

Summary
Porphyromonas gingivalis, a Gram-negative oral anaerobe, is associated with periodontitis, a disease that in some form affects up to 80% of the adult population in the United States. The organism interacts with gingival epithelium and surrounding tissue, and in this study we analyzed interactions initiated by P. gingivalis and by a peptide derived from the adhesin domain of arg-gingipain A, a member of a family of surface cysteine proteinases. Recombinant peptide A44 blocked adherence of bacteria to host cell monolayers, and bound to components of the cell membrane fraction. In pull-down assays A44 associated with proteins involved in a clathrin-dependent endocytosis pathway. Inhibitor studies confirmed a role for clathrin, and confocal microscopy demonstrated that both A44-coated beads and intact bacteria colocalized with GFP-clathrin in host cells. Finally, we used siRNA to determine whether clathrin or caveolin-1 was involved in association of peptide and intact bacteria with host cells. Again, the results of these assays indicated that association of both A44 and P. gingivalis depended on the presence of clathrin, and support a working model in which A44 initiates a clathrin-dependent pathway that potentially leads to internalization of peptide or bacteria by host epithelial cells.
Introduction
Porphyromonas gingivalis, a Gram-negative oral anaerobe, is associated with periodontal disease. It is estimated that up to 80% of the adult population in the United States has some form of infection ranging from gingivitis to severe refractory disease (http://www.nidcr.nih.gov/HealthInformation/DiseasesAndConditions/GumPeriodontalDiseases/PeriodontalDiseases.htm). The ecological niche of P. gingivalis is the subgingival biofilm where it interacts with other bacteria and epithelial cells that line the gingival sulcus. High disease prevalence justifies the need for effective therapeutic interventions, and an understanding of how bacterial and host proteins interact will open up a new inventory of targets for interventions that prevent or cure infection.
An early step in infection is colonization of the gingival junctional epithelium, and within the last decade numerous studies have implicated P. gingivalis surface structures, i.e. fimbriae and gingipain-associated adhesins, in adherence processes. Studies with either native fimbriae or recombinant fimbrilin (FimA, the subunit protein of fimbriae) focused on both its pathogenic effects and host cell receptors. In mouse embryo calvariae, native fimbriae induced bone resorption that was inhibited by fibronectin, suggesting that fimbriae use β1 integrins as host cell receptors (Kawata et al., 1997). In further support of this association, the adherence of recombinant type II fimbrilin-coated microspheres to HEp-2 cells was inhibited by anti-α5β1 antibody in a dose-dependent manner (Nakagawa et al., 2002). Binding of recombinant fimbrilin to β1 integrin in primary gingival epithelial cells was established by direct assay and subsequently fimbriae–integrin signaling was documented (Yilmaz et al., 2002; Yilmaz et al., 2003). Furthermore, it was demonstrated that P. gingivalis cells and fimbriae competed with extracellular matrix protein ligands for integrin receptors (Nakagawa et al., 2005). Pathways independent of fimbriae-integrin binding were also postulated since fimA mutants still showed (reduced) invasion of gingival epithelial cells (Yilmaz et al., 2002), and alternate receptors for fimbriae were detected (Weinberg et al., 1997).
Gingipains, a family of cysteine proteinases produced by P. gingivalis, are present on the cell surface, in extracellular vesicles, and in culture supernatants. Gingipains also interact with host epithelial cells, and a considerable body of work has characterized the damaging effects of their proteolytic activities on host proteins (Pike et al., 1996) and on the integrity of epithelial cell monolayers (Katz et al., 2000; Chen et al., 2001a; Katz et al., 2002). However, a key study suggested that gingipain adhesin domains may be involved in targeting their associated proteolytic activities to cell surface receptors, since colocalization of the arg-gingipain (RgpA) adhesin domain with fibronectin and the α5β1 integrin receptor for fibronectin was visualized (Scragg et al., 1999). Subsequently, the intracellular localization of the RgpA adhesin domain within KB oral epithelial cells was demonstrated (Scragg et al., 2002), indicating a potential role for adhesins in bacterial internalization.
Previously, using both genetic and biochemical approaches, we showed that adhesin domains of RgpA, lys-gingipain (Kgp), and possibly the related domains of hemagglutinin A (HagA) bound to epithelial cells (Chen et al., 2001b). From these data we hypothesized that bacterial adherence to host cells was also gingipain-mediated, supported by the demonstration that binding of both gingipain adhesins and P. gingivalis to HEp-2 (HeLa) cell monolayers, our in vitro epithelial cell model system, was blocked by antibodies to the adhesin domain of RgpA (Chen and Duncan, 2004). In the present study we focused on peptide A44 derived from the adhesin domain of RgpA to define its role in adherence to and internalization by epithelial cells. Using binding assays, inhibitor studies, confocal microscopy, and siRNA we demonstrated for the first time that entry of A44 and P. gingivalis into HEp-2 epithelial cells was dependent on host cell clathrin.
Results
Recombinant (r-) A44 binds to host proteins
The gingipain family comprises four proteins, three of which have catalytic activity. Arg-gingipains A and B (RgpA and RgpB) cleave proteins after arginine residues, while lys-gingipain (Kgp) cleaves after lysine. In addition to catalytic domains, RgpA and Kgp also possess adhesin domains, while a fourth related protein, HagA, contains only repeats of the adhesin domain. High molecular weight, unprocessed gingipains, and autoprocessed adhesin domains are located on the P. gingivalis outer membrane, in membrane vesicles, and are also found as free proteins in culture media (Potempa et al., 1995; Bhogal et al., 1997; Chen et al., 2001b; Veith et al., 2002; Takii et al., 2005). The order and approximate molecular weights of peptides within the adhesin domain of RgpA are shown in Fig. 1A. In order to characterize binding and biological properties, individual adhesin peptides were cloned, expressed as His-tag fusion proteins for detection purposes, and purified from Escherichia coli.
Fig. 1. Structure of arg-gingipain adhesin domain and binding properties of A44.

A. Peptides are named according to their RgpA source and approximate molecular weights. Individual peptides were expressed as recombinant His-tag fusions in E. coli BL21.
B. Binding of native and r-A44 peptides to fibronectin and fibrinogen was measured by ELISA using anti-A44 specific primary and HRP-conjugated secondary antibodies. Key:  r-A44 binding to fibrinogen;
r-A44 binding to fibrinogen;  n-A44 binding to fibrinogen;
n-A44 binding to fibrinogen;  r-A44 binding to fibronectin;
r-A44 binding to fibronectin; 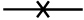 n-A44 binding to fibronectin.
n-A44 binding to fibronectin.
Previously, host proteins fibrinogen and fibronectin were shown to be in vitro substrates for native gingipain proteolytic and adhesin binding activities (Pike et al., 1996). We compared and quantified binding of native and r-A44 peptides to fibrinogen and fibronectin by ELISA. The data presented in Fig. 1B were obtained with low peptide concentrations in order to distinguish differences, and bound peptides were detected with rabbit anti-A44 primary and horse radish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies. Recombinant and native peptides had nearly identical binding affinities for fibronectin, while binding of native A44 to fibrinogen was very similar to, but slightly better than the r-peptide. According to dissociation constants (KD) calculated from the ELISA data (Bozzini et al., 1992), that for r-A44 binding to fibrinogen was 2.4 × 10−9 M compared to 2.8 × 10−9 M for the native peptide, and values for rand n-A44 binding to fibronectin were 2.9 × 10−9 and 3.0 × 10−9, respectively. Thus, we concluded that binding properties of the recombinant and native peptides were very similar, and indicated high affinities of each peptide for each substrate.
Peptide A44 interacts with HEp-2 cells and blocks binding of P. gingivalis
To relate adhesin peptide-host protein interactions to the biological context of our in vitro infection model, we tested whether r-A44 could bind to HEp-2 epithelial cells in a modified capture assay (Chen et al., 2001b). The peptide was incubated with HEp-2 monolayers and after washing to remove unbound peptide, monolayers were lysed and extracts fractionated by SDS-PAGE and blotted to membranes for probing with anti-His-tag primary and HRP-conjugated secondary antibodies. In the SDS-PAGE gel shown in Fig. 2A, A44 was present in the HEp-2 cell lysate indicating that it bound to the monolayers. Next, we tested whether A44 blocked adherence of P. gingivalis cells to HEp-2 cells. The peptide significantly blocked P. gingivalis adherence with almost 40% inhibition achieved at a low concentration of 0.025 µM (Fig. 2B), however, the interaction appeared saturable since higher concentrations of peptide only inhibited adherence an additional 10%.
Fig. 2. A44 blocks binding of P. gingivalis to HEp-2 cells.

