

Abstract
Protein concentrations depend not only on the mRNA level, but also on the translation rate and the degradation rate. Prediction of mRNA's translation rate would provide valuable information for in-depth understanding of the translation mechanism and dynamic proteome. In this study, we developed a new computational model to predict the translation rate, featured by (1) integrating various sequence-derived and functional features, (2) applying the maximum relevance & minimum redundancy method and incremental feature selection to select features to optimize the prediction model, and (3) being able to predict the translation rate of RNA into high or low translation rate category. The prediction accuracies under rich and starvation condition were 68.8% and 70.0%, respectively, evaluated by jackknife cross-validation. It was found that the following features were correlated with translation rate: codon usage frequency, some gene ontology enrichment scores, number of RNA binding proteins known to bind its mRNA product, coding sequence length, protein abundance and 5′UTR free energy. These findings might provide useful information for understanding the mechanisms of translation and dynamic proteome. Our translation rate prediction model might become a high throughput tool for annotating the translation rate of mRNAs in large-scale.
Introduction
It is often assumed that genes with high mRNA levels also have high protein abundance. Thus, mRNA levels are used instead of protein abundance. However, the regulation of gene expression takes place at many levels, from transcription to translation and to the post-translational modification. Many studies either could not find the assumed correlation between mRNA level and protein abundance [1] or the correlation was very weak[2], [3]. By estimation, only 20%–40% of protein abundance is determined by the concentration of its corresponding mRNA [4], [5]. The reason for such weak correlation between protein and mRNA levels is that protein concentrations depend not only on the mRNA level, but also the translation rate and the degradation rate [6].
Translation is the third process of gene expression. In this stage, mRNA is decoded by the ribosome which binds to tRNAs with complementary anticodon sequences. The tRNAs carry specific amino acids that are synthesized into a polypeptide as the mRNA passes through the ribosome. Translation has three steps: initiation, elongation and termination [7]. Both empirical and theoretical studies showed that the bottleneck step in the translation process is the initiation of protein translation [8], [9], [10]. At the initiation step, the ribosome binds to the five prime untranslated region (5′UTR) of mRNA and moves along the mRNA until it gets to the translation start site (TSS). After initiation is completed, the ribosome enters the elongation stage. At elongation step, the ribosome waits until it intercepts an appropriate tRNA whose anticodon complements the codon at the A site of ribosome. Once the correct tRNA is intercepted by the ribosome, the amino acid from the tRNA is transferred to the ribosome associated peptide chain, and the ribosome moves forward one codon. The waiting for the correct tRNA limits the elongation process [10], [11]. Translational initiation rate determines protein production rate and elongation rate determines ribosome occupancy [8]. Therefore, ribosome density is proportional to translational initiation rate which determines protein production while it is inversely proportional to translational elongation rate.
The regulation of translation plays as important role as transcriptional regulation in the control of gene expression. Changes of the mRNAs translation rate have great influence on the actual protein abundance. Dysregulation of translation will result in various diseases, such as cancer and neurological disorders [12].
