

Abstract
The pairing of selection and screening methods with randomly mutated libraries can be an exceptionally powerful means for probing the functions of biological molecules and for developing novel regents from random libraries of peptides and oligonucleotides. The use of such approaches is beginning to permeate the ion channel field where they are being deployed to uncover fundamental aspects about ion channel structure and gating, small molecule–channel interactions, and the development of novel agents to control channel activity.
Introduction
Brains, hearts, senses, and muscles all run on bioelectrical signals that race along cell membranes on the millisecond timescale. To make these exceptionally rapid signals, cells rely on the activity of a large, diverse set of transmembrane macromolecular complexes known as ion channels. The hydrophobic nature of the cell membrane presents a significant barrier to the passage of charged particles such as ions. Cells expend a great deal of their ATP resources to drive a variety of pumps that establish asymmetric ion gradients across their cell membranes. When ion channel proteins open, energy stored in these ionic gradients is released as the ions flow down their electrochemical gradients and across cell membranes.1 This rapid transport of ions, catalyzed by ion channel proteins, is the fundamental process that creates the electrical signals that underlie the normal functioning of our cardiovascular and nervous systems. Without such activity, there would be no thoughts, no racing heart at the sight of a loved one, no feeling of pain, and no warm embraces. Further, ion channel misfunction is linked to an ever-growing range of human diseases including arrhythmias, migraine, diabetes, and movement disorders.2,3 Consequently, there is a great interest both in understanding the molecular basis for how channels work and in the development of new reagents that can control their functions.
Because ion channels are membrane proteins, the use of high-resolution biophysical techniques to elaborate the molecular architectures that underlie channel function remains very challenging.4 Thus, there has been a great deal of effort focused on other types of approaches that can enlighten the connections between ion channel molecules and their activities. In this regard, genetic methods constitute an exceedingly powerful means for querying biological systems and for establishing insights into how macromolecules function. One of the biggest strengths of genetic approaches is that they offer an assumption-free method in which a system can be probed to identify functional alterations that are rooted in mutational changes in specific macromolecules. Classical genetic studies in which functional defects in both multicellular and unicellular organisms were traced to ion channel gene mutations have played a large role in ion channel studies. These investigations have determined the identities of founding members of many important ion channel families, such as voltage-gated potassium channels,5–7 sensory transduction TRP channels,8–10 and centrally important proteins involved in channel regulation.11,12 Over the past ten years or so, a different sort of channel-focused genetics has been emerging, one that starts not with an investigation of a physiological process but with a molecule. This approach is typically termed ‘reverse genetics’. Rather than look for mutant genes in an organism to identify a specific channel that is key to some process, researchers have established a number of heterologous expression systems in which large numbers of mutant channels can be assayed directly for new or altered properties. The experimental advantage of such gene-based methods is that none require purification of the protein of interest. Thus, all of the power of molecular biology and molecular evolution methods can be brought to bear on discovery-oriented selections and screens that when paired with electrophysiological analysis lead to deep molecular insight into the mechanisms of ion channel function.
Basic considerations
Genetic systems that use unicellular organisms, such as bacteria (e.g., Escherichia coli) or yeast (e.g., Saccharomyces cerevisiae), have been one of the mainstays of biological investigation and provide a potent means to assay large numbers of variants, up to ∼1 million, in a parallel manner in a short period of time, typically within a week or so. The challenge of using genetic methods to study ion channels in unicellular systems is that one needs to establish a robust phenotype that can be the focus of either a selection or simple assay that can constitute a screen.† Systems in which expression of an ion channel gene overcomes a specific functional deficit that allows the cell to survive some external challenge are the strongest in this regard. Alternatively, fluorescence-based methods that monitor calcium signals resulting from the activity of ion channels constitute a second productive approach. Once a microorganism-based genetic system is established, one can readily examine the properties of libraries of large numbers of mutant channels.
Libraries of mutant channels can be generated using a variety of approaches: chemical mutagenesis,13,14 error-prone PCR,15–22 passage of the target gene through a bacterial mutator strain,23–25 designed mutant libraries made from synthetic oligonucleotides encoding whole gene or targeted to key gene portions,26–28 and DNA shuffling approaches.29,30 The method of library generation is less important than the coverage and amount of sequence diversity that it contains. Given a good library and a robust selection or screen, one can readily find a host of interesting mutants that merit characterization by other methods.
Ion channel subunits are generally medium to large sized proteins. Pore-forming subunits from members of the voltage-gated channel family are predominantly in the range of 300–500 amino acids and some are as large as 2500–3000 amino acids.31 Considering these subunit sizes and the limits imposed on the level of diversity by host organism transformation efficiencies, typically 105–106 individual clones, none of the current mutagenesis schemes can yield libraries that contain enough mutants to sample all possible variant sequences for a given subunit. For example, a 300-residue subunit has 20300 possible sequences, a number that surpasses all estimates of the total numbers of atoms in the universe. Given the paltry amount of sequence space that can be explored for a given subunit, one might imagine that the chance of discovering a mutation that changes function or that identifies a key functional residue by a completely blind mutagenesis approach might have little chance of success.
In spite of these seemingly insurmountable odds, there is ample evidence that the situation is not as dire as might be initially predicted. There are many experimental strategies for making the most of number limits that are inherent to the selection/screening process. An initial broad sweep in which an entire channel gene is targeted for mutation so that each position is likely to be changed at least once can lead to the identification of a particular region or set of residues that can be more intensely explored by subsequent focused libraries that more extensively test the amino acid restrictions of particular positions. For example, there are 6000 possible variants for a 300 amino acid subunit if each position is substituted with all 20 amino acids. In a well-made random library made by error-prone PCR there is a good chance that most positions would be changed to at least a few amino acids of very different chemical character and allow the investigator to uncover a few key regions that might affect function from the first pass selection or screen. Alternatively, if one has an interest in a region with known functional importance, one can directly employ focused libraries that target a particular channel element. To date, a combination of molecular evolution–selection approaches has been applied to four classes of channels: potassium channels, TRP channels, mechanosensitive bacterial channels from the MscL family, and voltage-gated calcium channels. These efforts have yielded a multitude of interesting channel mutants that have brought genuinely new insight into channel function.
Rescue of ion transport deficient microorganisms
The biggest challenge in establishing a genetic system to study a particular ion channel is to devise a situation in which activity of the channel of interest is intimately tied to cell survival or to a robust secondary assay. One powerful approach has been the use of systems in which expression of the channel of interest affects ion homeostasis. To this end, systems that rely on potassium uptake assays have been particularly fruitful.