A. Western blot of HEp-2 lysate after incubation with peptide A44. Presence of the peptide in HEp-2 lysates was detected with anti-His-tag primary and HRP-conjugated secondary antibodies.
B. Adherence of P. gingivalis to HEp-2 monolayers in the presence of A44. Data were obtained from at least three independent experiments in which each data point was obtained from triplicate assays. The number of bacteria that adhered to HEp-2 cells in the absence of A44 was designated as 100%, and the number of bacteria that adhered in the presence of peptide was expressed as relative to this control value (± standard deviation).
Finally, latex-beads were coated with A44, control beads with bovine serum albumin (BSA), and their interactions with HEp-2 monolayers were examined by scanning and transmission electron microscopy. Beads coated with BSA did not bind to HEp-2 cells (not shown) while A44-coated beads aggregated and remained mostly at the surface of cells (Figs. 3A and B). However, by transmission electron microscopy (Figs. 3C and D), two sections show single beads that were internalized and enclosed in vacuoles. These results suggested that contact of A44 with the epithelial cell membrane could initiate an entry pathway.
Fig. 3. Interactions of A44-coated latex beads with HEp-2 cells.

Images of HEp-2 cells were taken after 1 h incubation with A44-coated beads. Panels A and B show scanning, and C and D transmission microscopy of samples.
Interactions of A44 with HEp-2 proteins
To gain further insight into adherence of A44 we examined binding to HEp-2 cytoplasmic, membrane, and cytoskeletal protein fractions isolated by the method of Kenny and Finlay (1997). Proteins in each fraction were separated by SDS-PAGE and either Coomassie-stained or blotted to membranes and probed with peptides A44 (Fig. 4A). Binding of A44 to HEp-2 proteins was detected with anti-His-tag primary and HRP-labeled secondary antibodies and, as shown in Fig. 4A, A44 consistently interacted with a 175 kDa HEp-2 membrane protein.
Fig. 4. A44 binds to a HEp-2 membrane protein.

A. HEp-2 fractions were: Cp, cytoplasmic; M, membrane; and Sk, cytoskeleton. The first set of lanes shows Coomassie-stained SDS-PAGE of the fractions; the second shows a Western blot of the same fractions after overlay with A44. Binding of peptide A44 was detected with anti-His-tag primary and HRP-conjugated secondary antibodies.
B. Membrane protein(s) that bound to A44 were isolated by affinity purification using an A44-coupled Ni2+-NTA agarose column to isolate binding protein(s) from the membrane fraction of HEp-2 cells, as shown in the Coomassie-stained SDS-PAGE of HEp-2 proteins eluted from the column with 50 – 300 mM imidazole.
C. Coomassie-stained SDS-PAGE of the starting membrane fraction from HEp-2 cells (lane 1); an affinity purified fraction obtained after elution with 300 mM imidazole (lane 2); the 175 kDa band (the putative A44 binding protein) excised and electroeluted from SDS-PAGE gels of the HEp-2 cell membrane fraction (lane 3).
D. Far-Western blot of the samples depicted in C probed with A44. Peptide binding was detected with anti-His-tag primary and HRP-conjugated secondary antibodies.
The 175 kDa protein ligand of A44 was isolated from the membrane fraction of HEp-2 cells by pull-down assays in which the A44-His-tag peptide was coupled to Ni2+-agarose beads. Figure 4B shows a Coomassie-stained SDS-PAGE gel of membrane proteins of approximately 175 kDa that bound to A44 and were eluted from beads with increasing concentrations of imidazole. Figure 4C shows a Coomassie stained SDS-PAGE gel of the starting membrane protein fraction with an arrow indicating the 175 kDa region (lane 1), pull-down proteins eluted from agarose beads, including A44 itself (lane 2), and the excised and electroeluted 175 kDa protein region (lane 3). A duplicate of this gel was transferred to a nitrocellulose membrane and incubated with A44-His-tag peptide. Binding of A44 to the samples was detected with rabbit anti-His-tag primary and goat anti-rabbit HRP- conjugated secondary antibodies and the results indicated that A44 bound to the excised protein (Fig. 4D). Protein present in 175 kDa band was identified by MALDI-TOF mass spectrometry. Although the protein appeared to be a single band in the gel, two separate protein sequences were identified, clathrin and early endosome antigen 1 (EEA1), suggesting that A44 may be associated with clathrin-mediated endocytosis (CME).
To further understand the nature of A44-host cell interactions, we tested whether inhibitors of CME blocked the association of A44 and epithelial cells. We do not know the extent of A44 or P. gingivalis adherence to and internalization by HEp-2 cells in these experiments, therefore the term “association” is used to include both functions. Monolayers of HEp-2 cells were preincubated (45 min) in the absence or presence of inhibitors that prevented clathrin- or caveolae/lipid raft-mediated endocytosis, actin polymerization, microtubule formation, and macropinocytosis. The media were replaced with fresh medium containing A44 and incubation continued for a further 45 min. Aspirated media, and washed HEp-2 cell lysates were fractionated by SDS-PAGE, blotted to membranes and probed with anti-His-tag antibody to detect A44. Free peptide was present in media from all samples (Fig. 5A), however A44 was only detected in lysates of untreated HEp-2 cells and those pretreated with genistein, amiloride, nystatin, and filipin. Association of A44 with HEp-2 cells was inhibited by chlorpromazine, methyl-β-cyclodextrin, wortmannin (an inhibitor of PI3 kinase and autophagy), and cytochalasin D, suggesting a clathrin- and actin-dependent entry pathway, supported by the finding that genistein, an inhibitor of caveolae/raft endocytosis, did not block association. Nocodazole and paclitaxel, that disrupt microtubule formation, also blocked the association of peptide with HEp-2 cells. Association of A44 was not inhibited by genistein indicating that a caveolae/raft– dependent endocytic pathway was not involved, and lack of inhibition by nystatin and filipin support this notion. Resistance to amiloride indicated that the entry pathway did not involve macropinocytosis.
Fig. 5. Effects of inhibitors on association of A44 and P. gingivalis with HEp-2 cells.

A. Media (M) and lysates (L) of control and pretreated HEp-2 cells were fractionated by SDS-PAGE and blotted to membranes. Peptide A44 was detected with His-tag primary and HRP-conjugated secondary antibodies. Key: 0, no pretreatment; Cyt D, cytochalasin D; Cp, chlorpromazine; Gen, genistein; Mcd, methyl-β-cyclodextrin; Wor, wortmannin; Noc, nocodazole; Pac, paclitaxel; Amo, amiloride; Nys, nystatin; and Fil, filipin.
B. Monolayers were pretreated with inhibitors and washed before the addition of fresh media containing P. gingivalis. After incubation and washing, the relative numbers of bacteria associated with HEp-2 cells were quantified by Q-PCR. Key: as in A. Using Student’s t-test statistical significance in levels of bacterial association was obtained at p<0.0001 (*) and p<0.05 (**).
We extended these experiments by testing whether the same inhibitors prevented association of live P. gingivalis with HEp-2 cells. Monolayers were pretreated for 45 minutes with the inhibitors, as above. Upon removal of inhibitors, bacteria were added and incubation continued for a further 90 min. Relative numbers of bacteria in lysates of infected epithelial cells were determined by quantitative (Q) PCR (Hybiske and Stephens, 2007). With P. gingivalis genomic DNA as template, cell-associated bacteria were quantified by amplification of 16S rDNA using primers described previously (Hosogi and Duncan, 2005). As shown in Fig. 5B, the results were very similar to those obtained with A44, i.e. lysates of monolayers treated with chlorpromazine and methyl-β-cyclodextrin contained very low numbers of attached bacteria, approximately 5% of those associated with untreated control monolayers (100%). This result implied that P. gingivalis-HEp-2 association also depended on a clathrin-mediated pathway. In contrast, pretreatment of monolayers with genistein, nystatin, and filipin reduced the number of associated bacteria while binding of A44 was unaffected, and wortmannin, an inhibitor of A44 binding, reduced association of P. gingivalis by approximately 40%.