With ribosome-profiling technology, ribosome-protected mRNA fragments can be deep-sequenced and the translation rate can be monitored, but it is time-consuming, expensive and not helpful for understanding the translation mechanisms. Here we choose Saccharomyces cerevisiae, one of the most studied model organisms, to perform our study and predict the translation rate. We used the ribosome-profiling data from Ingolia's work [13] in which the read density of mRNA is measured by deep sequencing of ribosome-protected mRNA fragments under both rich and starvation conditions. According to Ingolia's work [13], the translation rate (or called as translation efficiency) is defined as the normalized read density of translation (footprints) divided by the normalized read density of transcription (mRNA). The ratio of ribosome footprints to mRNA fragments can roughly quantify the rate of protein synthesis [13] although further improvements could incorporate variations in the speed of elongation along the mRNA. Each mRNA is represented by various sequence-derived and functional features related to translation, such as codon usage frequencies, gene ontology enrichment scores, biochemical and physicochemical features, start codon features, coding sequence length, minimum free energy, 5′UTR length, 3′UTR length, number of transcription factors known to bind at the promoter region, number of RNA binding proteins known to bind its mRNA product, protein abundance, mRNA half life, protein half life and 5′UTR free energy. With this dataset, an efficient computational model to predict the translation rate was constructed with Nearest Neighbor Algorithm (NNA) and cross-validated. The prediction accuracies of jackknife cross-validation under rich and starvation condition were 68.8% and 70.0%, respectively. More specifically, to identify the most important features regulating translation rates under different conditions, we applied maximum relevance & minimum redundancy and incremental feature selection to select the important features for predicting the translation rate in rich and starvation conditions, respectively. Our results suggest that the following features are correlated with translation rate: codon usage frequency, some gene ontology enrichment scores, biochemical and physicochemical features of protein (such as amino acids composition, polarity, normalized Van Der Waals volume), number of RNA binding proteins known to bind its mRNA product, coding sequence length, protein abundance and 5′UTR free energy. Our findings might provide clues for understanding the mechanisms of translation. The translation rate prediction model could become a useful tool for annotating the translation rate of mRNAs in large-scale.
Materials and Methods
Dataset
The ribosome-profiling data we used are from Ingolia's work [13] and publicly available at GEOs http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13750. With ribosome-profiling technology, Ingolia et al. [13] deep-sequenced the ribosome-protected mRNA fragments and monitored the genome-wide translation with subcodon resolution in Saccharomyces cerevisiae under both rich and starvation conditions. To get the translation rate, we divided the normalized read density of translation (footprints) by the normalized read density of transcription (mRNA) [13]. The ratio of ribosome footprints to mRNA fragments represents the translation rate and according to their values [13], we characterize the translation rates into two groups which are: (1) smaller than median or equal to median, (2) greater than median. Open Reading Frames (ORFs) in the former group have low translation rate, while the ORFs in the latter group have high translation rate. We characterized the translation rates in rich condition and starvation condition, respectively. The number of ORFs with low translation rates and high translation rates in rich condition and starvation condition can found in Table 1 .
Table 1. The number of ORFs with low translation rates and high translation rates in rich condition and starvation condition.
| Starvation condition | The number of ORFs | |||
| The number of ORFs with Low translation rate | The number of ORFs with High translation rate | |||
| Rich condition | The number of ORFs with Low translation rate | 1125 | 209 | 1334 |
| The number of ORFs with High translation rate | 209 | 1124 | 1333 | |
| The number of ORFs | 1334 | 1333 | 2667 | |
Feature Construction
Codon usage frequency features
We downloaded the ORF coding sequences from Saccharomyces Genome Database (SGD) [14] and calculated the codon relative frequencies with seqinR [15]. It was reported that highly expressed genes have different codon preference with low expressed gene and the pattern of codon usage can be used to predict the gene expression level in yeast [16]. It is highly possible that ORFs with different translation rate have different codon usage pattern, too. There were 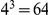 codon usage frequency features.
codon usage frequency features.
Gene Ontology features
Proteins are produced to achieve their biological functions. As demand determines production, the translation rate of ORF is definitely correlated with its biological functions. The function of one protein can be better described in protein interaction network, i.e. the network context will give a comprehensive and robust description of its function. In this study, the network context we used was STRING[17]. The Gene Ontology enrichment score of protein  on Gene Ontology term
on Gene Ontology term 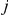 was defined as the –log10 of the hypergeometric test p value [18] of its neighbors on STRING network and can be computed by equation (1):
was defined as the –log10 of the hypergeometric test p value [18] of its neighbors on STRING network and can be computed by equation (1):
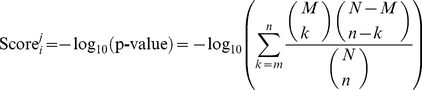 |
(1) |
where 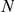 is the number of overall ORFs in yeast,
is the number of overall ORFs in yeast, 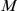 is the number of ORFs annotated to Gene Ontology term
is the number of ORFs annotated to Gene Ontology term 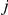 ,
,  is the number of ORFs in ORF set
is the number of ORFs in ORF set  which includes protein
which includes protein  and its neighbors on STRING network,
and its neighbors on STRING network,  is the number of ORFs from ORF set
is the number of ORFs from ORF set  that are annotated to Gene Ontology term
that are annotated to Gene Ontology term 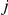 . The larger the enrichment score of one Gene Ontology term, the more overrepresented this term is. There were 4148 Gene Ontology (GO) enrichment score features.