All cells need potassium to survive. Bacteria and yeast have special uptake systems that harvest potassium from the environment.32,33 Deletion or inactivation of the genes responsible for potassium uptake (E. coli TK242034,35 and S. cerevisiae Δtrk1Δtrk236,37) yields strains that survive when bathed in high concentrations of potassium (∼100 mM) but not when subjected to low external potassium concentrations (0.5–2 mM). The activity of the plasma membrane proton-ATPase sets the membrane potentials of both microorganisms in a very negative range (∼ −300 mV).38 Because this range is below the equilibrium potential for potassium ions under low external potassium (−133 mV for 1 mM [K+]out/150 mM [K+]in at 37 °C), expression of a functional potassium channel can provide a route for potassium uptake under low external potassium conditions and rescue the growth of the potassium-starved strain. Initially, potassium-uptake deficient yeast strains were used to clone and study the properties of plant potassium channel genes (see for example ref. 37, 39–41). The demonstration of functional expression of a mammalian inward rectifier potassium channel in potassium transport deficient yeast42 ushered in a series of studies on inward rectifiers and voltage-dependent potassium channels in which large libraries of channel mutants were used to study basic channel architectural constraints,15,26 ion selectivity,30,43 and gating properties.16,29 In these studies, a central element was the pairing of the results of the yeast selections with the biophysical characterization of channel function using electrophysiological approaches. Such correlations are essential. The power of genetic screens is that one can find interesting mutants with relative ease. The potential drawback is that one never knows all of the parameters that are actually being tested by the system. Thus, it is crucial to follow the selections or screens with extensive characterization to understand how the isolated mutants might be working. That caution aside, the combination of mutant libraries with channel selections or screens has led to the isolation of a remarkable number of new channel mutants with very interesting properties.
Selections for new activities
Many channels move between a closed state that does not conduct ions, and an open state that passes ions as the result of some type of stimulus such as ligand binding or voltage change. Such channels are excellent candidates for gain-of-function (GOF) selections and screens in which one hunts for mutations that endow the target channel with a new functional property such as activation in the absence of the normal gating stimulus. This type of GOF investigation has proven instrumental for the identification of residues that have important roles in maintaining the integrity of the closed state or in stabilizing the open state and that therefore may constitute key moving parts of the channel.
A common type of GOF selection is one in which the investigator tries to bring a non-functional channel that is closed due to the absence of activating signal to life (Fig. 1A). In these life and death selections, only those bacteria or yeasts fortunate enough to receive a gene encoding a functional channel survive when the selective pressure is applied. Such selections are straightforward and require little sophisticated equipment. The library of channel genes is transformed into the host strain and plated under conditions where the channel is not required for survival. Subsequently, the colonies are transferred by replica plating onto plates that have selective conditions, such as low potassium, where only microbes carrying a plasmid that encodes for a functional channel survive. From these remaining colonies one can recover the plasmid, sequence the gene, retest the plasmid to verify the phenotype, and scrutinize the mutant by biophysical characterization.
Fig. 1.
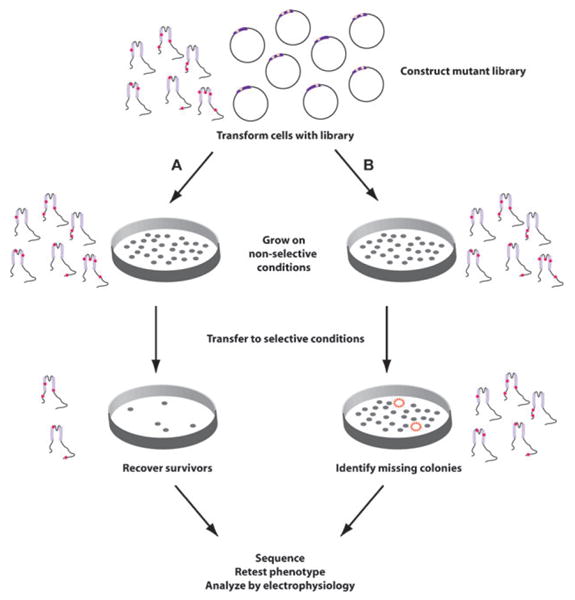
Schematic for selection of functional channels from mutant libraries. Top, depicts a collection of mutants of a two-transmembrane domain channel subunit encoded by plasmids. Path A shows the procedure when functional channels confer rescue under selective conditions. The library is first plated onto non-selective conditions. Under these conditions, channel function is not required for survival. After colonies are established they are replica-plated onto plates that have selective conditions in which channel activity is required for survival. All non-functional clones are lost at this step and only functional mutants remain. Path B shows the outcome when functional channels confer a lethal phenotype. In this case, all colonies survive the non-selective conditions, but only those colonies expressing active channels die on the selective plates (indicated by the red dashed circles). In both cases, recovery of the plasmid, retesting to verify phenotype, DNA sequencing to identify the mutations, and subsequent characterization by electrophysiological studies are essential.