A44 and P. gingivalis colocalize with clathrin
The interaction of A44-coated beads with HEp-2 cells expressing clathrin light chain-green fluorescent protein (GFP) fusion was examined by confocal microscopy. Beads that remained at the cell surface were stained with rabbit anti-His-tag primary antibody, followed by donkey anti-rabbit secondary antibody conjugated with Alexa Fluor 647 (blue). Aggregates of blue external beads are shown in the stacked image in Fig. 6A. To detect intracellular as well as extracellular beads, HEp-2 cells were permeabilized and treated with anti-His-tag antibody followed by Alexa Fluor 568 (red)-coupled anti-rabbit antibody (Fig. 6B). The distribution of GFP-clathrin within cells (Fig. 6C) was detected directly (excitation at 482–484 nm, emission 502–510 nm). The merged images (Fig. 6D) show an aggregate of beads that are clearly undergoing internalization and are surrounded by punctate clusters of GFP-clathrin. Another merged image (Fig. 6E) shows a cluster of A44-coated beads inside a cell (red) surrounded by clathrin–GFP, colocalization of beads and clathrin is depicted in yellow. Similar experiments were carried out to examine whether intact P. gingivalis also interacted with clathrin-GFP. After incubation, infected monolayers were permeabilized and aggregates of bacteria were detected by treatment with anti-RgpA primary followed by Alexa Fluor 568 (red)-coupled anti-rabbit secondary antibody (Fig. 6F). Again, the pattern of GFP-clathrin expression was detected directly as above (Fig. 6G). The merged image showed the close association of clathrin and bacteria (yellow) as depicted in Fig 6H. These experiments provided direct evidence that peptide A44 and intact P. gingivalis cells associated with clathrin in epithelial cells.
Fig. 6. Colocalization of A44 and P. gingivalis with GFP-clathrin in HEp-2 cells.

A–E. Double-immunofluorescence of A44-coated latex beads and HEp-2 cells expressing GFP-clathrin.
A: Beads adhering to the cell surface were detected with rabbit anti-His-tag primary and Alexa Fluor 647 (blue)-coupled secondary antibodies.
B: HEp-2 cells were permeabilized and adherent and internalized beads were stained with rabbit anti-His-tag primary and Alexa Fluor 568 (red)-coupled secondary antibodies.
C: GFP-clathrin was detected directly at excitation wavelength range 482–488 nm and emission wavelength 502–510 nm.
D: Merged figures showing an aggregate of beads associated with clathrin, areas of colocalization are yellow.
E: Another merged image showing convergence of clathrin (green) and beads (red) with colocalization shown in yellow.
F–H. Colocalization of P. gingivalis 33277 and HEp-2 cells expressing GFP-clathrin.
F: P. gingivalis 33277 was detected in permeabilized HEp-2 cells with anti-RgpA primary and Alexa Fluor 568 (red)-coupled secondary antibodies.
G: GFP-clathrin expression was detected as described above.
H: Merged images from panels E and F demonstrate colocalization of P. gingivalis cells and GFP-clathrin.
Also by confocal microscopy, we obtained supporting evidence that actin polymerization was associated with P. gingivalis adherence and internalization by HEp-2 cells. After P. gingivalis infection of HEp-2 cells, bacteria were detected with rabbit anti-RgpA adhesin domain primary antibody and Alexa Fluor 488 (green)-coupled anti-rabbit secondary antibody. Actin in HEp-2 cells was detected by staining with rhodamine phalloidin (red). Figure 7 shows a field of cells in which clusters of bacteria that colocalize with actin appear yellow; these clusters of P. gingivalis surrounded by foci of polymerized actin are indicated by white arrows. External bacteria in the field are green.
Fig. 7. Actin polymerization associated with P. gingivalis adherence and internalization by HEp-2 cells.

After infection P. gingivalis was detected with rabbit anti-RgpA adhesin domain primary antibody and Alexa Fluor 488 (green)-coupled anti-rabbit secondary antibody. Actin in HEp-2 cells was detected by staining with rhodamine phalloidin (red). Adherent external P. gingivalis cell clusters are green, internalized cells are yellow and several are surrounded by foci of polymerized actin (indicated by arrows).
A44 and P. gingivalis interactions with HEp-2 cells depend on clathrin
We used a third approach of gene silencing (siRNA) to determine whether uptake of A44 and P. gingivalis by epithelial cells depended on clathrin or caveolin-1. A double-stranded oligonucleotide containing a target sequence from human clathrin heavy chain was cloned into pSUPER, resulting in plasmid pSUPER-Cla. HEp-2 cells were transfected with pSUPER-Cla, and control cells were with the pSUPER empty vector. After 36 h, transfected cells were replated in fresh medium and examined for expression of clathrin mRNA and protein. Figure 8A shows the reduced expression of clathrin transcript in cells transfected with pSUPER-Cla, relative to expression of β-globin, a control gene, and caveolin-1 that are both expressed at the 100% relative level by the cells. The reduced expression of clathrin was confirmed by Western blot (Fig. 8B) since cells transfected with pSUPER-Cla did not produce detectable levels of clathrin in comparison with cells transfected with the pSUPER empty vector or pSUPER-Cav.
Fig. 8. Effect of clathrin depletion on association of A44 and P. gingivalis with HEp-2 cells.

A. Relative expression, assessed by QRT-PCR, of β-globin (Glo), clathrin (Cla), and caveolin-1 (Cav) mRNAs in HEp-2 cells transfected with pSUPER-Cla. The differences in clathrin expression in cells containing pSUPER and pSUPER-Cla were statistically significant at p<0.0001 (*) by Student’s t-test.
B. Western blot of clathrin production in HEp-2 cells transfected with pSUPER empty vector, pSUPER-Cla (Cla-ko), and pSUPER-Cav (Cav-ko), respectively.
C. Association of peptide A44 with control HEp-2 cells transfected with pSUPER empty vector and experimental cells transfected with pSUPER-Cla. Peptide in culture media (M) or cell lysates (L) was detected with anti-His-tag primary and HRP-conjugated secondary antibodies.
D. Merged confocal image of A44-coated beads associated with HEp-2 cells expressing clathrin-GFP.
E. Merged image of A44-coated beads with HEp-2 cells expressing clathrin-GFP transfected with pSuper-Cla.
F. Association of P. gingivalis with control HEp-2 cells transfected with pSUPER empty vector and pSUPER-Cla. The number of bacteria associated with HEp-2 cells containing pSUPER was was designated as 100%, and the number of bacteria associated with HEp-2 cells containing pSUPER-Cla was expressed as relative to this control value (± standard deviation). Using Student’s t-test statistical significance in the relative bacterial association was obtained at p<0.0001 (*).
We examined association of peptide A44 with control and clathrin-depleted cells by Western blot (Fig 8C). Peptide was found in the medium and lysate of cells containing the pSUPER empty vector, and also in the medium and lysate of cells containing pSUPER-Cla. We reasoned that although clathrin was depleted in the latter, the surface receptor for A44 may still be present permitting A44 binding, however, progression to cell entry should be blocked because of clathrin ablation. If this is the case, bound A44 may be removed from the cells by a washing procedure such as that to remove antibody from tumor cells (Liu et al., 2007). Indeed, after a brief acid wash, A44 could be removed from lysates of clathrin-depleted cells, but not from control cells infected with pSUPER empty vector or with pSUPER-Cav (Fig. 8C), supporting our reasoning and signifying that A44 entry into cells was dependent on the presence of clathrin. To further investigate these steps, we examined the interaction of A44-coated beads with clathrin-depleted HEp-2 cells by confocal microscopy. Peptide-coated latex beads were incubated with control HEp-2 cells expressing clathrin light chain–GFP transfected with pSUPER empty vector. Using a double immunofluorescence protocol, external A44 beads were stained with Alexa Fluor 647 (blue), HEp-2 cells were then permeabilized and all beads, inside and out, were stained with Alexa Fluor 568 (red). In the overlay image (Fig. 8D), external beads appear cerise and internalized beads red, and clathrin (green) colocalized with clusters of beads that appear to be undergoing internalization. The experiment was carried out with the same HEp-2 cell line transfected with pSUPER-Cla to knockdown clathrin expression (Fig. E). After the same staining protocol, all the A44 beads appeared cerise indicating that they were not internalized, and expression of clathrin-GFP was ablated in most cells except for a small focus of expression in the image. These data indicate that only in the presence of clathrin can the peptide enter HEp-2 cells. In Fig. 8E, external beads appear to associate with a cell in the field, supporting evidence for the above experiments indicating that peptide can still bind to clathrin-depleted cells, possibly through acid-sensitive A44-membrane receptor binding.
Association of P. gingivalis with transfected cells was quantified by Q-PCR and the results are presented in Fig. 8F. Taking the relative number of bacteria associated with control pSUPER- transfected cells as 100%, the number of bacteria associated with clathrin knockdown cells was less than 10%, clearly indicating a dependence on the presence of clathrin.
A44 and P. gingivalis interactions with HEp-2 cells do not depend on caveolin-1
Similar analyses were carried out with HEp-2 cells transfected with pSUPER-Cav containing the target sequence for caveolin-1 (Gonzalez et al., 2004). Transfected cells were examined for expression of caveolin-1 mRNA and protein. In pSUPER-Cav transfected cells expression of caveolin-1 mRNA was reduced by approximately 70% compared to expression of β-globin; expression of clathrin was unaffected (Fig. 9A). At the protein level, HEp-2 cells transfected with either pSUPER empty vector or pSUPER-Cla expressed caveolin-1, while the protein was not detected in pSUPER-Cav-transfected cells, as shown in the Western presented in Fig. 9B.
