science.2194289. [DOI] [PubMed] [Google Scholar]
- 5.La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. doi:10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 6.Shen Y., Peterson A.S. Atrophins' emerging roles in development and neurodegenerative disease. Cell Mol. Life Sci. 2009;66:437–446. doi: 10.1007/s00018-008-8403-9. doi:10.1007/s00018-008-8403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L., Tsai C.C. Atrophin proteins: an overview of a new class of nuclear receptor corepressors. Nucl. Recept. Signal. 2008;6:e009. doi: 10.1621/nrs.06009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D.W., Amos C., Dobyns W.B., Subramony S.H., Zoghbi H.Y., Lee C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. doi:10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 9.Helmlinger D., Hardy S., Sasorith S., Klein F., Robert F., Weber C., Miguet L., Potier N., Van-Dorsselaer A., Wurtz J.-M., et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. doi:10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 10.Kerkhofs S., Denayer S., Haelens A., Claessens F. Androgen receptor knockout and knock-in mouse models. J. Mol. Endocrinol. 2009;42:11–17. doi: 10.1677/JME-08-0122. doi:10.1677/JME-08-0122. [DOI] [PubMed] [Google Scholar]
- 11.Zeitlin S., Liu J.P., Chapman D.L., Papaioannou V.E., Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat. Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. doi:10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 12.Zuccato C., Tartari M., Crotti A., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B.R., Hayden M.R., Timmusk T., et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. doi:10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 13.Auerbach W., Hurlbert M.S., Hilditch-Maguire P., Wadghiri Y.Z., Wheeler V.C., Cohen S.I., Joyner A.L., MacDonald M.E., Turnbull D.H. The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum. Mol. Genet. 2001;10:2515–2523. doi: 10.1093/hmg/10.22.2515. doi:10.1093/hmg/10.22.2515. [DOI] [PubMed] [Google Scholar]
- 14.Van Raamsdonk J.M., Murphy Z., Slow E.J., Leavitt B.R., Hayden M.R. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. doi:10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- 15.Lim J., Hao T., Shaw C., Patel A.J., Szabó G., Rual J.-F., Fisk C.J., Li N., Smolyar A., Hill D.E., et al. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. doi:10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Becker E.B., Oliver P.L., Glitsch M.D., Banks G.T., Achilli F., Hardy A., Nolan P.M., Fisher E.M., Davies K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice; Proc. Natl Acad. Sci. USA; 2009. pp. 6706–6711. doi:10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee L.C., Chen C.M., Chen F.L., Lin P.Y., Hsiao Y.C., Wang P.R., Su M.T., Hsieh-Li H.M., Hwang J.C., Wu C.H., et al. Altered expression of HSPA5, HSPA8 and PARK7 in spinocerebellar ataxia type 17 identified by 2-dimensional fluorescence difference in gel electrophoresis. Clin. Chim. Acta. 2009;400:56–62. doi: 10.1016/j.cca.2008.10.013. doi:10.1016/j.cca.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A.B., Richman R., Hill D.E., Orr H.T., Zoghbi H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. doi:10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukong K.E., Richard S. Motor coordination defects in mice deficient for the Sam68 RNA-binding protein. Behav. Brain Res. 2008;189:357–363. doi: 10.1016/j.bbr.2008.01.010. doi:10.1016/j.bbr.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Parfitt D.A., Michael G.J., Vermeulen E.G., Prodromou N.V., Webb T.R., Gallo J.M., Cheetham M.E., Nicoll W.S., Blatch G.L., Chapple J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009;18:1556–1565. doi: 10.1093/hmg/ddp067. doi:10.1093/hmg/ddp067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt W.M., Kraus C., Hoger H., Hochmeister S., Oberndorfer F., Branka M., Bingemann S., Lassmann H., Muller M., Macedo-Souza L.I., et al. Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep. 2007;8:691–697. doi: 10.1038/sj.embor.7401001. doi:10.1038/sj.embor.7401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sone M., Uchida A., Komatsu A., Suzuki E., Ibuki I., Asada M., Shiwaku H., Tamura T., Hoshino M., Okazawa H., et al. Loss of yata, a novel gene regulating the subcellular localization of APPL, induces deterioration of neural tissues and lifespan shortening. PLoS ONE. 2009;4:e4466. doi: 10.1371/journal.pone.0004466. doi:10.1371/journal.pone.0004466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X., Liu Q., Jiang R. Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics. 2009;25:98–104. doi: 10.1093/bioinformatics/btn593. doi:10.1093/bioinformatics/btn593. [DOI] [PubMed] [Google Scholar]
- 24.Lam Y.C., Bowman A.B., Jafar-Nejad P., Lim J., Richman R., Fryer J.D., Hyun E.D., Duvick L.A., Orr H.T., Botas J., et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. doi:10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 25.Al-Ramahi I., Perez A.M., Lim J., Zhang M., Sorensen R., de Haro M., Branco J., Pulst S.M., Zoghbi H.Y., Botas J. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007;3:e234. doi: 10.1371/journal.pgen.0030234. doi:10.1371/journal.pgen.0030234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canterini S., Bosco A., De Matteis V., Mangia F., Fiorenza M.T. THG-1pit moves to nucleus at the onset of cerebellar granule neurons apoptosis. Mol. Cell Neurosci. 2009;40:249–257. doi: 10.1016/j.mcn.2008.10.013. doi:10.1016/j.mcn.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Sampath J., Long P.R., Shepard R.L., Xia X., Devanarayan V., Sandusky G.E., Perry W.L., 3rd, Dantzig A.H., Williamson M., Rolfe M., et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am. J. Pathol. 2003;163:1781–1790. doi: 10.1016/S0002-9440(10)63538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rappsilber J., Ryder U., Lamond A.I., Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–1245. doi: 10.1101/gr.473902. doi:10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watase K., Barrett C.F., Miyazaki T., Ishiguro T., Ishikawa K., Hu Y., Unno T., Sun Y., Kasai S., Watanabe M., et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels; Proc. Natl Acad. Sci. USA; 2008. pp. 11987–11992. doi:10.1073/pnas.0804350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kordasiewicz H.B., Thompson R.M., Clark H.B., Gomez C.M. C-termini of P/Q-type Ca2+ channel alpha1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum. Mol. Genet. 2006;15:1587–1599. doi: 10.1093/hmg/ddl080. doi:10.1093/hmg/ddl080. [DOI] [PubMed] [Google Scholar]
- 31.David G., Abbas N., Stevanin G., Dürr A., Yvert G., Cancel G., Weber C., Imbert G., Saudou F., Antoniou E., et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. doi:10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 32.Gouw L.G., Kaplan C.D., Haines J.H., Digre K.B., Rutledge S.L., Matilla A., Leppert M., Zoghbi H.Y., Ptacek L.J. Retinal degeneration characterizes a spinocerebellar ataxia mapping to chromosome 3p. Nat. Genet. 1995;10:89–93. doi: 10.1038/ng0595-89. doi:10.1038/ng0595-89. [DOI] [PubMed] [Google Scholar]
- 33.Mushegian A.R., Vishnivetskiy S.A., Gurevich V.V. Conserved phosphoprotein interaction motif is functionally interchangeable between ataxin-7 and arrestins. Biochemistry. 2000;39:6809–6813. doi: 10.1021/bi992694y. doi:10.1021/bi992694y. [DOI] [PubMed] [Google Scholar]
- 34.Scheel H., Tomiuk S., Hofmann K. Elucidation of ataxin-3 and ataxin-7 function by integrative bioinformatics. Hum. Mol. Genet. 2003;12:2845–2852. doi: 10.1093/hmg/ddg297. doi:10.1093/hmg/ddg297. [DOI] [PubMed] [Google Scholar]
- 35.Helmlinger D., Hardy S., Eberlin A., Devys D., Tora L. Both normal and polyglutamine-expanded ataxin-7 are components of TFTC-type GCN5 histone acetyltransferase- containing complexes. Biochem. Soc. Symp. 2006;73:155–163. doi: 10.1042/bss0730155. [DOI] [PubMed] [Google Scholar]