. The larger the enrichment score of one Gene Ontology term, the more overrepresented this term is. There were 4148 Gene Ontology (GO) enrichment score features.
Biochemical and physicochemical features of proteins
To encode proteins of different sequence lengths with a uniform dimensional vector, we adopted the notion of pseudo amino acid composition (PseAAC) [19], [20]. Each protein sequence was represented by 132 biochemical and physicochemical features which can be categorized into seven groups: (1) amino acid composition [21], [22], (2) solvent accessibility, (3) normalized van der Waals volume, (4) polarizability, (5) secondary structure, (6) hydrophobicity, and (7) polarity [23]. Except for amino acid composition, all the other six ones are generated by integrating the pseudo properties of amino acid in the protein sequence and each amino acid can be classified into two or three pseudo groups. For secondary structure, each amino acid can be predicted by SSpro [24] as: helix, strand or coil. For solvent accessibility, each amino acid is predicted by ACCpro [25] as: exposed or buried to solvent. In terms of hydrophobicity, there are three groups of amino acid: hydrophobic (C, V, F, L, I, M,W), neutral (G, P, H, A, S, T,Y) and polar (Q, E, R, K, D, N)[26]. For polarizability: {Y, M, K, R,H, F,W}, {C, Q, I, P, N, V, E L} and {S, D, G, A, T} [27]. For normalized van der Waals volume: {K, F, M, H, R, Y, W}, {E, Q, N, V, I, L} and {S, C, G, A,T, P, D} [28]. For polarity: {K, N, H, Q, R,E, D}, {T, G, P, A, S} and {W, C, L, I, F,M, V, Y} [29].
To generate the global protein features by integrating the local quantities of amino acid over the entire protein sequence, the following three quantities are calculated: 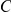 (composition),
(composition), 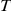 (transition), and
(transition), and 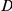 (distribution). The detailed computational procedures and a well illustrated example can be found in our previous work [30]. Generally speaking,
(distribution). The detailed computational procedures and a well illustrated example can be found in our previous work [30]. Generally speaking, 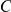 refers to the percent of each pseudo group in the sequence;
refers to the percent of each pseudo group in the sequence; 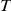 to the frequencies with which one pseudo group changes to another; and
to the frequencies with which one pseudo group changes to another; and 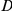 to the relative position where the first, twenty-five percent, fifty-percent, seventy-five percent, and last of each kind of pseudo letters occur.
to the relative position where the first, twenty-five percent, fifty-percent, seventy-five percent, and last of each kind of pseudo letters occur.
For polarity, secondary structure, polarizability, hydrophobicity and normalized van der Waals volume, each amino acid has three pseudo groups and would generate 21 protein features. For solvent accessibility, each amino acid has two pseudo groups and would only generate 7 protein features.
Now for the amino acid composition we have 20 features; for solvent accessibility, 7 features; and for the other five properties, each has 21 features. Combining them together, each protein has 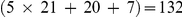 features. The detailed explanation of each biochemical and physicochemical feature can be found in our previous work [30].
features. The detailed explanation of each biochemical and physicochemical feature can be found in our previous work [30].