Fig. 9. Effect of caveolin-1 depletion on association of A44 and P. gingivalis with HEp-2 cells.

A. Relative expression, assessed by QRT-PCR, of β-globin, clathrin, and caveolin-1 mRNAs in HEp-2 cells transfected with pSUPER-Cav. The differences in caveolin-1 expression in cells containing pSUPER and pSUPER-Cav were statistically significant at p<0.0001 (*) by Student’s t-test.
B. Western of caveolin-1 production in HEp-2 transfected with pSUPER empty vector, pSUPER-Cla, and pSUPER-Cav, respectively.
C. Association of peptide A44 with control HEp-2 cells transfected with pSUPER empty vector and experimental cells transfected with pSUPER-Cav. Peptide in culture media (M) or cell lysates (L) was detected with anti-His-tag primary and HRP-conjugated secondary antibodies.
D. Association of P. gingivalis with control HEp-2 cells transfected with pSUPER empty vector or with pSUPER-Cav. The number of bacteria associated with HEp-2 cells containing pSUPER was designated as 100%, and the number of bacteria associated with HEp-2 cells containing pSUPER-Cav was expressed as relative to this control value (± standard deviation). Differences between the numbers of bacteria associated with cells containing pSUPER or pSUPER-Cav were not statistically significant by Student’s t-test.
Association of peptide A44 with cells containing control pSUPER and experimental pSUPER-Cav was examined by Western blot and in each case A44 was found in culture media and in HEp-2 cell lysates (Fig. 9C) suggesting that association was not dependent on caveolin-1. Depletion of caveolin-1 apparently reduced association of P. gingivalis by approximately 40% (Fig. 9D), however, by Student’s t-test differences between the numbers of bacteria associated with cells containing pSUPER or pSUPER-Cav were not statistically significant.
Discussion
Current views on P. gingivalis-host interactions are that given the appropriate environmental conditions, this opportunistic pathogen dominates its ecological niche, triggering disruption of host cell function and cycles of tissue damage manifested as periodontitis, a disease exacerbated in hosts with certain genetic backgrounds or underlying systemic pathologies (Van Dyke and Tohme, 2000).
Several reports provide direct in vivo evidence that P. gingivalis can be found inside oral epithelial cells. In one, the organism was detected by fluorescent in situ hybridization within buccal cells from a majority of human subjects (Rudney et al., 2001). In another, diseased gingival biopsies were immunostained with gingipain-derived monoclonal antibodies and aggregates of P. gingivalis cells were detected around cell nuclei of surface epithelial cells (Rautemaa et al., 2004). These observations were consistent with an earlier in vitro demonstration that RgpA translocated to the nuclear region of both KB and HeLa cells as visualized by immunostaining with monoclonal antibodies to the RgpA adhesin domain (Scragg et al., 2002). Other significant findings from the same study were that perinuclear localization occurred within one hour of infection; the gingipain catalytic domain or activity did not participate in targeting; and the adhesin domain alone could traffic to the peri-nuclear location. In previous work, we showed that gingipain adhesin domains were involved in adherence of P. gingivalis to epithelial cells and this adherence was blocked by antibodies to the RgpA adhesin domain (Chen et al., 2001b; Chen and Duncan, 2004). Our data, together with the demonstration that the RgpA adhesin domain was internalized by epithelial cells (Scragg et al., 2002) indicated a complex interaction between adhesins and host cells, prompting our work to functionally dissect adhesin component peptides. In this study we investigated the molecular mechanisms associated with the early stages of infection i.e. adherence and entry into host cells by an adhesin peptide and intact bacteria.
Hemagglutination is a rapid and simple assay for both cell-associated and extracellular gingipain activity, with the implication that, in addition to erythrocytes, hemagglutinins would also adhere to other host cells and proteins. Hemagglutinating activity was shown to reside within gingipain adhesin domains; was inhibited by exogenous proteins fibrinogen, fibronectin, and laminin; and binding of native gingipains to these proteins was confirmed by ELISA (Pike et al., 1996). In the present study, we obtained similar binding data with r- and native A44 peptide (Fig. 1), and in a more biological context, we established that A44 blocked adherence of P. gingivalis cells to HEp-2 monolayers in vitro (Fig. 2). This is consistent with earlier work showing that antibody to the RgpA adhesin domain also blocked P. gingivalis adherence while preimmune serum has no effect (Chen and Duncan, 2004).
Scanning and transmission electron microscopy provided more information on how A44 interacted with cells in that latex beads coated with the peptide were observed mostly attached to cell surfaces, although beads were also internalized (Fig. 3). Peptide A44 bound to a 175 kDa protein that was isolated by affinity binding from the membrane fraction of HEp-2 epithelial cells, and binding of A44 to the eluted protein was verified by Western overlay (Fig. 4). However, by mass spectrometry two proteins were identified in the excised band, EEA1 (162 kDa) a marker for clathrin-dependent endocytosis, and clathrin itself (191.5 kDa). Cofractionation of these proteins may result from poor resolution of large proteins in the gel system or a consequence of high binding affinities within the membrane extract. So far the actual membrane receptor has not been identified, nevertheless, it was intriguing that both proteins participate in a clathrin-dependent pathway in which transmembrane receptors and their ligands are concentrated at the cell membrane into pits formed by the assembly of clathrin and cystolic proteins (Seto et al., 2002; Conner and Schmid, 2003). During clathrin-dependent endocytosis, receptors and ligands become internalized via clathrin–coated vesicles that fuse with the early endosome in a step requiring the small GTPase Rab5 (Chavrier et al., 1990) and its effector protein EEA1, which also localizes with endosome membranes (Christoforidis et al., 1999; Stenmark et al., 1996).
Chlorpromazine blocks recycling of clathrin and adaptor protein AP2 from the endosome, and neither A44 nor P. gingivalis associated with chlorpromazine-treated HEp-2 cells, supporting a role for clathrin in their adherence and potential entry (Figs.5A and B). Pretreatment with methyl-β-cyclodextrin also reduced A44 and P. gingivalis association, and it is well-documented that methyl-β-cyclodextrin-induced cholesterol depletion also inhibits formation of clathrin-coated pits as well as lipid rafts (Rodal et al., 1999; Subtil et al., 1999). Inhibition of phosphatidylinositol 3-kinase by wortmannin leads to decreased association of clathrin with endosome vesicles and disruption of the pathway (Sachse et al., 2002; Huang et al., 2007), and with wortmannin-pretreated HEp-2 cells there was also decreased association of A44 and P. gingivalis. Taken together, these results point to a role for clathrin in adherence and entry of A44 and P. gingivalis into epithelial cells. Genistein, an inhibitor of tyrosine kinase involved in the formation of caveosomes, did not prevent A44 from binding to HEp-2 cells suggesting that lipid rafts were not involved in uptake, but did reduce association of intact P. gingivalis. This may imply that entry of intact bacteria, with their full complement of surface proteins, is more complex in that they can use more than one pathway to enter host cells. In addition, there is precedence for lipid raft-mediated clathrin-dependent endocytosis as reported for anthrax toxin (Abrami et al., 2003), that may also be utilized by bacteria. By confocal microscopy we obtained direct evidence of the association of GFP-clathrin with A44-coated latex beads and with P. gingivalis cells (Fig.6), and that foci of polymerized actin surrounded aggregates of bacteria (Fig. 7).
To test directly whether clathrin is involved in adherence and entry of A44 and P. gingivalis into HEp-2 cells, we examined these functions in cells that were depleted of clathrin by siRNA. The clathrin-specific oligonucleotide sequence we used had previously been shown to decrease clathrin heavy chain expression in HeLa cells (Motley et al., 2003), and our data indicated that the sequence efficiently reduced expression of clathrin mRNA and protein in HEp-2 cells (Fig. 8). By Western, A44 was detected in lysates of clathrin-depleted (knockdown) cells (Fig. 8C), and in the confocal micrograph shown in Fig 8E at least one aggregate of A44-coated beads was associated with the surface of a clathrin-depleted cell. We argued that although the receptor for A44 is still present in clathrin-depleted cells, A44-receptor binding may be more labile to acid washing than binding which progresses to clathrin-mediated cell entry, and this proved to be the case. The confocal micrographs suggest that when A44 or P. gingivalis “association” with clathrin does occur, entry into host cells is initiated (Figs. 6 and Fig. 8D). Reduction in caveolin expression did not affect association indicating that caveolae/lipid raft pathways may not be involved.