- 36.Helmlinger D., Hardy S., Abou-Sleymane G., Eberlin A., Bowman A.B., Gansmüller A., Picaud S., Zoghbi H.Y., Trottier Y., Tora L., et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. doi:10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helmlinger D., Tora L., Devys D. Transcriptional alterations and chromatin remodeling in polyglutamine diseases. Trends Genet. 2006;22:562–570. doi: 10.1016/j.tig.2006.07.010. doi:10.1016/j.tig.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Taylor J., Grote S.K., Xia J., Vandelft M., Graczyk J., Ellerby L.M., La Spada A.R., Truant R. Ataxin-7 can export from the nucleus via a conserved exportin-dependent signal. J. Biol. Chem. 2006;281:2730–2739. doi: 10.1074/jbc.M506751200. doi:10.1074/jbc.M506751200. [DOI] [PubMed] [Google Scholar]
- 39.Yoo S.Y., Pennesi M.E., Weeber E.J., Xu B., Atkinson R., Chen S., Armstrong D.L., Wu S.M., Sweatt J.D., Zoghbi H.Y. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/s0896-6273(02)01190-x. doi:10.1016/S0896-6273(02)01190-X. [DOI] [PubMed] [Google Scholar]
- 40.Cancel G., Duyckaerts C., Holmberg M., Zander C., Yvert G., Lebre A.S., Ruberg M., Faucheux B., Agid Y., Hirsch E., et al. Distribution of ataxin-7 in normal human brain and retina. Brain. 2000;123:2519–2530. doi: 10.1093/brain/123.12.2519. doi:10.1093/brain/123.12.2519. [DOI] [PubMed] [Google Scholar]
- 41.Mauger C., Del-Favero J., Ceuterick C., Lubke U., van Broeckhoven C., Martin J. Identification and localization of ataxin-7 in brain and retina of a patient with cerebellar ataxia type II using anti-peptide antibody. Brain Res. Mol. Brain Res. 1999;74:35–43. doi: 10.1016/s0169-328x(99)00256-9. doi:10.1016/S0169-328X(99)00256-9. [DOI] [PubMed] [Google Scholar]
- 42.Garden G.A., La Spada A.R. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum. 2008;7:138–149. doi: 10.1007/s12311-008-0027-y. doi:10.1007/s12311-008-0027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yvert G., Lindenberg K.S., Picaud S., Landwehrmeyer G.B., Sahel J.A., Mandel J.L. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum. Mol. Genet. 2000;9:2491–2506. doi: 10.1093/hmg/9.17.2491. doi:10.1093/hmg/9.17.2491. [DOI] [PubMed] [Google Scholar]
- 44.Yvert G., Lindenberg K.S., Devys D., Helmlinger D., Landwehrmeyer G.B., Mandel J.L. SCA7 mouse models show selective stabilization of mutant ataxin-7 and similar cellular responses in different neuronal cell types. Hum. Mol. Genet. 2001;10:1679–1692. doi: 10.1093/hmg/10.16.1679. doi:10.1093/hmg/10.16.1679. [DOI] [PubMed] [Google Scholar]
- 45.La Spada A.R., Fu Y.H., Sopher B.L., Libby R.T., Wang X., Li L.Y., Einum D.D., Huang J., Possin D.E., Smith A.C., et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31:913–927. doi: 10.1016/s0896-6273(01)00422-6. doi:10.1016/S0896-6273(01)00422-6. [DOI] [PubMed] [Google Scholar]
- 46.Lindenberg K.S., Yvert G., Müller K., Landwehrmeyer G.B. Expression analysis of ataxin-7 mRNA and protein in human brain: evidence for a widespread distribution and focal protein accumulation. Brain Pathol. 2000;10:385–394. doi: 10.1111/j.1750-3639.2000.tb00270.x. doi:10.1111/j.1750-3639.2000.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holmberg M., Duyckaerts C., Durr A., Cancel G., Gourfinkel-An I., Damier P., Faucheux B., Trottier Y., Hirsch E.C., Agid Y., et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum. Mol. Genet. 1998;7:913–918. doi: 10.1093/hmg/7.5.913. doi:10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- 48.Boxem M., Maliga Z., Klitgord N., Li N., Lemmens I., Mana M., de Lichtervelde L., Mul J.D., van de Peut D., Devos M., et al. A protein domain-based interactome network for C. elegans early embryogenesis. Cell. 2008;134:534–545. doi: 10.1016/j.cell.2008.07.009. doi:10.1016/j.cell.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young J.E., Gouw L., Propp S., Sopher B.L., Taylor J., Lin A., Hermel E., Logvinova A., Chen S.F., Chen S., et al. Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcriptional dysregulation. J. Biol. Chem. 2007;282:30150–30160. doi: 10.1074/jbc.M705265200. doi:10.1074/jbc.M705265200. [DOI] [PubMed] [Google Scholar]