GOF selection experiments have a distinct advantage as a means to find mutants that confer a new activity to the channel of interest. There are many ways a mutation can kill a channel but far fewer in which a mutational change can impart a new functionality. The types of mutations that constitute the bulk of a randomly mutated library (neutral mutations and mutations that result in channels that are non-functional due to premature stop codons, missense mutations in the active site, misfolding or faulty membrane insertion) result in cell death under selective conditions and as a consequence are readily culled from the library. After application of selective pressure, one is left with a small number of colonies, which should bear channels with increased activity. Thus, rare mutations that have strong effects on function can be found. A good example of the power of this type of selection approach was a pair of studies that aimed to isolate mutants of G-protein activated inward rectifier channels (Kir3.1 and Kir3.2) that had enhanced activity in the absence of their normal ligand, the Gβγ complex.16,29 Independent work in the Jan and Reuveny groups identified a number of Kir3.1 and Kir3.2 mutants that suggested a crucial role for the movement and bending of the inner pore-lining transmembrane segment in gating. These ideas were derived prior to the advancement of similar potassium channel gating hypotheses based on structural studies.44 Similar GOF studies with the bacterial channel KcsA have identified mutants that increase channel opening28 and a set of activatory mutations that cluster in the portion of the structure that is essential for maintaining the closed state.22
A different type of GOF selection searches for mutant channels that kill the host (Fig. 1B). This type of GOF selections is more technically challenging than GOF rescue selections as the colonies that do not survive transfer to selective conditions will be far fewer than those that do. Nevertheless, selections that look for activated channels that are lethal have proved to be an effective way to find interesting gating mutants. In one of the first reported lethal GOF screens, Loukin and colleagues uncovered gating mutants in likely pore-lining segments of the yeast potassium channel TOK1 (YKC1) that have an important role in closed state stabilization.13 A related study of the E. coli potassium channel Kch identified GOF mutations resident in a key cytoplasmic element that is thought to control channel gating.21 More recently, Myers et al. have used a lethal GOF screen to isolate mutants of the heat and capsaicin sensitive TRP channel, TRPV1, that increase basal activity and discovered that the pore-helix segment likely plays an important role in channel activation.17
Instead of a selection, GOF experiments can also be conducted as a screen in which channel function is coupled to a secondary assay. One productive approach has been to use fluorescence-based detection to monitor intracellular calcium changes that are linked to the activity of the channel of interest. The Kung and Saimi groups have developed such an assay for yeast and employed it to identify mutants that increase the activity of the native yeast TRP channel TRPY1,24 an intracellular ion channel that resides in the yeast vacuole. Expression of a calcium-sensor protein, apoaqueorin, allowed the measurement of a calcium-dependent luminescence signal resulting from the release of calcium from the vacuole into the cytoplasm through TRPY1 following osmotic shock. By screening ∼4000 TRPY1 mutants in a multiwell format, the investigators identified a set of mutants with increased calcium signals. Follow-up studies by patch clamp electrophysiology showed that the mutants have increased activity and identify key elements of the pore domain that are excellent candidates for part of the channel gate.23,24
Genetic selections have also been important for isolating interesting mutants of a bacterial mechanosensitive ion channel MscL.45 Two different approaches have been reported: one looked for GOF mutants that impair host growth,14,25 and the other used dyes that allow one to asses cell viability following an osmotic challenge20,46 and that could distinguish GOF and loss of function (LOF) mutants by exploiting differences in vital dyes, propidium iodide and SYTO 9, that report on cell viability. One important difference highlighted by these MscL studies is that screens, while powerful, are still more labor intensive and limited in scope than selections. The vital dye screen examined ∼400 mutants whereas the functional selections were able to sort through two orders of magnitude more mutants (∼50 000) and correctly identify a residue that is centrally important for maintaining the integrity of the closed state.14,45
High throughput functional screens
A different way to deploy the power of reverse genetics is to establish a screen in which particular functional properties of a large number of mutants can be measured in cultured mammalian cells. By coupling calcium imaging in transfected mammalian cell lines with studies of channels that are both temperature and ligand gated, TRPM818 and TRPV3,19 the Patapoutian lab was able to examine pools of ∼14 000 mutants of each channel and isolate mutants that displayed selective alterations in either ligand or thermal sensitivity. Together with the TRPV1 study,17 the results of screening of TRPM8 and TRPV3 channel mutants indicate that at least some of the activation modes of TRP channels can be uncoupled (ligand vs. temperature, protons vs. ligand). Even though the exact molecular mechanisms remain to be resolved, these studies provide further examples of the power of unbiased screens as in all three cases the investigators identified key regions of the channels that are likely to be important for gating responses.
Taken together, the results from these varied investigations of different channel types demonstrate that genetic screens of a modest number of mutants (in the thousands to 10 000s) can be an exceptionally productive means to uncover mutants that identify key elements that are likely to be core elements of the gating mechanism. This linkage is further reinforced by the cases where activating mutants reside in essential gating elements that are characterized by high-resolution structural studies.16,21,28,29 Overall, the relatively high frequency with which GOF mutants occur in ion channels bodes well for future screens for other channels as it appears that one does not have to rely on sifting through massive numbers of mutants to find interesting and informative ones.
Feeling around in the dark—random mutagenesis and selection as a means for defining architectural elements and testing protein–protein interactions
Many membrane proteins are assembled from helical bundles that are largely perpendicular to the membrane. This situation facilitates the use of extensive mutagenesis as a means to derive constraints that inform ideas about membrane protein architecture. Patterns of the physicochemical properties of allowed amino acid substitutions at specific positions can be used to identify secondary structures, lipid-facing residues, and protein–protein interaction surfaces. Such indirect structural analysis of membrane proteins has a long history best exemplified by the work done on the transmembrane dimer glycophorin.47–49
Access to a genetically tractable system enables another powerful type of experiment that can yield structural constraints, the search for intragenic suppressors that rescue a defect that destroys channel function (Fig. 2A). The strength of the approach is that in the complete absence of structural data one can identify residue pairs that are functionally coupled. The search for suppressors can also be enlightening when set in the context of testing a structural model as one can focus the mutagenesis efforts towards candidate segments that might interact. Work on structurally well-characterized systems indicates that, in general, suppressor pairs are the most effective at restoring protein function when the suppressor lies in close proximity to the original perturbation.50 While it is not strictly necessary for suppressor and defect mutations to interact directly to restore function, second site suppressor experiments can provide constraints that can vet models for how transmembrane segments might be arranged.
Fig. 2.
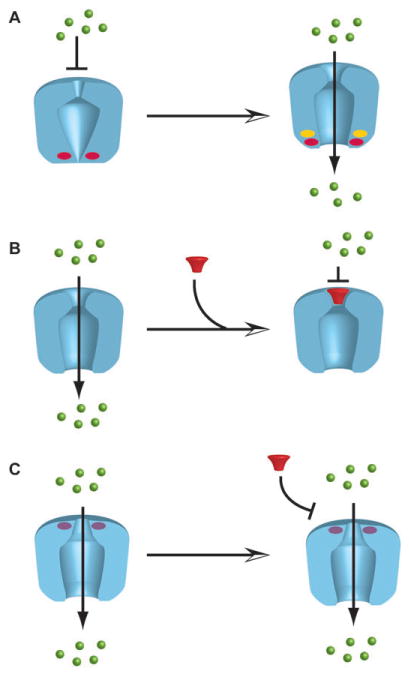
Cartoons depicting the principles behind second site suppressor experiments. A, a channel bearing a mutation that prevents opening (indicated by the red oval) is converted to an open state by a second mutation (yellow oval) in the vicinity of the primary mutation (red oval). B, cartoon depicting channel inhibition by a pore blocker. C, a channel bearing a mutation in the pore blocker binding site (purple oval) is resistant to block and retains function.