The versatility of bacteria in exploiting multiple host pathways to promote infection is best exemplified with Listeria monocytogenes that uses different receptors and internalization mechanisms depending on which surface ligand it presents to host cells (reviewed by Bonazzi and Cossart, 2006). Surface proteins internalin A and B (InlA and InlB) induce internalization of Listeria by the zipper mechanism. The receptor for InlA is E-cadherin found at adherens junctions. This transmembrane protein is linked to the cytoskeleton, providing a mechanism to change host cell shape and initiate engulfment. New work has revealed that InlB is internalized by a different pathway (Veiga and Cossart, 2005; Li et al., 2005). Through binding to Met, the tyrosine kinase receptor and ligand for hepatocyte growth factor, InlB initiates Met-dependent phosphorylation of β-catenin leading to opening of adherens junctions. InlB is also found in culture media (like P. gingivalis gingipain adhesin peptides) and is speculated to interact with host cells faster than, and independently from intact bacteria via disrupted adherens junctions. Entry of InlB and the Met receptor is mediated by clathrin-coated pits, as visualized by immunofluoresence colocalization, and confirmed by siRNA knockdown experiments (Veiga and Cossart, 2006). This demonstration that internalization of bacteria can occur by clathrin-dependent process challenged the dogma that this mechanism was limited to smaller particles. Most recently it was established that several bacteria internalized by the zippering mechanism, i.e. L. monocytogenes, Yersinia pseudotuberculosis, and Staphylococcus aureus, utilize clathrin-dependent entry as a general pathway for invasion of host cells, although the exact mechanism(s) are not yet understood (Veiga et al., 2007). Our results with peptide A44 and P. gingivalis cells also suggest this mechanism, the focus of further work.
Recently, Tsuda et al., (2005; 2008) examined the interactions of HeLa and CHO cells with fluorescent beads coated with P. gingivalis membrane vesicles, a rich source of gingipains (Potempa et al., 1995). Data obtained by confocal microscopy and inhibitor studies indicated that uptake of the vesicle-coated beads involved the recruitment of α5β1 integrin at the internalization site; was dynamin-dependent; possibly involved lipid rafts, but did not require clathrin. In another study, Tamai et al., (2005) used microscopy and knockdown experiments and showed that ICAM-1 and caveolae played a role in the internalization of intact P.gingivalis by KB cells (a HeLa subclone). In endothelial cells, P. gingivalis colocalized with small GTPase Rab5 in vacuoles that resembled autophagosomes (Dorn et al., 2001), and later it was demonstrated that P. gingivalis internalization by endothelial cells was mediated by lipid rafts (Belanger et al., 2006). These studies, and our own, use different host cells, either bacterial products or whole bacteria, and different incubation conditions, and implicate diverse host pathways for uptake of P. gingivalis. Nevertheless, these pathways may not be mutually exclusive since the associations observed may be part of a continuum and represent different steps in a common trafficking pathway. Alternatively, as exemplified by L. monocytogenes, bacteria can be internalized by different pathways predicated by the bacterial ligand presented to the host cell (Veiga and Cossart, 2006). Based on the data presented in this report we show for the first time that both A44- and P. gingivalis-host epithelial cell binding recruits a clathrin-dependent entry pathway leading to uptake of peptide and bacteria, one strategy used by this pathogen.
Experimental Procedures
Bacterial strains and culture conditions
Porphyromonas gingivalis ATCC 33277 was cultured as described by Nishikawa et al., (2004). As host cells, we used HEp-2 epithelial cells that were cultured as described previously (Hosogi and Duncan, 2005) except that 10% fetal calf serum was used instead of bovine calf serum. Based on karyotyping, the HEp-2 cell line has been reclassified as HeLa (Chen, 1988). Both cell lines have been used extensively to study host–bacterial interactions, including those with P. gingivalis (Viega et al., 2007; Scragg et al., 2002; Tsuda et al., 2005; 2008), and in the present study we used HEp-2 cells to maintain consistency with previous work.
Chemicals, antibodies and proteins
Unless stated otherwise, chemicals, human proteins, and commercial antibodies were from the Sigma Chemical Company, and restriction enzymes were obtained from New England Biolabs. Enhanced Chemiluminescence detection kits were purchased from GE Life Sciences.
Native A44 peptide was purified using a modified immunoblot protocol (Smith and Fisher, 1984). Anti-A44 specific antibodies were purified from rabbit polyclonal antibody to the complete RgpA adhesin domain using r-A44 as bait in the filter purification method. Native A44 was prepared from a cell surface extract of P. gingivalis according to a published protocol (Chen et al., 2001b). Briefly, 48 hr cultures of P. gingivalis strain ATCC 33277 from blood plates were resuspended in phosphate buffered saline (PBS) and washed once. After resuspension in PBS, cultures were vortexed vigorously to release adhesin peptides from the bacterial surface, and after centrifugation (7000 rpm, 2 min) the supernatant was incubated with a membrane spotted with specific anti-A44 antibody for several hours at 4°C. The membrane was washed three times with PBS and bound native A44 peptide was eluted with 100 mM glycine (pH 2.5). The protein solution was neutralized with 1 M Tris (pH 9.0). Western blots confirmed that the specific antibody recognized the native peptide in a cell surface extract from strain ATCC 33277 (data not shown).
Construction of plasmids for production of adhesin peptide fusion protein
For the production of A44 peptide-hexahistidyl-tagged fusions, specific gene fragments were cloned into vector pET22b (Novagen). The A44-encoding region was amplified from chromosomal DNA of P. gingivalis ATCC 33277 by PCR with forward primer (F) 5’-TCATATGAGCGGTCAGGCCGAGATTG-3’ and reverse primer (R) 5’-TCCTCGAGCTTGCCGTTGGCCTTGATCT-3’, where the underlined sequences are NdeI and XhoI sites, respectively. PCR amplicons were ligated directly into pTOPO using the TOPO TA cloning kit (Invitrogen). The resulting pTOPO plasmids were digested with NdeI and XhoI, the inserts were gel-purified using the Qiaquick kit (Qiagen) then ligated with NdeI- and XhoI-digested pET22b vector and transformed into Escherichia coli BL21 (Novagen). For production of recombinant A44, E. coli BL21 was grown to late exponential growth phase (OD500nm 1.0) and peptide expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 3–4 h. Cells were lysed using 5 ml lysis buffer per gram wet weight (100 mM NaH2PO4, 10 mM Tris, 6 M guanidine HCl [pH 8.0]). The peptide was purified using Ni2+-NTA-agarose according to the supplier’s instructions (Qiagen).
Detection of peptide binding to human proteins by enzyme-linked immunosorbent assay (ELISA)
The wells of 96-well microtiter plates were coated with 2 µg fibrinogen or fibronectin in 300 µl of PBS, at 4°C, overnight. To quantify peptide binding by ELISA, wells were washed three times with 300 µl phosphate-buffered saline (PBS), and non-specific binding-sites were blocked with 10% (w/v) bovine serum albumin (BSA) in PBS for 1 h at room temperature (RT). After three washes with PBS (300 µl), recombinant or native A44 were added at several concentrations (0 to 50 µg protein/well) and incubation continued for 1 h, RT. Wells were washed three times with 300 µl PBS and incubated with rabbit anti-A44 antibodies at 1:20,000 dilution in PBS for 1 h, RT, followed by addition of HRP-labeled goat anti-rabbit antibodies (1:10,000 in PBS). After three further PBS washes, the peroxidase reaction was started with the addition of 100 µl 3,3’,5,5’-tetramethyl-benzidine. The colour reaction was stopped with 100 µl 2 M H2SO4 and the absorption of the solution was quantified at 450 nm in an HTS 7000 plus Bio Assay Reader (Perkin Elmer). As controls, one set of wells was not coated with proteins, a second was coated with bovine serum albumin (BSA) only, and a third with either native or r-adhesin peptides omitting incubation with anti-A44 primary antibody to control for non-specific binding of the secondary antibody.
Adherence of A44 to HEp-2 cells
To examine binding of A44 to epithelial cells, HEp-2 cells were seeded into tissue culture plates (1 × 107 cells per plate) and cultivated over night in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal calf serum (FCS). The medium was replaced with 10 ml fresh DMEM, 2 µg/ml purified A44 was added and the plates were incubated for 45 min at 37°C in 5% CO2. Several controls that were used in these experiments included: cells incubated without peptide to control for non-specific binding of primary and secondary antibodies, and A15, A27, and K39 that also bound to HEp-2 cells (data not shown). After incubation, the plates were washed three times with PBS. One ml PBS was added and HEp-2 cells were scraped from the plates and transferred to sample tubes. After centrifugation (7000 rpm, 2 min) the supernatant was discarded, the pellet resuspended in Laemmli buffer, and the samples boiled for 5 min. Proteins in the sample were size-separated by SDS-PAGE, then electro-transferred to nitrocellulose membranes. After overnight blocking at RT with 5% (w/v) milk in PBS, the presence of A44 was detected by probing with anti-His-tag primary and HRP-conjugated secondary antibodies.