Start codon features
During the translation initiation, the 40S subunit of ribosome binds to a site upstream of start codon. It proceeds downstream until it encounters the start codon and form the initiation complex of translation. The start codon is typically AUG (or ATG in DNA) and related with translation initiation. We extracted sequences in untranslated region 3 bp upstream of the initial ATG and sequences in coding region 3 bp downstream of the initial ATG. We encoded the 6 bp DNA sequences up/downstream of start codon ATG binarily and each base pair was represented by a 4-dementional vector: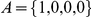 ,
,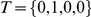 ,
, 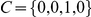 and
and 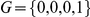 .
.
Coding sequence length
We calculated the coding sequence length of each ORF as a potential feature for translation rate prediction.
Free energy of 42 nucleotide cross translation start site
Kudla et al. [31] identified a region, from nucleotide (nt) –4 to +37 relative to translation start site, for which predicted folding energy can explain some of the of the variation to differences in protein levels. So we calculated the minimum free energy of 42 nucleotide (nt) –4 to +37 relative to translation start site, with Vienna [32].
Various parameters of untranslated regions from Tuller's study
Tuller et al.[33] collected various properties of untranslated regions of the S. cerevisiae genome and we used the following 8 features from Tuller's study: 5′UTR length, 3′UTR length, Number of transcription factors known to bind at the promoter region, Number of RNA binding proteins known to bind its mRNA product, Protein abundance, mRNA half life [34], Protein half life and 5′UTR free energy[35]. Unlike the above free energy, here the 5′UTR free energy is calculated with 5′-UTR 100 nt [33], [35].
Feature space of ORF
As mentioned above, there are 64 codon usage frequency features, 4148 Gene Ontology (GO) enrichment score features, 132 biochemical and physicochemical features, 24start codon features and 10 other features. The total featuresused in this study to represent an ORF sample would be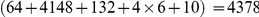 .
.
mRMR method
In this study, we used the Maximum Relevance and Minimum Redundancy (mRMR) feature selection method [36], [37] to rank 4378 features of each ORF considering both their relevance to translation rates and the redundancy among them. The mRMR selected features have maximum relevance with the translation rates and meanwhile minimally redundant, i.e., maximally dissimilar to each other. Both relevance and redundancy are measured with mutual information (MI), which is defined as follows:
| (2) |
where  and
and 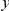 are two vectors,
are two vectors, 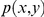 is the joint probabilistic density,
is the joint probabilistic density, 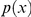 and
and 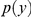 are the marginal probabilistic densities.
are the marginal probabilistic densities.
Let 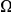 denotes the whole vector set containing all 4378 features,
denotes the whole vector set containing all 4378 features, 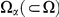 denotes the selected feature set with
denotes the selected feature set with  feature vectors, and
feature vectors, and 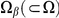 denotes the to-be-selected feature set with
denotes the to-be-selected feature set with 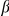 feature vectors. The relevance
feature vectors. The relevance 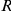 of a feature
of a feature 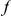 in
in 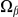 with the translation rate class
with the translation rate class  can be computed by equation (3):
can be computed by equation (3):
| (3) |
The redundancy 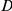 of a feature
of a feature 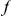 in
in 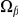 with all the features in
with all the features in 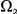 can be computed by equation (4):
can be computed by equation (4):
| (4) |
To select a feature 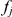 from
from 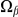 with maximum relevance with translation rates and minimum redundancy with selected features in
with maximum relevance with translation rates and minimum redundancy with selected features in 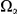 , the mRMR function which integrates equation (3) and equation (4) is defined:
, the mRMR function which integrates equation (3) and equation (4) is defined:
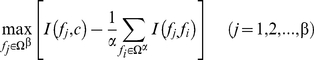 |
(5) |
For a feature pool containing 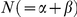 features, feature evaluation will be executed in
features, feature evaluation will be executed in 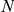 rounds. After these evaluations, a feature set
rounds. After these evaluations, a feature set 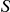 will be obtained:
will be obtained:
| (6) |
where each feature has an mRMR order, representing at which round the feature is selected. The smaller order means more important.