Based on a genetic selection that characterized large numbers of mutant channels, analysis of transmembrane amino acid substitution patterns together with second site suppressor experiments was used to probe the transmembrane architecture of the mammalian inward rectifier Kir2.1. This study established the overall helix packing arrangement of mammalian inward rectifiers,26 which was subsequently validated by crystallographic studies,51 and uncovered an intrasubunit intramembrane hydrogen bond that is essential for channel assembly. Suppressor studies have also given important insight into regarding residues that may interact in the open state of MscL.25 The most extensive use of suppressors to extract functional constraints comes from a combined modeling–selection study on the hyperpolarization activated channel KAT1 that helped to establish the likely range of voltage sensor motion.15,52 These studies complement the ongoing crystallographic work in the field and provide new ideas for testing key questions about what sorts of conformational transitions underlie channel gating.
Selections and libraries meet channel blockers—second site suppressor to map sites and mechanisms of action
There are many diverse classes of ion channels that are now known from extensive gene characterization efforts. Unfortunately, the ability to identify ion channel genes has far surpassed the ability to define novel pharmacological agents for particular channels. Consequently, many ion channels have poor to no pharmacology. This situation limits the ability of investigators to make the connections between a particular ion channel gene and its exact biological function. Thus, one of the key challenges for ion channel research is to develop means to identify new agents that can control channel activity. Genetic selections offer a novel, unbiased way to identify channel-modifying compounds. A number of studies have used the Δtrk yeast system to screen for and map the sites of action of ion channel blockers. Studies by Zaks-Makhina and colleagues identified a novel potassium channel blocker using a yeast genetic screen based on Kir2.1 rescue. Surprisingly, the compound turned out to be a better inhibitor of the voltage-gated potassium channel Kv2.1,53 a result that may be related to the high degree of structural conservation present in potassium channel pore domains.54
Identification of a new channel modulator is only a first step. One of the immediate questions that a researcher faces once a new channel blocker or activator is identified is: ‘How does the compound act?’ The use of genetic selections to find suppressors of channel blockers is a potent approach for addressing this question as one demands two stringent criteria: the channel must become insensitive to the blocker as a result of the mutational change but still function as an ion channel (Fig. 2B and C). My laboratory used a combination of blocker screening and the selection of blocker resistant mutants from pore-domain libraries to examine whether the selection system would be a fruitful way not only to find blockers but also to map their sites of action.27 By focusing on the well-known potassium channel blocker barium and selecting for barium resistant Kir2.1 channels, we uncovered an unusual mutation located very near the barium binding site that could make the channels resistant to the blocker without perturbing other functional properties. The mutation placed a positively charged residue in close proximity to the ion conduction pathway at a position that should effectively cancel any effects from a putative helix macrodipole that was thought to be important for ion conduction.55 Extensive biophysical characterization and computational studies established that the barium resistance was electrostatic in origin and showed that the helix macrodipole could not be an important factor for ion conduction.27
Recently, an elegant set of studies reported the identification and characterization of a new voltage-gated calcium channel blocker through the use of a genetic selection based on the roundworm Caenorhabditis elegans.56,57 Roy and colleagues initially searched a ∼14 000 compound library for new small molecules that could be used to explore the biology of C. elegans. One of the ∼300 hits yielded a novel compound, nemadipine-A, that caused a variety of growth and egg-laying defects in the worms. Nemadipine-A is related to the class of drugs known as 1,4-dihydropyridines (DHPs) that affect voltage-gated calcium channel function and are used to treat hypertension. Subsequent studies for suppressors of nemadipine-A activity identified the target as the sole C. elegans voltage-gated calcium channel α1-subunit, Egl-19.56 This channel is homologous to the human L-type CaV1 family. CaV1 subunits are large (∼2500 amino acids) and might seem to be an unlikely candidate for a productive unbiased screen. Nevertheless, a follow-up study in which chemically mutagenized worms were used to look for suppressors of nemadipine-A identified a number of mutants in the worm CaV1 channel.57 Remarkably, the mutants identified eleven residues that had been previously shown to be critical for DHP binding in mammalian CaV1 channels and a new set of eight mutants at previously uncharacterized positions. When tested in the context of the electrophysiologically well-characterized rat CaV1.2 channel, six of the novel mutants altered DHP sensitivity and convincingly demonstrated the potential for using this system as a means for finding new important elements of drug sensitivity and channel gating. The set of studies by Roy and colleagues is a fantastic demonstration of the power of organism based genetic screens to identify novel small molecules and to gain important and unexpected insights into the mechanism of action. Together, the yeast and worm-based channel blocker identification and suppressor studies establish important proof-of-concept examples that will hopefully inspire further development of channel selection systems that can further enrich channel pharmacology and extend our understanding of drug–channel interactions.
In vitro evolution methods and channels, breaking over the horizon
The evolution of new traits that arise from the combination of individual variation in a population and application of selective pressure is the fundamental principle that underpins all of modern biology. This principle is not limited to living biological systems but can also be harnessed to shape molecules. In vitro evolution experiments have been among the most powerful ones deployed by biochemists for finding molecules with novel properties and have been a robust area of biochemical research with a more than 40 year history.58 These experiments use Darwinian selection to cull polynucleotides or polypeptides having novel properties from large libraries of variants through multiple rounds of competition, selection, and amplification (Fig. 3). One major advantage of in vitro evolution methods is that one can access exceptionally large libraries that contain up to 1013–1015 unique molecules.
Fig. 3.