In certain experiments we tested binding of A44 to HEp-2 cells that had been pretreated with chemical inhibitors. Cells were preincubated for 45 min at 37°C in DMEM with 10% FCS containing each of the inhibitors: 1 µg/ml cytochlasin D (Rosenshine et al., 1994; Tang et al., 1998); 10 µg/ml chlorpromazine (Contamin et al., 2000); 200 µM genistein (Rosenshine et al., 1994); 10 mM methyl-β-cyclodextrin (Zuhorn et al., 2002); 100 nM wortmannin (Tang et al., 1998); 20 µM nocodazole (Rosenshine et al., 1994); 1 µg/ml paclitaxel (Hertle and Schwarz, 2004); 100 µM amiloride (Contamin et al., 2000); 25 µg/ml nystatin (Ricci et al., 2000); and 1 µg/ml filipin (Zuhorn et al., 2002). The inhibitors were then washed away and the cells were incubated with A44 (2 µg/ml) for 45 min. Association of A44 with cells was detected as described above.
Adherence of P. gingivalis to epithelial cells
The adherence of P. gingivalis ATCC 33277 to epithelial cells in the presence or absence of peptide A44 was measured by viable counting as described previously (Chen et al., 2001b). Briefly, HEp-2 epithelial cells were seeded to 24-well tissue culture plates (approximately 4 × 105 cells per well) and cultivated over night in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal calf serum (FCS). As controls, we used peptide A27 and K39 that did not block adherence of P. gingivalis, and A15 that did block by a mechanism currently under investigation (data not shown). After replacement with fresh medium, the monolayers were infected with P. gingivalis at a multiplicity of infection of 100:1 (based on approximately 1× 106 HEp-2 cells) together with peptides at 0.025 to 0.4 µM, and incubated for 90 min, 37°C in 5% CO2. Each infection was carried out in triplicate. Infected host cells were washed three times with PBS to remove unattached bacteria and unbound peptides, then lysed with distilled water. Appropriate dilutions of the lysate, containing adherent bacteria, were spread on agar plates for viable counting. The number of adherent bacteria was expressed as a percentage of the number input bacteria added to the monolayers, and the data were obtained from at least three independent experiments.
Relative numbers of P. gingivalis that adhered to inhibitor-pretreated monolayers was measured by Q-PCR using P. gingivalis-specific 16S rDNA primers (Hosogi and Duncan, 2005). For Q-PCR, each infected HEp-2 lysate (1 µl) was used to quantify P. gingivalis chromosomal DNA in 20 µl iQ SYBR green reaction mix (Biorad) according to the manufacturer’s instructions. Real-time cycling conditions were: 3 min, 95°C, initial activation step; followed by 50 cycles at 95°C, 15s; and annealing-elongation at 60°C, 15s, in an iCycler (BioRad). Melting curve analyses to confirm the formation of a single PCR product were carried out as follows: 95°C for 1 min, 55°C for 1 min, and 55°C to 95°C with a heating rate of 0.5°C per 10s. A sequence specific standard curve was generated using 10-fold serial dilutions (100-10− 4) of P. gingivalis genomic DNA prepared from the same number bacteria as added to wells in the infection assays. The quantity of P. gingivalis DNA from each infection lysate was extrapolated from the DNA standard curve. Each experiment was carried out in triplicate and the data were obtained from at least three independent experiments.
Electron microscopy of adhesin peptide-coated latex beads
Approximately 109 1 µm diameter latex beads (Sigma) were washed three times in 25 mM 2-(N-morpholino) ethanesulfonic acid (MES) buffer (pH 6.1) and resuspended in 1 ml of the same buffer containing either A44 peptide (500 µg/ml) or BSA (10 mg/ml), then incubated with end-over-end rotation, overnight, at 4°C. Beads were washed once with buffer, blocked with BSA (10 mg/ml), 1h, RT, washed twice with buffer, and once with DMEM containing 10% FCS. Confluent HEp-2 cells in 24-well plates (1×106 cells /ml /well) were inoculated with 1× 108 beads and incubated for 45 min, 37°C, with 5% CO2. For scanning electron microscopy, cells were washed four times with PBS and fixed with 3% (v/v) paraformaldehyde and 4% (v/v) glutaraldehyde in 100 mM phosphate buffer (pH 7.4). Cell membranes were stained with 1% osmium tetroxide (OsO4), and scanning electron microscopy was performed with a JEOL 6400 electron microscope (JEOL, Japan). For transmission electron microscopy, HEp-2-cells were incubated for 45 min with A44-coupled latex beads, washed five times with PBS, then scraped from the plate and fixed with 2% paraformaldehyde and 2% glutaraldehyde. Membranes were stained with 2% OsO4, and then cells were embedded in LR White (London Resin Company, Ltd.). After sectioning and counterstaining with uranyl acetate and lead citrate, sections were viewed with a JEOL 1200 EX electron microscope (JEOL) operating at 100 kV.
Confocal laser scanning microscopy
For some experiments, a HEp-2 cell line was constructed that expressed GFP-tagged clathrin light chain. The pEGFP-LCa plasmid (Gaidarov et al., 1999) was a generous gift from Dr. J. H. Keen (Thomas Jefferson University, Philadelphia, PA) and transient transfection of HEp-2 cells was carried out using Lipofectamine ™ LTX (Invitrogen), according to the manufacturer’s protocol. HEp-2 or HEp-2(pEGFP-Lca) cells were seeded to confluence (1 × 106 cells) on cover slips in 24-well plates, and incubated overnight in DMEM containing 10% FCS. Either P. gingivalis 33277 (MOI:100) or r-adhesin-coupled latex beads (1 × 108 beads/well) were added and incubated at 37°C for 20–45 min. Cells were washed three times with PBS, and then fixed with 3% paraformaldehyde and 4% glutaraldehyde in PBS at RT. Cells were washed twice with PBS, and extracellular beads or bacteria were stained with primary anti-His-tag or anti-RgpA antibodies (1:1000 dilution in PBS) for 45 min, RT. Wells were washed three times with PBS and incubated with Alexa Fluor 647 (blue)-coupled secondary anti-rabbit IgG (Molecular Probes). Following three washes with PBS, cells were permeabilized with 0.1% (v/v) Triton-X-100 in PBS, 2 min, RT, and washed twice with PBS. Permeabilized cells were treated with either anti-RgpA or anti-His-tag antibodies (1:100 dilution in PBS) for 45 min, RT. After washing, cells were treated with Alexa Fluor 568 (red)-coupled anti-rabbit antibody (45 min, RT). Staining of extracellular and intracellular bacteria and beads was observed with a Leica TCS SP2 microscope (Leica Microsystems GmbH, Germany).
To detect actin-rearrangement during infection, P. gingivalis was incubated with HEp-2-cells grown on coverslips (as above) for 20 min, 37°C, with 5% CO2. Monolayers were washed three times with PBS and fixed and permeabilized as above. The cells were washed again and incubated with a rabbit anti-RgpA adhesin domain primary antibody followed by Alexa 488 (green)-coupled anti-rabbit secondary antibody. F-actin in HEp-2 cells was stained with rhodamine phalloidin (Molecular Probes) diluted 1:50 in PBS and incubation for 20 min at RT.
Fractionation of HEp2-cells
HEp-2 cell fractions were prepared as described previously (Kenny and Finlay, 1997). Briefly, HEp-2 monolayers were washed with ice-cold PBS and cells were permeabilized with 1% (v/v) saponin buffer containing 50 mM Tris/HCl (pH 7.5), 1 mM phenylmethylsulfonyl fluoride. After 5 min incubation on ice, the samples were centrifuged (5000 × g, 5 min, 4°C), and the soluble cytoplasmic protein fraction removed. The insoluble pellet was washed in saponin buffer, and membrane proteins separated from insoluble components by the addition of 1% Triton-X-100 in saponin buffer. The remaining cytoskeletal protein fraction was dissolved in distilled water. Proteins in each cell fraction were separated by SDS-PAGE and probed with peptide A44 in far-Western assays.