Nearest Neighbor Algorithm
To classify ORFs into different translation rate categories, the Nearest Neighbor Algorithm (NNA) was applied. Its basic idea is to predict a new ORF into its translation rate categories by comparing the features of this ORF with the features of those with known translation rate categories. The distance between two ORF vectors 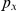 and
and 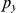 is defined as [30], [38]:
is defined as [30], [38]:
| (7) |
where 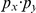 is the inner product of
is the inner product of 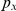 and
and 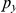 , and
, and 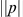 is the module of vector
is the module of vector 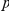 .
. 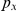 and
and 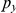 are consider to be more similar if
are consider to be more similar if 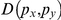 is smaller.
is smaller.
In NNA, an ORF with feature vector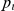 will be predicted as having the same translation rate class as its nearest neighbor which has the smallest
will be predicted as having the same translation rate class as its nearest neighbor which has the smallest 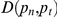 . That is
. That is
| (8) |
where 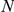 represents the number of training ORFs with known translation rates.
represents the number of training ORFs with known translation rates.
Jackknife Cross-Validation Method
We used Jackknife Cross-Validation Method [38], [39], one of the most objective methods, to evaluate the performance of prediction. During Jackknife Cross-Validation, each ORF in the dataset is tested in turn by the translation rate predictor, which is trained by the other ORFs in the data set. Each ORF is involved in training 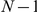 times and is tested exactly once. To evaluate the performance of the translation rate predictor, the prediction accuracy for the overall ORFs can be calculated as:
times and is tested exactly once. To evaluate the performance of the translation rate predictor, the prediction accuracy for the overall ORFs can be calculated as:
| (9) |
where 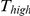 and
and 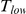 stand for the number of correctly predicted ORFs with high and low translation rate, respectively;
stand for the number of correctly predicted ORFs with high and low translation rate, respectively; 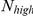 and
and 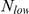 are the number of ORFs with high and low translation rate, respectively.
are the number of ORFs with high and low translation rate, respectively.
Incremental Feature Selection (IFS)
When the mRMR step was completed, we obtained an ordered feature list but still do not know how many fore features in the list should be chosen. To determine the optimal number of features, Incremental Feature Selection (IFS) [30], [38] was applied by constructing 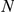 feature subsets of the feature list
feature subsets of the feature list 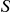 provided by mRMR. Starting from only the first feature
provided by mRMR. Starting from only the first feature 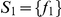 , the feature subset
, the feature subset 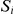 is defined as:
is defined as:
| (10) |
by adding feature 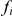 to the previous subset
to the previous subset 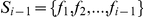 .
.
For each feature subset 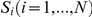 , we calculated the prediction accuracy elevated by Jackknife Cross-Validation. The prediction accuracies with different feature numbers form an IFS curve with feature number
, we calculated the prediction accuracy elevated by Jackknife Cross-Validation. The prediction accuracies with different feature numbers form an IFS curve with feature number as its x-axis and the prediction accuracy as its y-axis.
as its x-axis and the prediction accuracy as its y-axis.
The correlation between features and translation rate
To identify the direction of the correlation between features selected by mRMR and IFS with translation rate, we calculated the point-biserial correlation coefficient between them. The point biserial correlation [40] is a measure of association between a continuous variable and a binary variable. Assume that X is the selected feature which is a continuous variable and Y is the translation rate which is binary. The point biserial correlation is calculated as
| (11) |
where 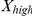 is the mean of
is the mean of 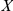 with high translation rate,
with high translation rate, 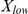 is the mean of
is the mean of 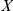 with low translation rate,
with low translation rate, 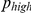 is the proportion of
is the proportion of 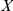 with high translation rate,
with high translation rate, 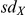 is the standard deviation of
is the standard deviation of 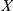 . The point biserial correlation is positive when large values of
. The point biserial correlation is positive when large values of  are associated with high translation rate and small values of
are associated with high translation rate and small values of 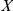 are associated with low translation rate.
are associated with low translation rate.