- 50.Li S., Armstrong C.M., Bertin N., Ge H., Milstein S., Boxem M., Vidalain P.O., Han J.D., Chesneau A., Hao T., et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. doi:10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rual J.-F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., Ayivi-Guedehoussou N., et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. doi:10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 52.Barabasi A.L., Oltvai Z.N. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. doi:10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 53.Gulbahce N., Lehmann S. The art of community detection. Bioessays. 2008;30:934–938. doi: 10.1002/bies.20820. doi:10.1002/bies.20820. [DOI] [PubMed] [Google Scholar]
- 54.Barabasi A.L., Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. doi:10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 55.Yu H., Braun P., Yildirim M.A., Lemmens I., Venkatesan K., Sahalie J., Hirozane-Kishikawa T., Gebreab F., Li N., Simonis N., et al. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322:104–110. doi: 10.1126/science.1158684. doi:10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu H., Kim P.M., Sprecher E., Trifonov V., Gerstein M. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007;3:e59. doi: 10.1371/journal.pcbi.0030059. doi:10.1371/journal.pcbi.0030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chuang H.Y., Lee E., Liu Y.T., Lee D., Ideker T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han J.D., Bertin N., Hao T., Goldberg D.S., Berriz G.F., Zhang L.V., Dupuy D., Walhout A.J., Cusick M.E., Roth F.P., et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. doi:10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 59.Taylor I.W., Linding R., Warde-Farley D., Liu Y., Pesquita C., Faria D., Bull S., Pawson T., Morris Q., Wrana J.L. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009;27:199–204. doi: 10.1038/nbt.1522. doi:10.1038/nbt.1522. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J., Zhang C., Lamb J., Edwards S., Sieberts S.K., et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. doi:10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ideker T., Sharan R. Protein networks in disease. Genome Res. 2008;18:644–652. doi: 10.1101/gr.071852.107. doi:10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hidalgo C.A., Blumm N., Barabasi A.L., Christakis N.A. A dynamic network approach for the study of human phenotypes. PLoS Comput. Biol. 2009;5:e1000353. doi: 10.1371/journal.pcbi.1000353. doi:10.1371/journal.pcbi.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park J., Lee D.S., Christakis N.A., Barabasi A.L. The impact of cellular networks on disease comorbidity. Mol. Syst. Biol. 2009;5:262. doi: 10.1038/msb.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rzhetsky A., Wajngurt D., Park N., Zheng T. Probing genetic overlap among complex human phenotypes; Proc. Natl Acad. Sci. USA; 2007. pp. 11694–11699. doi:10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blackshaw S., Harpavat S., Trimarchi J., Cai L., Huang H., Kuo W.P., Weber G., Lee K., Fraioli R.E., Cho S.-H., et al. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:E247. doi: 10.1371/journal.pbio.0020247. doi:10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hublitz P., Kunowska N., Mayer U.P., Muller J.M., Heyne K., Yin N., Fritzsche C., Poli C., Miguet L., Schupp I.W., et al. NIR is a novel INHAT repressor that modulates the transcriptional activity of p53. Genes Dev. 2005;19:2912–2924. doi: 10.1101/gad.351205. doi:10.1101/gad.351205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piri N., Kwong J.M., Song M., Elashoff D., Caprioli J. Gene expression changes in the retina following optic nerve transection. Mol. Vis. 2006;12:1660–1673. [PubMed] [Google Scholar]
- 68.Sarthy P.V., Fu M. Localization of laminin B1 mRNA in retinal ganglion cells by in situ hybridization. J. Cell Biol. 1990;110:2099–2108. doi: 10.1083/jcb.110.6.2099. doi:10.1083/jcb.110.6.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brady M.E., Ozanne D.M., Gaughan L., Waite I., Cook S., Neal D.E., Robson C.N. Tip60 is a nuclear hormone receptor coactivator. J. Biol. Chem. 1999;274:17599–17604. doi: 10.1074/jbc.274.25.17599. doi:10.1074/jbc.274.25.17599. [DOI] [PubMed] [Google Scholar]