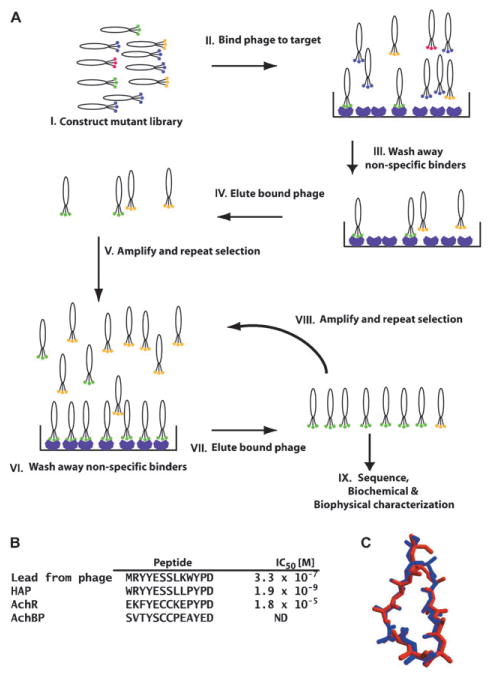
A, schematic of the in vitro selection cycle using phage-display. I: depicts a collection of phage variants in which the library of peptides or proteins is displayed as a fusion to a phage coat protein. Colors indicate individual variants. II: the phage library is mixed with an immobilized purified target protein. III: phages that do not bind are washed away. Some sequences that bind non-specifically, indicated by the yellow hexagons, may remain. IV: recovery of bound phage by elution with ligand or low pH. V: eluted phages are amplified by passage through E. coli. The amplified library of recovered variants is then used in a second round of selection, steps VI through VIII. IX: progress of the experiment is usually monitored by sequencing some fraction of the selected clones. As the cycles of selection progress, the sequence variation of the library should decrease. Once the rounds of selection are finished (generally three–ten rounds), the selected peptide or protein product is made and characterized. In outline, the depicted selection cycle is similar to the procedures used for in vitro selection of nucleic acid aptamers by SELEX. B, left, comparison of α-BXT binding peptides discovered using phage display and subsequent design (HAP) with the sequences of the binding site from the channel (AChR) and AChBP. IC50 values for blocking α-BXT binding to AChR. C, comparison of the structure of the backbone and Cβ positions of the HAP peptide from the HAP–α-BXT complex (red) and AChBP residues (blue). Panels B and C are adapted from ref. 74.
The main in vitro evolution technologies focus on the two types of biopolymers that have well-known sequence-dependent folding and self-assembly properties: oligonucleotides, both DNA and RNA, and peptides and proteins. Nucleic acid polymers have the advantage that the molecule contains both the information for folding and the information for direct amplification (using enzymes). Peptide and protein display methods require a means to link the functional molecule (the polypeptide) with the information required for directing its synthesis (a piece of DNA). A wide variety of in vitro evolution systems that link these two together using bacteriophage,59–61 ribosome display,60–62 and mRNA display61–63 are now widely used. All of these methods work best when they are directed against a purified target. As the expression, purification, and biochemical isolation of ion channels is still not routine these technologies have not yet been fully harnessed in the service of studying ion channels. Nevertheless, it has been demonstrated that one can run selections using membranes or cells that bear the target receptor to isolate target-specific polymers. Thus, the ability to isolate a purified target is not absolutely essential.
Aptamers are nucleic acid polymers that act as high-affinity binders for a particular target,64 such as a protein or small molecule, and are evolved by an in vitro selection method SELEX (systematic evolution of ligands by exponential enrichment).65 The concept is straightforward. One starts with a large library of randomized nucleic acid sequences flanked by fixed sequences that can be used for enzymatic amplification. Typically, aptamer libraries are made from DNA or RNA polymers of 20–100 nucleotides and can contain up to a trillion unique members. The library is then subjected to a selection procedure that involves incubation with the target, some procedure to separate the bound from unbound molecules, and capture of the few molecules that bind. Following recovery, binders are amplified, for example by PCR, and the process is repeated multiple times in order to isolate sequences that have a high-affinity interaction with the target. One of the biggest challenges with such approaches is coming up with a good strategy to squelch the background binding. Successful approaches include elution by competition with a known ligand of the target or counterselections against decoy targets to eliminate background binders.
A number of groups have succeeded at evolving channel-directed nucleic acid aptamers by employing approaches that target a channel that is not a purified protein, but that is presented in a cell membrane environment. The Hess group has used the fact that the Torpedo electric organ is an exceptionally enriched source of nicotinic acetylcholine receptors (nAChRs) and conducted SELEX experiments using a combination of gel-shifts and high-affinity binder displacement experiments to isolate aptamer sequences that bind to nAChRs and inhibit AChR activity in isolated muscle cells.66,67 A similar approach using picrotoxin displacement of aptamers from rat forebrain preparations has led to the isolation of RNA aptamers that bind GABAA receptors with nanomolar affinity and inhibitory activity against heterologously expressed channels.68 The apparent success at isolating aptamers that are specific for a target displayed in a very heterogeneous environment indicates that there may be a great potential for using similar approaches for other ion channels.
To date, few of the ever-growing numbers of channels and channel domains that have been purified and expressed for crystallographic studies have been exploited as selection targets. This situation is starting to change. Two recent reports make use of the soluble, ligand-binding extracellular domain of the glutamate receptor subtype GluR2 in SELEX experiments that are no doubt a harbinger of the near future of this exciting area of research. The Niu group has recently reported the isolation of an RNA aptamer having nanomolar affinity for GluR2 by using SELEX on HEK cells that expressed glutamate receptors following transient transfection.69 The authors show that the RNA aptamer can inhibit channel function and also characterize its binding properties against the soluble version of the GluR2 extracellular domain. In an approach that exploited binding to the structurally well-characterized S1/S2 soluble domain,70 the Jayaraman group was also able to isolate an RNA aptamer that is a competitive antagonist of GluR2 and that displays subtype specificity as it is inactive against the related glutamate receptor GluR6.71 Together, these reports highlight the exciting possibilities for developing novel molecules that may prove useful for studies of ion channel function.
Phage display libraries offer a useful platform for the isolation and evolution of peptides and proteins with unique properties (Fig. 3A). In this format, randomized sequences are displayed in the form of fusion proteins that are linked to particular phage coat proteins. Such formats have been extremely useful for the evolution of antibodies59 and antibody-like molecules.72 Selection involves binding, washing, and elution steps having the same possible pitfalls of non-specific binding as the SELEX experiments. Library construction and phage amplification and propagation are done through steps that require E. coli and as a result the library sizes are a good deal smaller than what one can work with in SELEX (the best being 109). Nevertheless, recent work shows that this is not a serious limitation as specific molecules have been evolved that can bind a variety of targets.
α-Bungarotoxin (α-BXT) is a peptide toxin found in elapid snake venom and is a potent inhibitor of nAChRs (EC50 ≈ 10−11 M). Using phage display of random fifteen residue peptides, Fuchs and colleagues identified a peptide that bound to α-BXT with micromolar affinity, could prevent toxin binding to the receptor, and that had a sequence that resembled the sequence found in the agonist binding site73 (Fig. 3B). By incorporating a few amino acid changes, the investigators were able to turn this lead peptide into one having almost two orders of magnitude higher affinity for α-BXT. Comparison of the structure of a designed higher affinity version of the α-BXT inhibitory peptide complexed with α-BXT and the conformation of the agonist binding loop of a soluble homolog of the extracellular domain of nAChR revealed a remarkable structural similarity74 (Fig. 3C). This work provides an elegant example of the power of phage display to discover new reagents and new biological insights. Peptides such as these that are discovered by phage display may prove to be particularly useful new reagents for controlling channel function.