Far-Western blot analysis
Purified target proteins or cell lysates were size separated by SDS-PAGE, then electro-blotted to nitrocellulose membranes (Bio-Rad). Membranes were blocked overnight with 5% (w/v) milk in PBS, washed three times with PBS, and incubated with A44 at 2 µg/ml in 5% (w/v) milk in PBS, 1 hr, RT. Membranes were washed three times with PBS to remove unbound peptide, and then incubated for 1 h, RT, with rabbit anti-His-tag antibody (1:20,000 dilution in 5% milk). Afterwards, membranes were washed three times with PBS, then incubated with HRP-labeled anti-rabbit IgG antibody (1:10,000 in 5% milk) for 1 h, RT. To remove unbound antibody, membranes were washed twice with PBS, three times with PBS containing 0.1% (v/v) Tween 20, and twice with PBS. Finally, HRP-labeled antibodies were detected by chemiluminescence, using the ECL system.
Affinity Purification of A44 binding proteins
Recombinant A44 was coupled to Ni2+-NTA-agarose overnight at 4°C. Unbound protein was removed by washing with buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). The membrane fraction from HEp-2 cells was added to the A44-coupled agarose and incubated with end-over-end rotation at 4°C, 4–6 h. Control assays with uncoupled beads and RgpA adhesin peptides A15, A27 that either do not bind (uncoupled beads and A15) or pull down a different protein (A27). The agarose was washed and bound proteins were eluted with buffer (50 mM NaH2PO4, 300 mM NaCl, 300 mM imidazole, pH 8.0). Samples were dialysed against PBS and analyzed in SDS-PAGE gels. Overlay Western Blots were performed to identify a receptor for A44. A protein band was excited from a corresponding coomasie stained gel (ImperialTM Stain, Pierce) and send for MALDI-TOF analysis to the Taplin Biological Mass Spectrometry Facility, Harvard Medical School, Boston.
SiRNA knockdown experiments
Oligonucleotides (64mers) containing either the human clathrin heavy chain target sequence 5’-GTAATCCAATTCGAAGACC-3’ or the caveolin-1 target sequence 5’-CCAGAAGGGACACACAGTT-3 were synthesized (Integrated DNA technologies). Each oligonucleotide pair (25 µg) was annealed in 1× MRB buffer (10 mM Tris/ HCl (pH 7.5), 50 mM NaCl, 100 mM MgCl2, 1 mM DTT) at 68°C, and after cooling down to room temperature ligated into pSUPER (OligoEngine) resulting in plasmids pSUPER-Cla and pSUPER-cav-1. Vector pSUPER without target sequence was used as a negative control. HEp-2 cells were seeded the day prior transfection in 6-well plates at a density of 3–6× 105 cells/well in DMEM with 10% FCS. Transfections were performed using the FuGENE HD transfection reagent from Roche. For each culture well, 2 µg plasmid DNA/100 µl DMEM was incubated with 5 µl FuGENE HD transfection reagent for 15 minutes, RT; subsequently mixes were added drop-wise to the monolayer.
After 36 hrs incubation cells were replated, incubated overnight, and then used for different analyses. Expression of clathrin, caveolin-1 and β-globin mRNA in experimental (knockdown) and control cells was quantified by QRT-PCR using the clathrin, caveolin-1, and β-globin specific primers described by Hybiske and Stephens (2007). Briefly, RNA (0.5 µg) from transfected cells was added to a reaction mix containing 1 µl forward primer (10 µM stock), 1 µl reverse primer (10 µM stock), 10 µl 2 × SYBR Green RT-PCR mix, 0.5 µl iScript RT for one-step RT-PCR (BioRad) and adjusted to 20 µl total volume with water. Reactions were run in an iCycler under the following conditions: 10 min at 50°C for cDNA synthesis followed by 10 min at 95°C to inactivate the iScript RT reaction; 50 cycles each of denaturation at 95°C, 15s, annealing at 55°C, 15s, and elongation at 72°C, 1 min. Expression ratios were calculated according to the method of Pfaffl (2001) for relative quantification using β-globin values as internal control.
In addition, protein production was assessed by Western blot using α-clathrin HC (heavy chain) and α-caveolin-1 primary antibodies (Abcam) and anti-rabbit-HRP secondary antibody. Adherence/internalization of A44-coated latex beads to knockdown and control cells was examined by confocal microscopy, as described above. We also tested binding of peptide A44 (2 µg/ml) to transfected cells after 45 min incubations. After washing the cells three times with PBS, 1 ml acid buffer (0.2 M acetic acid in 0.5 M NaCl, pH 2.5) was added for 5 min. After removal of the buffer, the cells were washed once more with PBS, and A44 association with the cells was detected as described above. Lastly, the adherence of P. gingivalis to experimental and control cells was quantified by quantitative PCR, as described above.
Acknowledgements
We thank Drs Ziedonis Skobe and Elia Beniash, and Justine Dobeck for advice and help with electron and confocal microscopy, and Dr. Roland Zahn, Beth Israel Deaconess Medical Center, Boston, for help with siRNA experiments. This work was supported by the US National Institutes of Health grant R01-DE10510.
References
- Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Rodrigues PH, Dunn WA, Jr, Progulske-Fox A. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2:165–170. doi: 10.4161/auto.2828. [DOI] [PubMed] [Google Scholar]
- Bhogal PS, Slakeski N, Reynolds EC. A cell-associated protein complex of Porphyromonas gingivalis W50 composed of Arg- and Lys-specific cysteine proteinases and adhesins. Microbiology. 1997;143:2485–2495. doi: 10.1099/00221287-143-7-2485. [DOI] [PubMed] [Google Scholar]
- Bonazzi M, Cossart P. Bacterial entry into cells: a role for the endocytic machinery. FEBS Lett. 2006;580:2962–2967. doi: 10.1016/j.febslet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Bozzini S, Visai L, Pignatti P, Petersen TE, Speziale P. Multiple binding sites in fibronectin and the staphylococcal fibronectin receptor. Eur J Biochem. 1992;207:327–333. doi: 10.1111/j.1432-1033.1992.tb17054.x. [DOI] [PubMed] [Google Scholar]
- Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- Chen Z, Casiano CA, Fletcher HM. Protease-active extracellular protein preparations from Porphyromonas gingivalis W83 induce N-cadherin proteolysis, loss of cell adhesion, and apoptosis in human epithelial cells. J Periodontol. 2001a;72:641–650. doi: 10.1902/jop.2001.72.5.641. [DOI] [PubMed] [Google Scholar]
- Chen T, Nakayama K, Belliveau L, Duncan MJ. Porphyromonas gingivalis gingipains and adhesion to epithelial cells. Infect Immun. 2001b;69:3048–3056. doi: 10.1128/IAI.69.5.3048-3056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Duncan MJ. Gingipain adhesin domains mediate Porphyromonas gingivalis adherence to epithelial cells. Microb Pathog. 2004;36:205–209. doi: 10.1016/j.micpath.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Chen TR. Re-evaluation of HeLa, HeLa S3, and HEp-2 karyotypes. Cytogenet Cell Genet. 1988;48:19–24. doi: 10.1159/000132579. [DOI] [PubMed] [Google Scholar]
- Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Contamin S, Galmiche A, Doye A, Flatau G, Benmerah A, Boquet P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol Biol Cell. 2000;11:1775–1787. doi: 10.1091/mbc.11.5.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69:5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated pit dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Nagiel A, Lin AJ, Golan DE, Michel T. Small interfering RNA-mediated down-regulation of caveolin-1 differentially modulates signaling pathways in endothelial cells. J Biol Chem. 2004;279:40659–40669. doi: 10.1074/jbc.M407051200. [DOI] [PubMed] [Google Scholar]