- 70.Ström A.L., Jonasson J., Hart P., Brännström T., Forsgren L., Holmberg M. Cloning and expression analysis of the murine homolog of the spinocerebellar ataxia type 7 (SCA7) gene. Gene. 2002;285:91–99. doi: 10.1016/s0378-1119(02)00399-2. doi:10.1016/S0378-1119(02)00399-2. [DOI] [PubMed] [Google Scholar]
- 71.Zhang C.L., Zou Y., Yu R.T., Gage F.H., Evans R.M. Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev. 2006;20:1308–1320. doi: 10.1101/gad.1413606. doi:10.1101/gad.1413606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schulz H.L., Goetz T., Kaschkoetoe J., Weber B.H.F. The Retinome—defining a reference transcriptome of the adult mammalian retina/retinal pigment epithelium. BMC Genomics. 2004;5:50. doi: 10.1186/1471-2164-5-50. doi:10.1186/1471-2164-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barbet F., Gerber S., Hakiki S., Perrault I., Hanein S., Ducroq D., Tanguy G., Dufier J.L., Munnich A., Rozet J.M., et al. A first locus for isolated autosomal recessive optic atrophy (ROA1) maps to chromosome 8q. Eur. J. Hum. Genet. 2003;11:966–971. doi: 10.1038/sj.ejhg.5201070. doi:10.1038/sj.ejhg.5201070. [DOI] [PubMed] [Google Scholar]
- 74.Barbet F., Hakiki S., Orssaud C., Gerber S., Perrault I., Hanein S., Ducroq D., Dufier J.L., Munnich A., Kaplan J., et al. A third locus for dominant optic atrophy on chromosome 22q. J. Med. Genet. 2005;42:e1. doi: 10.1136/jmg.2004.025502. doi:10.1136/jmg.2004.025502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuchshofer R., Stephan D.A., Russell P., Tamm E.R. Gene expression profiling of TGFbeta2- and/or BMP7-treated trabecular meshwork cells: identification of Smad7 as a critical inhibitor of TGF-beta2 signaling. Exp. Eye Res. 2009;88:1020–1032. doi: 10.1016/j.exer.2009.01.002. doi:10.1016/j.exer.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Keen T.J., Mohamed M.D., McKibbin M., Rashid Y., Jafri H., Maumenee I.H., Inglehearn C.F. Identification of a locus (LCA9) for Leber's congenital amaurosis on chromosome 1p36. Eur. J. Hum. Genet. 2003;11:420–423. doi: 10.1038/sj.ejhg.5200981. doi:10.1038/sj.ejhg.5200981. [DOI] [PubMed] [Google Scholar]
- 77.Kylstra J.A., Aylsworth A.S. Cone-rod retinal dystrophy in a patient with neurofibromatosis type 1. Can. J. Ophthalmol. 1993;28:79–80. [PubMed] [Google Scholar]
- 78.Lovicu F.J., Kolle G., Yamada T., Little M.H., McAvoy J.W. Expression of Crim1 during murine ocular development. Mech. Dev. 2000;94:261–265. doi: 10.1016/s0925-4773(00)00292-6. doi:10.1016/S0925-4773(00)00292-6. [DOI] [PubMed] [Google Scholar]
- 79.Michaelides M., Johnson S., Tekriwal A.K., Holder G.E., Bellmann C., Kinning E., Woodruff G., Trembath R.C., Hunt D.M., Moore A.T. An early-onset autosomal dominant macular dystrophy (MCDR3) resembling North Carolina macular dystrophy maps to chromosome 5. Invest. Ophthalmol. Vis. Sci. 2003;44:2178–2183. doi: 10.1167/iovs.02-1094. doi:10.1167/iovs.02-1094. [DOI] [PubMed] [Google Scholar]
- 80.Pal B., Mohamed M.D., Keen T.J., Williams G.A., Bradbury J.A., Sheridan E., Inglehearn C.F. A new phenotype of recessively inherited foveal hypoplasia and anterior segment dysgenesis maps to a locus on chromosome 16q23.2–24.2. J. Med. Genet. 2004;41:772–777. doi: 10.1136/jmg.2004.020040. doi:10.1136/jmg.2004.020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pennisi D.J., Wilkinson L., Kolle G., Sohaskey M.L., Gillinder K., Piper M.J., McAvoy J.W., Lovicu F.J., Little M.H. Crim1KST264/KST264 mice display a disruption of the Crim1 gene resulting in perinatal lethality with defects in multiple organ systems. Dev. Dyn. 2007;236:502–511. doi: 10.1002/dvdy.21015. doi:10.1002/dvdy.21015. [DOI] [PubMed] [Google Scholar]
- 82.Pentao L., Lewis R.A., Ledbetter D.H., Patel P.I., Lupski J.R. Maternal uniparental isodisomy of chromosome 14: association with autosomal recessive rod monochromacy. Am. J. Hum. Genet. 1992;50:690–699. [PMC free article] [PubMed] [Google Scholar]
- 83.Siegert S., Scherf B.G., Del Punta K., Didkovsky N., Heintz N., Roska B. Genetic address book for retinal cell types. Nat. Neurosci. 2009;12:1197–1204. doi: 10.1038/nn.2370. doi:10.1038/nn.2370. [DOI] [PubMed] [Google Scholar]