The types of protein and peptide molecules that can be displayed on phage are enormous. Peptide toxins from the venoms of snakes, insects, and marine snails have been indispensable for ion channel research and have even led to new therapeutics.75–77 It may be possible to display libraries of these types of molecules on a phage and evolve new toxins with altered specificities or that interact with ion channels that presently lack such modulators.
Conclusions and perspectives
The use of genetic selections in cellular and in vitro systems is becoming an important strategy for dissecting the ion channel functional mechanisms and holds great promise for the discovery of new biopolymers and small molecules that affect channel function. The initial reports using in vitro evolution experiments to develop channel-directed reagents offer a promising view of the types of applications that are well within reach for a variety of targets. As more and more channels and channel domains are produced for structural studies, one natural byproduct is likely to be the use of phage or RNA display methods to create new agents. Such applications offer an exciting new avenue for the intersection of channels and molecular evolution methods.
Finally, one wonders how far such laboratory-based evolution experiments can be pushed. One intriguing question is how did nature invent the various folds that became the ion channels we now know. The microorganism-based channel selection methods have thus far only been used to explore questions about the structure gating properties of existing channels. The application of molecular evolution approaches has yielded exciting new prospects for evolving soluble proteins with new functions.78 One can anticipate that similar exciting discoveries await those who can develop a system for it allows the directed evolution of ion channels with completely new functions or the evolution of an ion channel from scratch. Such research directions would greatly enhance our ability to turn channels into novel devices and to address fundamental questions regarding ion channel evolution.
Acknowledgments
I thank S. Bagriantsev, K. Brejc, B. Myers, A. Moroni, E. Reuveny, and G. Thiel for comments on the manuscript. This work was supported by grants to DLM from NIH-NINDS and American Heart Association. DLM is an AHA Established Investigator.
Biography

Daniel L. Minor, Jr., is an Associate Professor at the University of California, San Francisco, in the Cardiovascular Research Institute, Departments of Biochemistry and Biophysics, and Cellular and Molecular Pharmacology, and California Institute for Quantitative Biosciences. He also holds a position as Faculty Scientist at the Lawrence Berkeley National Laboratory. Minor received his BA in Biophysics and Biochemistry magna cum laude from the University of Pennsylvania. He earned his PhD in chemistry at the Massachusetts Institute of Technology for studies on protein structure and design. He began his studies of ion channels during postdoctoral training at the MRC Laboratory of Molecular Biology with Nigel Unwin, and UCSF Department of Physiology with Lily Jan. He was named a Beckman Young Investigator, McKnight Scholar, Rita Allen Scholar, Searle Scholar, and Sloan Fellow, and is currently an American Heart Association Established Investigator. His laboratory applies multidisciplinary approaches including selection methods, electrophysiology, and X-ray crystallography to dissect ion channel structure and function.
Footnotes
Definitions: classical genetics (forward genetics), a procedure that connects a phenotype to a particular genotype; reverse genetics, identification of phenotypes that result from specific mutations in a gene of interest; selection, a protocol in which functional molecules (in this case channels) are required for cell survival or yield a toxic phenotype; screen, application of an assay to a pool of mutant channels. In a screen every mutant must be examined; GOF, gain of function; LOF, loss of function.
References
- 1.Hille B. Ion Channels of Excitable Membranes. 3rd. Sinauer Associates, Inc.; Sunderland, MA: 2001. [Google Scholar]
- 2.Ashcroft FM. Ion Channels and Disease. Academic Press; San Diego, CA: 2000. [Google Scholar]
- 3.Ashcroft FM. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 4.Minor DL., Jr The neurobiologist's guide to structural biology: a primer on why macromolecular structure matters and how to evaluate structural data. Neuron. 2007;54:511–533. doi: 10.1016/j.neuron.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237:770–775. doi: 10.1126/science.2441471. [DOI] [PubMed] [Google Scholar]
- 6.Papazian DM, Schwarz TL, Tempel BL, Jan YN, Jan LY. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science. 1987;237:749–753. doi: 10.1126/science.2441470. [DOI] [PubMed] [Google Scholar]
- 7.Jan LY, Jan YN. Cloned potassium channels from eukaryotes and prokaryotes. Annu Rev Neurosci. 1997;20:91–123. doi: 10.1146/annurev.neuro.20.1.91. [DOI] [PubMed] [Google Scholar]
- 8.Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 9.Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 10.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annu Rev Physiol. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 12.Kink JA, Maley ME, Preston RR, Ling KY, Wallen-Friedman MA, Saimi Y, Kung C. Mutations in paramecium calmodulin indicate functional differences between the C-terminal and N-terminal lobes in vivo. Cell. 1990;62:165–174. doi: 10.1016/0092-8674(90)90250-i. [DOI] [PubMed] [Google Scholar]
- 13.Loukin SH, Vaillant B, Zhou XL, Spalding EP, Kung C, Saimi Y. Random mutagenesis reveals a region important for gating of the yeast K+ channel Ykc1. EMBO J. 1997;16:4817–4825. doi: 10.1093/emboj/16.16.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ou X, Blount P, Hoffman RJ, Kung C. One face of a transmembrane helix is crucial in mechanosensitive channel gating. Proc Natl Acad Sci U S A. 1998;95:11471–11475. doi: 10.1073/pnas.95.19.11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai HC, Grabe M, Jan YN, Jan LY. The S4 voltage sensor packs against the pore domain in the KAT1 voltage-gated potassium channel. Neuron. 2005;47:395–406. doi: 10.1016/j.neuron.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Sadja R, Smadja K, Alagem N, Reuveny E. Coupling Gbetagamma-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron. 2001;29:669–680. doi: 10.1016/s0896-6273(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 17.Myers BR, Bohlen CJ, Julius D. A yeast genetic screen reveals a critical role for the pore helix domain in TRP channel gating. Neuron. 2008;58:362–373. doi: 10.1016/j.neuron.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bandell M, Dubin AE, Petrus MJ, Orth A, Mathur J, Hwang SW, Patapoutian A. High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nat Neurosci. 2006;9:493–500. doi: 10.1038/nn1665. [DOI] [PubMed] [Google Scholar]