- Hertle R, Schwarz H. Serratia marcescens internalization and replication in human bladder epithelial cells. BMC Infect Dis. 2004;4:16. doi: 10.1186/1471-2334-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogi Y, Duncan MJ. Gene expression in Porphyromonas gingivalis after contact with human epithelial cells. Infect Immun. 2005;73:2327–2335. doi: 10.1128/IAI.73.4.2327-2335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Chang SC, Yu IC, Tsay YG, Chang MF. Large hepatitis delta antigen is a novel clathrin adaptor-like protein. J Virol. 2007;81:5985–5994. doi: 10.1128/JVI.02809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Mechanisms of Chlamydia trachomatis entry into nonphagocytic cells. Infect Immun. 2007;75:3925–3934. doi: 10.1128/IAI.00106-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J, Sambandam V, Wu JH, Michalek SM, Balkovetz DF. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun. 2000;68:1441–1449. doi: 10.1128/iai.68.3.1441-1449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J, Yang QB, Zhang P, Potempa J, Travis J, Michalek SM, Balkovetz DF. Hydrolysis of epithelial junctional proteins by Porphyromonas gingivalis gingipains. Infect Immun. 2002;70 doi: 10.1128/IAI.70.5.2512-2518.2002. 2512-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata Y, Iwasaka H, Kitano S, Hanazawa S. Porphyromonasgingivalis fimbria-stimulated bone resorption is inhibited through binding of the fimbriae to fibronectin. Infect Immun. 1997;65:815–817. doi: 10.1128/iai.65.2.815-817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny B, Finlay BB. Intimin-dependent binding of enteropathogenic Escherichia coli to host cells triggers novel signaling events, including tyrosine phosphorylation of phospholipase C-gamma1. Infect Immun. 1997;65:2528–2536. doi: 10.1128/iai.65.7.2528-2536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Xiang GS, Dokainish H, Ireton K, Elferink LA. The Listeria protein internalin B mimics hepatocyte growth factor-induced receptor trafficking. Traffic. 2005;6:459–473. doi: 10.1111/j.1600-0854.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- Liu G, Dou S, Yin D, Squires S, Liu X, Wang Y, Rusckowski M, Hnatowich DJ. A novel pretargeting method for measuring antibody internalization in tumor cells. Cancer Biother Radiopharm. 2007;22:33–39. doi: 10.1089/cbr.2006.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Kuboniwa M, Nakamura T, Kawabata S, Hamada S. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect Immun. 2002;70:277–285. doi: 10.1128/IAI.70.1.277-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Inaba H, Kawai S, Hamada S. Inhibitory effects of Porphyromonas gingivalis fimbriae on interactions between extracellular matrix proteins and cellular integrins. Microbes Infect. 2005;7:157–163. doi: 10.1016/j.micinf.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Yoshimura F, Duncan MJ. A regulation cascade controls expression of Porphyromonas gingivalis fimbriae via the FimR response regulator. Mol Microbiol. 2004;54:546–560. doi: 10.1111/j.1365-2958.2004.04291.x. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike RN, Potempa J, McGraw W, Coetzer TH, Travis J. Characterization of the binding activities of proteinase-adhesin complexes from Porphyromonas gingivalis. J Bacteriol. 1996;178:2876–2882. doi: 10.1128/jb.178.10.2876-2882.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potempa J, Pike R, Travis J. The multiple forms of trypsin-like activity present in various strains of Porphyromonas gingivalis are due to the presence of either Arg-gingipain or Lys-gingipain. Infect Immun. 1995;63:1176–1182. doi: 10.1128/iai.63.4.1176-1182.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautemaa R, Jarvensivu A, Kari K, Wahlgren J, DeCarlo A, Richardson M, Sorsa T. Intracellular localization of Porphyromonas gingivalis thiol proteinase in periodontal tissues of chronic periodontitis patients. Oral Dis. 2004;10:298–305. doi: 10.1111/j.1601-0825.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- Ricci V, Galmiche A, Doye A, Necchi V, Solcia E, Boquet P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol Biol Cell. 2000;11:3897–3909. doi: 10.1091/mbc.11.11.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal SK, Skretting G, Garred O, Vilhardt F, van Deurs B, Sandvig K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol Biol Cell. 1999;10:961–974. doi: 10.1091/mbc.10.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenshine I, Ruschkowski S, Finlay BB. Inhibitors of cytoskeletal function and signal transduction to study bacterial invasion. Methods Enzymol. 1994;236:467–476. doi: 10.1016/0076-6879(94)36035-9. [DOI] [PubMed] [Google Scholar]
- Rudney JD, Chen R, Sedgewick GJ. Intracellular Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in buccal epithelial cells collected from human subjects. Infect Immun. 2001;69:2700–2707. doi: 10.1128/IAI.69.4.2700-2707.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachse M, Urbé S, Oorschot V, Strous GJ, Klumperman J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol Biol Cell. 2002;13:1313–1328. doi: 10.1091/mbc.01-10-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scragg MA, Alsam A, Rangarajan M, Slaney JM, Shepherd P, Williams DM, Curtis MA. Nuclear targeting of Porphyromonas gingivalis W50 protease in epithelial cells. Infect Immun. 2002;70:5740–5750. doi: 10.1128/IAI.70.10.5740-5750.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scragg MA, Cannon SJ, Rangarajan M, Williams DM, Curtis MA. Targeted disruption of fibronectin-integrin interactions in human gingival fibroblasts by the RI protease of Porphyromonas gingivalis W50. Infect Immun. 1999;67:1837–1843. doi: 10.1128/iai.67.4.1837-1843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto ES, Bellen HJ, Lloyd TE. When cell biology meets development: endocytic regulation of signaling pathways. Genes Dev. 2002;16:1314–5736. doi: 10.1101/gad.989602. [DOI] [PubMed] [Google Scholar]
- Smith DE, Fisher PA. Identification, developmental regulation, and response to heat shock of two antigenically related forms of a major nuclear envelope protein in Drosophila embryos: application of an improved method for affinity purification of antibodies using polypeptides immobilized on nitrocellulose blots. J Cell Biol. 1984;99:20–28. doi: 10.1083/jcb.99.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Aasland R, Toh BH, D'Arrigo A. Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem. 1996;27:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- Subtil A, Gaidarov I, Kobylarz K, Lampson MA, Keen JH, McGraw TE. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc Natl Acad Sci USA. 1999;96:6775–6780. doi: 10.1073/pnas.96.12.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takii R, Kadowaki T, Baba A, Tsukuba T, Yamamoto K. A functional virulence complex composed of gingipains, adhesins, and lipopolysaccharide shows high affinity to host cells and matrix proteins and escapes recognition by host immune systems. Infect Immun. 2005;73:883–893. doi: 10.1128/IAI.73.2.883-893.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai R, Asai Y, Ogawa T. Requirement for intercellular adhesion molecule 1 and caveolae in invasion of human oral epithelial cells by Porphyromonas gingivalis. Infect Immun. 2005;73:6290–6298. doi: 10.1128/IAI.73.10.6290-6298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang P, Sutherland CL, Gold MR, Finlay BB. Listeria monocytogenes invasion of epithelial cells requires the MEK-1/ERK-2 mitogen-activated protein kinase pathway. Infect Immun. 1998;66:1106–1112. doi: 10.1128/iai.66.3.1106-1112.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda K, Amano A, Umebayashi K, Inaba H, Nakagawa I, Nakanishi Y, Yoshimori T. Molecular dissection of internalization of Porphyromonas gingivalis by cells using fluorescent beads coated with bacterial membrane vesicle. Cell Struct Funct. 2005;30:81–91. doi: 10.1247/csf.30.81. [DOI] [PubMed] [Google Scholar]
- Tsuda K, Furuta N, Inaba H, Kawai S, Hanada K, Yoshimori T, Amano A. Functional analysis of α5β1 integrin and lipid rafts in invasion of epithelial cells by Porphyromonas gingivalis using fluorescent beads coated with bacterial membrane vesicle. Cell Struct Funct. 2008 doi: 10.1247/csf.08012. PMID: 18388398; doi 10.1247/csf08012. [DOI] [PubMed] [Google Scholar]
- Van Dyke TE, Tohme ZN. Periodontal diagnosis: evaluation of current concepts and future needs. J Int Acad Periodontol. 2000;2:71–78. [PubMed] [Google Scholar]
- Veiga E, Cossart P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol. 2005;7:894–900. doi: 10.1038/ncb1292. [DOI] [PubMed] [Google Scholar]
- Veiga E, Cossart P. The role of clathrin-dependent endocytosis in bacterial internalization. Trends Cell Biol. 2006;16:499–504. doi: 10.1016/j.tcb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga E, Guttman JA, Bonazzi M, Boucrot E, Toledo-Arana A, Lin AE, Enninga J, Pizarro-Cerdá J, Finlay BB, Kirchhausen T, Cossart P. Invasive and adherent bacterial pathogens co-Opt host clathrin for infection. Cell Host Microbe. 2007;2:340–351. doi: 10.1016/j.chom.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veith PD, Talbo GH, Slakeski N, Dashper SG, Moore C, Paolini RA, Reynolds EC. Major outer membrane proteins and proteolytic processing of RgpA and Kgp of Porphyromonas gingivalis W50. Biochem J. 2002;363:105–115. doi: 10.1042/0264-6021:3630105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg A, Belton CM, Park Y, Lamont RJ. Role of fimbriae in Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1997;65:313–316. doi: 10.1128/iai.65.1.313-316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- Yilmaz O, Young PA, Lamont RJ, Kenny GE. Gingival epithelial cell signalling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology. 2003;149:2417–2426. doi: 10.1099/mic.0.26483-0. [DOI] [PubMed] [Google Scholar]
- Zuhorn IS, Kalicharan R, Hoekstra D. Lipoplex-mediated transfection of mammalian cells occurs through the cholesterol-dependent clathrin-mediated pathway of endocytosis. J Biol Chem. 2002;277:18021–18028. doi: 10.1074/jbc.M111257200. [DOI] [PubMed] [Google Scholar]