- 84.Stone E.M., Kimura A.E., Folk J.C., Bennett S.R., Nichols B.E., Streb L.M., Sheffield V.C. Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy to chromosome 11q13. Hum. Mol. Genet. 1992;1:685–689. doi: 10.1093/hmg/1.9.685. doi:10.1093/hmg/1.9.685. [DOI] [PubMed] [Google Scholar]
- 85.Stone E.M., Lotery A.J., Munier F.L., Héon E., Piguet B., Guymer R.H., Vandenburgh K., Cousin P., Nishimura D., Swiderski R.E., et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999;22:199–202. doi: 10.1038/9722. doi:10.1038/9722. [DOI] [PubMed] [Google Scholar]
- 86.Weigell-Weber M., Sarra G.M., Kotzot D., Sandkuijl L., Messmer E., Hergersberg M. Genomewide homozygosity mapping and molecular analysis of a candidate gene located on 22q13 (fibulin-1) in a previously undescribed vitreoretinal dystrophy. Arch. Ophthalmol. 2003;121:1184–1188. doi: 10.1001/archopht.121.8.1184. doi:10.1001/archopht.121.8.1184. [DOI] [PubMed] [Google Scholar]
- 87.Xu H.P., Zhao J.W., Yang X.L. Cholinergic and dopaminergic amacrine cells differentially express calcium channel subunits in the rat retina. Neuroscience. 2003;118:763–768. doi: 10.1016/s0306-4522(03)00022-8. doi:10.1016/S0306-4522(03)00022-8. [DOI] [PubMed] [Google Scholar]
- 88.Yang Z., Kitsos G., Tong Z., Payne M., Gorezis S., Psilas K., Grigoriadou M., Zhao Y., Kamaya S., Aperis G., et al. A novel locus on 19q13 associated with autosomal-dominant macular dystrophy in a large Greek family. J. Med. Genet. 2006;43:e57. doi: 10.1136/jmg.2005.040188. doi:10.1136/jmg.2005.040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yego E.C.K., Mohr S. siah-1 Protein is necessary for high glucose-induced glyceraldehyde-3-phosphate dehydrogenase nuclear accumulation and cell death in Muller cells. J. Biol. Chem. 2010;285:3181–3190. doi: 10.1074/jbc.M109.083907. doi:10.1074/jbc.M109.083907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aleman T.S., Cideciyan A.V., Volpe N.J., Stevanin G., Brice A., Jacobson S.G. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp. Eye Res. 2002;74:737–745. doi: 10.1006/exer.2002.1169. doi:10.1006/exer.2002.1169. [DOI] [PubMed] [Google Scholar]
- 91.Assink J.J., Tijmes N.T., ten Brink J.B., Oostra R.J., Riemslag F.C., de Jong P.T., Bergen A.A. A gene for X-linked optic atrophy is closely linked to the Xp11.4–Xp11.2 region of the X chromosome. Am. J. Hum. Genet. 1997;61:934–939. doi: 10.1086/514884. doi:10.1086/514884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benomar A., Krols L., Stevanin G., Cancel G., LeGuern E., David G., Ouhabi H., Martin J.J., Durr A., Zaim A., et al. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12–p21.1. Nat. Genet. 1995;10:84–88. doi: 10.1038/ng0595-84. doi:10.1038/ng0595-84. [DOI] [PubMed] [Google Scholar]
- 93.Del-Favero J., Krols L., Michalik A., Theuns J., Lofgren A., Goossens D., Wehnert A., Van den Bossche D., Van Zand K., Backhovens H., et al. Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA type II) caused by CAG triplet repeat expansion. Hum. Mol. Genet. 1998;7:177–186. doi: 10.1093/hmg/7.2.177. doi:10.1093/hmg/7.2.177. [DOI] [PubMed] [Google Scholar]
- 94.Fingert J.H., Honkanen R.A., Shankar S.P., Affatigato L.M., Ehlinger M.A., Moore M.D., Jampol L.M., Sheffield V.C., Stone E.M., Alward W.L. Familial cavitary optic disk anomalies: identification of a novel genetic locus. Am. J. Ophthalmol. 2007;143:795–800. doi: 10.1016/j.ajo.2007.01.042. doi:10.1016/j.ajo.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Francis P.J., Johnson S., Edmunds B., Kelsell R.E., Sheridan E., Garrett C., Holder G.E., Hunt D.M., Moore A.T. Genetic linkage analysis of a novel syndrome comprising North Carolina-like macular dystrophy and progressive sensorineural hearing loss. Br. J. Ophthalmol. 2003;87:893–898. doi: 10.1136/bjo.87.7.893. doi:10.1136/bjo.87.7.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Honkanen R.A., Jampol L.M., Fingert J.H., Moore M.D., Taylor C.M., Stone E.M., Alward W.L. Familial cavitary optic disk anomalies: clinical features of a large family with examples of progressive optic nerve head cupping. Am. J. Ophthalmol. 