- 19.Grandl J, Hu H, Bandell M, Bursulaya B, Schmidt M, Petrus M, Patapoutian A. Pore region of TRPV3 ion channel is specifically required for heat activation. Nat Neurosci. 2008;11:1007–1013. doi: 10.1038/nn.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maurer JA, Dougherty DA. Generation and evaluation of a large mutational library from the Escherichia coli mechanosensitive channel of large conductance, MscL: implications for channel gating and evolutionary design. J Biol Chem. 2003;278:21076–21082. doi: 10.1074/jbc.M302892200. [DOI] [PubMed] [Google Scholar]
- 21.Kuo MM, Saimi Y, Kung C. Gain-of-function mutations indicate that Escherichia coli Kch forms a functional K+ conduit in vivo. EMBO J. 2003;22:4049–4058. doi: 10.1093/emboj/cdg409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paynter JJ, Sarkies P, Andres-Enguix I, Tucker SJ. Genetic selection of activatory mutations in KcsA. Channels (Austin) 2008;2:413–418. doi: 10.4161/chan.2.6.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su Z, Zhou X, Haynes WJ, Loukin SH, Anishkin A, Saimi Y, Kung C. Yeast gain-of-function mutations reveal structure-function relationships conserved among different subfamilies of transient receptor potential channels. Proc Natl Acad Sci U S A. 2007;104:19607–19612. doi: 10.1073/pnas.0708584104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou X, Su Z, Anishkin A, Haynes WJ, Friske EM, Loukin SH, Kung C, Saimi Y. Yeast screens show aromatic residues at the end of the sixth helix anchor transient receptor potential channel gate. Proc Natl Acad Sci U S A. 2007;104:15555–15559. doi: 10.1073/pnas.0704039104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Wray R, Blount P. Intragenic suppression of gain-of-function mutations in the Escherichia coli mechanosensitive channel, MscL. Mol Microbiol. 2004;53:485–495. doi: 10.1111/j.1365-2958.2004.04150.x. [DOI] [PubMed] [Google Scholar]
- 26.Minor DL, Jr, Masseling SJ, Jan YN, Jan LY. Transmembrane structure of an inwardly rectifying potassium channel. Cell. 1999;96:879–891. doi: 10.1016/s0092-8674(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 27.Chatelain FC, Alagem N, Xu Q, Pancaroglu R, Reuveny E, Minor DL., Jr The pore helix dipole has a minor role in inward rectifier channel function. Neuron. 2005;47:833–843. doi: 10.1016/j.neuron.2005.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irizarry SN, Kutluay E, Drews G, Hart SJ, Heginbotham L. Opening the KcsA K+ channel: tryptophan scanning and complementation analysis lead to mutants with altered gating. Biochemistry. 2002;41:13653–13662. doi: 10.1021/bi026393r. [DOI] [PubMed] [Google Scholar]
- 29.Yi BA, Lin YF, Jan YN, Jan LY. Yeast screen for constitutively active mutant G protein-activated potassium channels. Neuron. 2001;29:657–667. doi: 10.1016/s0896-6273(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 30.Bichet D, Lin YF, Ibarra CA, Huang CS, Yi BA, Jan YN, Jan LY. Evolving potassium channels by means of yeast selection reveals structural elements important for selectivity. Proc Natl Acad Sci U S A. 2004;101:4441–4446. doi: 10.1073/pnas.0401195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev. 2005;57:387–395. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- 32.Epstein W. The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol. 2003;75:293–320. doi: 10.1016/s0079-6603(03)75008-9. [DOI] [PubMed] [Google Scholar]
- 33.Ko CH, Gaber RF. TRK1 and TRK2 encode structurally related K+ transporters in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:4266–4273. doi: 10.1128/mcb.11.8.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buurman ET, McLaggan D, Naprstek J, Epstein W. Multiple paths for nonphysiological transport of K+ in Escherichia coli. J Bacteriol. 2004;186:4238–4245. doi: 10.1128/JB.186.13.4238-4245.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Epstein W, Buurman E, McLaggan D, Naprstek J. Multiple mechanisms, roles and controls of K+ transport in Escherichia coli. Biochem Soc Trans. 1993;21:1006–1010. doi: 10.1042/bst0211006. [DOI] [PubMed] [Google Scholar]
- 36.Sentenac H, Bonneaud N, Minet M, Lacroute F, Salmon JM, Gaymard F, Grignon C. Cloning and expression in yeast of a plant potassium ion transport system. Science. 1992;256:663–665. doi: 10.1126/science.1585180. [DOI] [PubMed] [Google Scholar]
- 37.Anderson JA, Nakamura RL, Gaber RF. Heterologous expression of K+ channels in Saccharomyces cerevisiae: strategies for molecular analysis of structure and function. Symp Soc Exp Biol. 1994;48:85–97. [PubMed] [Google Scholar]
- 38.Rodriguez-Navarro A. Potassium transport in fungi and plants. Biochim Biophys Acta. 2000;1469:1–30. doi: 10.1016/s0304-4157(99)00013-1. [DOI] [PubMed] [Google Scholar]
- 39.Rubio F, Gassmann W, Schroeder JI. Sodium-driven potassium uptake by the plant potassium transporter HKT1 and mutations conferring salt tolerance. Science. 1995;270:1660–1663. doi: 10.1126/science.270.5242.1660. [DOI] [PubMed] [Google Scholar]
- 40.Uozumi N, Gassmann W, Cao Y, Schroeder JI. Identification of strong modifications in cation selectivity in an Arabidopsis inward rectifying potassium channel by mutant selection in yeast. J Biol Chem. 1995;270:24276–24281. doi: 10.1074/jbc.270.41.24276. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura RL, Gaber RF. Studying ion channels using yeast genetics. Methods Enzymol. 1998;293:89–104. doi: 10.1016/s0076-6879(98)93009-9. [DOI] [PubMed] [Google Scholar]
- 42.Tang W, Ruknudin A, Yang W, Shaw S, Knickerbocker A, Kurtz S. Functional expression of a vertebrate inwardly rectifying K+ channel in yeast. Mol Biol Cell. 1995;6:1231–1240. doi: 10.1091/mbc.6.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura RL, Anderson JA, Gaber RF. Determination of key structural requirements of a K+ channel pore. J Biol Chem. 1997;272:1011–1018. doi: 10.1074/jbc.272.2.1011. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 45.Perozo E. Gating prokaryotic mechanosensitive channels. Nat Rev Mol Cell Biol. 2006;7:109–119. doi: 10.1038/nrm1833. [DOI] [PubMed] [Google Scholar]