2007;143:788–794. doi: 10.1016/j.ajo.2007.01.029. doi:10.1016/j.ajo.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 97.Hughes A.E., Lotery A.J., Silvestri G. Fine localisation of the gene for central areolar choroidal dystrophy on chromosome 17p. J. Med. Genet. 1998;35:770–772. doi: 10.1136/jmg.35.9.770. doi:10.1136/jmg.35.9.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kinna G., Kolle G., Carter A., Key B., Lieschke G.J., Perkins A., Little M.H. Knockdown of zebrafish crim1 results in a bent tail phenotype with defects in somite and vascular development. Mech. Dev. 2006;123:277–287. doi: 10.1016/j.mod.2006.01.003. doi:10.1016/j.mod.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 99.Lotery A.J., Ennis K.T., Silvestri G., Nicholl S., McGibbon D., Collins A.D., Hughes A.E. Localisation of a gene for central areolar choroidal dystrophy to chromosome 17p. Hum. Mol. Genet. 1996;5:705–708. doi: 10.1093/hmg/5.5.705. doi:10.1093/hmg/5.5.705. [DOI] [PubMed] [Google Scholar]
- 100.Einum D.D., Townsend J.J., Ptácek L.J., Fu Y.H. Ataxin-7 expression analysis in controls and spinocerebellar ataxia type 7 patients. Neurogenetics. 2001;3:83–90. doi: 10.1007/s100480000100. doi:10.1007/s100480000100. [DOI] [PubMed] [Google Scholar]
- 101.Bloor A.J., Kotsopoulou E., Hayward P., Champion B.R., Green A.R. RFP represses transcriptional activation by bHLH transcription factors. Oncogene. 2005;24:6729–6736. doi: 10.1038/sj.onc.1208828. doi:10.1038/sj.onc.1208828. [DOI] [PubMed] [Google Scholar]
- 102.Shimono Y., Murakami H., Hasegawa Y., Takahashi M. RET finger protein is a transcriptional repressor and interacts with enhancer of polycomb that has dual transcriptional functions. J. Biol. Chem. 2000;275:39411–39419. doi: 10.1074/jbc.M006585200. doi:10.1074/jbc.M006585200. [DOI] [PubMed] [Google Scholar]
- 103.Cao T., Borden K.L., Freemont P.S., Etkin L.D. Involvement of the rfp tripartite motif in protein–protein interactions and subcellular distribution. J. Cell Sci. 1997;110:1563–1571. doi: 10.1242/jcs.110.14.1563. [DOI] [PubMed] [Google Scholar]
- 104.Grabiner B.C., Blonska M., Lin P.-C., You Y., Wang D., Sun J., Darnay B.G., Dong C., Lin X. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-{kappa}B activation. Genes Dev. 2007;21:984–996. doi: 10.1101/gad.1502507. doi:10.1101/gad.1502507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ishikawa K., Fujigasaki H., Saegusa H., Ohwada K., Fujita T., Iwamoto H., Komatsuzaki Y., Toru S., Toriyama H., Watanabe M., et al. Abundant expression and cytoplasmic aggregations of [alpha]1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6. Hum. Mol. Genet. 1999;8:1185–1193. doi: 10.1093/hmg/8.7.1185. doi:10.1093/hmg/8.7.1185. [DOI] [PubMed] [Google Scholar]
- 106.McMahon S.J., Pray-Grant M.G., Schieltz D., Yates J.R., 3rd, Grant P.A. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity; Proc. Natl Acad. Sci. USA; 2005. pp. 8478–8482. doi:10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marmorstein L.Y., Munier F.L., Arsenijevic Y., Schorderet D.F., McLaughlin P.J., Chung D., Traboulsi E., Marmorstein A.D. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration; Proc. Natl Acad. Sci. USA; 2002. pp. 13067–13072. doi:10.1073/pnas.202491599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roybal C.N., Marmorstein L.Y., Vander Jagt D.L., Abcouwer S.F. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest. Ophthalmol. Vis. Sci. 2005;46:3973–3979. doi: 10.1167/iovs.05-0070. doi:10.1167/iovs.05-0070. [DOI] [PubMed] [Google Scholar]
- 109.Marmorstein L.Y., McLaughlin P.J., Peachey N.S., Sasaki T., Marmorstein A.D. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum. Mol. Genet. 2007;16:2423–2432. doi: 10.1093/hmg/ddm199. doi:10.1093/hmg/ddm199. [DOI] [PubMed] [Google Scholar]