- 46.Maurer JA, Dougherty DA. A high-throughput screen for MscL channel activity and mutational phenotyping. Biochim Biophys Acta, Biomembr. 2001;1514:165. doi: 10.1016/s0005-2736(01)00390-x. [DOI] [PubMed] [Google Scholar]
- 47.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 48.Lemmon MA, Treutlein HR, Adams PD, Brunger AT, Engelman DM. A dimerization motif for transmembrane alpha-helices. Nat Struct Biol. 1994;1:157–163. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 49.Lemmon MA, Engelman DM. Specificity and promiscuity in membrane helix interactions. Q Rev Biophys. 1994;27:157–218. doi: 10.1017/s0033583500004522. [DOI] [PubMed] [Google Scholar]
- 50.Schreiber G, Fersht AR. Energetics of protein–protein interactions: analysis of the barnase–barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 51.Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 52.Grabe M, Lai HC, Jain M, Jan YN, Jan LY. Structure prediction for the down state of a potassium channel voltage sensor. Nature. 2007;445:550–553. doi: 10.1038/nature05494. [DOI] [PubMed] [Google Scholar]
- 53.Zaks-Makhina E, Kim Y, Aizenman E, Levitan ES. Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol Pharmacol. 2004;65:214–219. doi: 10.1124/mol.65.1.214. [DOI] [PubMed] [Google Scholar]
- 54.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 55.Roux B, MacKinnon R. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 1999;285:100–102. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- 56.Kwok TC, Ricker N, Fraser R, Chan AW, Burns A, Stanley EF, McCourt P, Cutler SR, Roy PJ. A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature. 2006;441:91–95. doi: 10.1038/nature04657. [DOI] [PubMed] [Google Scholar]
- 57.Kwok TC, Hui K, Kostelecki W, Ricker N, Selman G, Feng ZP, Roy PJ. A genetic screen for dihydropyridine (DHP)-resistant worms reveals new residues required for DHP-blockage of mammalian calcium channels. PLoS Genet. 2008;4:e1000067. doi: 10.1371/journal.pgen.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joyce GF. Forty years of in vitro evolution. Angew Chem, Int Ed. 2007;46:6420–6436. doi: 10.1002/anie.200701369. [DOI] [PubMed] [Google Scholar]
- 59.Sidhu SS, Koide S. Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol. 2007;17:481–487. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 60.Dufner P, Jermutus L, Minter RR. Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 2006;24:523–529. doi: 10.1016/j.tibtech.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 61.Levin AM, Weiss GA. Optimizing the affinity and specificity of proteins with molecular display. Mol BioSyst. 2006;2:49. doi: 10.1039/b511782h. [DOI] [PubMed] [Google Scholar]
- 62.Lipovsek D, Pluckthun A. In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods. 2004;290:51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 63.Gold L. mRNA display: diversity matters during in vitro selection. Proc Natl Acad Sci U S A. 2001;98:4825–4826. doi: 10.1073/pnas.091101698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breaker RR. Natural and engineered nucleic acids as tools to explore biology. Nature. 2004;432:838–845. doi: 10.1038/nature03195. [DOI] [PubMed] [Google Scholar]
- 65.Shamah SM, Healy JM, Cload ST. Complex target SELEX. Acc Chem Res. 2008;41:130–138. doi: 10.1021/ar700142z. [DOI] [PubMed] [Google Scholar]
- 66.Cui Y, Ulrich H, Hess GP. Selection of 2′-fluoro-modified RNA aptamers for alleviation of cocaine and MK-801 inhibition of the nicotinic acetylcholine receptor. J Membr Biol. 2004;202:137–149. doi: 10.1007/s00232-004-0725-4. [DOI] [PubMed] [Google Scholar]
- 67.Ulrich H, Ippolito JE, Pagan OR, Eterovic VA, Hann RM, Shi H, Lis JT, Eldefrawi ME, Hess GP. In vitro selection of RNA molecules that displace cocaine from the membrane-bound nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A. 1998;95:14051–14056. doi: 10.1073/pnas.95.24.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui Y, Rajasethupathy P, Hess GP. Selection of stable RNA molecules that can regulate the channel-opening equilibrium of the membrane-bound gamma-aminobutyric acid receptor. Biochemistry. 2004;43:16442–16449. doi: 10.1021/bi048667b. [DOI] [PubMed] [Google Scholar]
- 69.Huang Z, Pei W, Jayaseelan S, Shi H, Niu L. RNA aptamers selected against the GluR2 glutamate receptor channel. Biochemistry. 2007;46:12648–12655. doi: 10.1021/bi701036p. [DOI] [PubMed] [Google Scholar]
- 70.Mayer ML. Glutamate receptor ion channels. Curr Opin Neurobiol. 2005;15:282–288. doi: 10.1016/j.conb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 71.Du M, Ulrich H, Zhao X, Aronowski J, Jayaraman V. Water soluble RNA based antagonist of AMPA receptors. Neuropharmacology. 2007;53:242–251. doi: 10.1016/j.neuropharm.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sennhauser G, Grutter MG. Chaperone-assisted crystallography with DARPins. Structure. 2008;16:1443–1453. doi: 10.1016/j.str.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 73.Balass M, Katchalski-Katzir E, Fuchs S. The alpha-bungarotoxin binding site on the nicotinic acetylcholine receptor: analysis using a phage-epitope library. Proc Natl Acad Sci U S A. 1997;94:6054–6058. doi: 10.1073/pnas.94.12.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harel M, Kasher R, Nicolas A, Guss JM, Balass M, Fridkin M, Smit AB, Brejc K, Sixma TK, Katchalski-Katzir E, Sussman JL, Fuchs S. The binding site of acetylcholine receptor as visualized in the X-Ray structure of a complex between alpha-bungarotoxin and a mimotope peptide. Neuron. 2001;32:265–275. doi: 10.1016/s0896-6273(01)00461-5. [DOI] [PubMed] [Google Scholar]
- 75.Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- 76.Swartz KJ. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 2007;49:213–230. doi: 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Catterall WA, Cestele S, Yarov-Yarovoy V, Yu FH, Konoki K, Scheuer T. Voltage-gated ion channels and gating modifier toxins. Toxicon. 2007;49:124–141. doi: 10.1016/j.toxicon.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 78.Bershtein S, Tawfik DS. Advances in laboratory evolution of enzymes. Curr Opin Chem Biol. 2008;12:151–158. doi: 10.1016/j.cbpa.2008.01.027. [DOI] [PubMed] [Google Scholar]