- 110.Fu L., Garland D., Yang Z., Shukla D., Rajendran A., Pearson E., Stone E.M., Zhang K., Pierce E.A. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum. Mol. Genet. 2007;16:2411–2422. doi: 10.1093/hmg/ddm198. doi:10.1093/hmg/ddm198. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Z., Alpert D., Francis R., Chatterjee B., Yu Q., Tansey T., Sabol S.L., Cui C., Bai Y., Koriabine M., et al. Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy; Proc. Natl Acad. Sci. USA; 2009. pp. 3219–3224. doi:10.1073/pnas.0813400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Der Kaloustian V.M., Jarudi N.I., Khoury M.J., Afifi A.K., Bahuth N.B., Deeb M.E., Shammas J., Mikati M.A. Familial spinocerebellar degeneration with corneal dystrophy. Am. J. Med. Genet. 1985;20:325–339. doi: 10.1002/ajmg.1320200216. doi:10.1002/ajmg.1320200216. [DOI] [PubMed] [Google Scholar]
- 113.Hagberg B., Haltia M., Sourander P., Svennerholm L., Eeg-Olofsson O. Polyunsaturated fatty acid lipidosis infantile form of so-called neuronal ceroidlipofuscinosis. Acta Paediatr. Scand. 1974;63:753–763. doi: 10.1111/j.1651-2227.1974.tb17001.x. doi:10.1111/j.1651-2227.1974.tb17001.x. [DOI] [PubMed] [Google Scholar]
- 114.Jbour A.K., Mubaidin A.F., Till M., El-Shanti H., Hadidi A., Ajlouni K.M. Hypogonadotrophic hypogonadism, short stature, cerebellar ataxia, rod-cone retinal dystrophy, and hypersegmented neutrophils: a novel disorder or a new variant of Boucher-Neuhauser syndrome? J. Med. Genet. 2003;40:e2. doi: 10.1136/jmg.40.1.e2. doi:10.1136/jmg.40.1.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mousa A.R., Al-Din A.S., Al-Nassar K.E., Al-Rifai K.M., Rudwan M., Sunba M.S., Behbehani K. Autosomally inherited recessive spastic ataxia, macular corneal dystrophy, congenital cataracts, myopia and vertically oval temporally tilted discs. Report of a Bedouin family—a new syndrome. J. Neurol. Sci. 1986;76:105–121. doi: 10.1016/0022-510x(86)90145-0. doi:10.1016/0022-510X(86)90145-0. [DOI] [PubMed] [Google Scholar]
- 116.Mullen R.J., Eicher E.M., Sidman R.L. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl Acad. Sci. USA. 1976;73:208–212. doi: 10.1073/pnas.73.1.208. doi:10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Porto F.B., Mack G., Sterboul M.J., Lewin P., Flament J., Sahel J., Dollfus H. Isolated late-onset cone-rod dystrophy revealing a familial neurogenic muscle weakness, ataxia, and retinitis pigmentosa syndrome with the T8993G mitochondrial mutation. Am. J. Ophthalmol. 2001;132:935–937. doi: 10.1016/s0002-9394(01)01187-4. doi:10.1016/S0002-9394(01)01187-4. [DOI] [PubMed] [Google Scholar]
- 118.Berrebi A.S., Oberdick J., Sangameswaran L., Christakos S., Morgan J.I., Mugnaini E. Cerebellar Purkinje cell markers are expressed in retinal bipolar neurons. J. Comp. Neurol. 1991;308:630–649. doi: 10.1002/cne.903080409. doi:10.1002/cne.903080409. [DOI] [PubMed] [Google Scholar]
- 119.Lamesch P., Li N., Milstein S., Fan C., Hao T., Szabo G., Hu Z., Venkatesan K., Bethel G., Martin P., et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307–315. doi: 10.1016/j.ygeno.2006.11.012. doi:10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. doi:10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Katz D., Baptista J., Azen S.P., Pike M.C. Obtaining confidence intervals for the risk ratio in cohort studies. Biometrics. 1978;34:469–474. doi:10.2307/2530610. [Google Scholar]
- 122.Carson J.P., Thaller C., Eichele G. A transcriptome atlas of the mouse brain at cellular resolution. Curr. Opin. Neurobiol. 2002;12:562–565. doi: 10.1016/s0959-4388(02)00356-2. doi:10.1016/S0959-4388(02)00356-2. [DOI] [PubMed] [Google Scholar]
- 123.Fyffe S.L., Neul J.L., Samaco R.C., Chao H.T., Ben-Shachar S., Moretti P., McGill B.E., Goulding E.H., Sullivan E., Tecott L.H., et al. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. doi:10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Visel A., Thaller C., Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–D556. doi: 10.1093/nar/gkh029. doi:10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yaylaoglu M.B., Titmus A., Visel A., Alvarez-Bolado G., Thaller C., Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev. Dyn. 2005;234:371–386. doi: 10.1002/dvdy.20441. doi:10.1002/dvdy.20441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.