Results
Identification of relevant features and construct translation rate prediction model
Using mRMR method, we ranked and analyzed the top 500 relevant features to translation rate with Maximum Relevance Minimum Redundancy method. Each of them has the maximal relevance with translation rate and the minimal redundancy with other features. Then in Incremental Feature Selection (IFS) procedure, 500 prediction models were constructed using nearest neighbor algorithm with 1, 2, 3… 499 and 500 features respectively and tested by jackknife cross-validations as described above. The IFS results of rich and starvation condition were shown in Figure 1 (A) and Figure 1 (B), respectively. It can be seen from Figure 1 (A) that the translation rate prediction model of rich condition achieved the peak accuracy at 68.8% with 37 features. These 37 features formed the optimal feature set for translation rate prediction model of rich condition and are provided in Table S1. Similarly, in Figure 1 (B), the translation rate prediction model of starvation condition achieved the highest accuracy at 70.0% with 86 features. These 86 features formed the optimal feature set for translation rate prediction model of starvation condition and can be found in Table S2.
Figure 1. The IFS curves of translation rate prediction in rich and starvation condition.

The IFS curves for (A) the translation rate prediction model of rich condition achieved the peak accuracy at 68.8% with 37 features and (B) the translation rate prediction model of starvation condition achieved the highest accuracy at 70.0% with 86 features.
Factors correlated with translation rate
We compared the optimal 37-feature set of rich condition and the optimal 86-feature set of starvation condition and found there were 27 common features between them. These 27 common features are provided in Table 2 . To identify what kinds of features are important for translation rate prediction, we calculated the numbers of each kind of features in the optimal feature set. Figure 2 shows the numbers of each kind of features in (A) the optimal 37-feature set of rich condition, (B) the optimal 86-feature set of starvation condition. As we can see from Figure 2 , Table S1, Table S2 and Table 2 , the following kinds of features are correlated with translation rate: (1) Codon usage frequency, (2) some Gene Ontology (GO) enrichment scores, (3) protein features (such as amino acids composition, polarity, normalized Van Der Waals volume) and (4) other features (such as Number of RNA binding proteins known to bind its mRNA product, Coding sequence length, Protein abundance and 5′UTR free energy).
Table 2. The common features for translation rate prediction in both rich condition and starvation condition.
| Name | Feature Type | Point-Biserial Correlation (rich) | Point-Biserial Correlation (starvation) |
| ATA | Codon usage frequency | −0.3641809 | −0.320724134 |
| V123 | Amino acids composition | 0.217345654 | 0.249518281 |
| CGA | Codon usage frequency | −0.297473206 | −0.244839127 |
| TCC | Codon usage frequency | 0.251689274 | 0.234058044 |
| NoofRNABindingProteins | Other (Number of RNA binding proteins known to bind its mRNA product) | 0.22353164 | 0.194339726 |
| GCT | Codon usage frequency | 0.279887045 | 0.266483213 |
| V126 | Amino acids composition | −0.180096048 | −0.149802124 |
| GGA | Codon usage frequency | −0.208428434 | −0.176300373 |
| cds.length | Other (Coding sequence length) | 0.097429773 | −0.03025402 |
| V72 | Polarity | 0.279590151 | 0.307614177 |
| CGG | Codon usage frequency | −0.189139955 | −0.147269889 |
| PA | Other (Protein abundance) | 0.141561548 | 0.120850079 |
| AGG | Codon usage frequency | −0.199042873 | −0.154301709 |
| CCA | Codon usage frequency | 0.282776605 | 0.283726919 |
| ACC | Codon usage frequency | 0.24618065 | 0.230897941 |
| TGC | Codon usage frequency | −0.220759013 | −0.173512017 |
| GO:0005737 | GO (GO:0005737_cytoplasm) | 0.242558032 | 0.206209243 |
| GCC | Codon usage frequency | 0.268835706 | 0.270918872 |
| GTA | Codon usage frequency | −0.212847373 | −0.20408338 |
| GO:0042277 | GO (GO:0042277_peptide binding) | 0.137845496 | 0.139232871 |
| CTT | Codon usage frequency | −0.203855194 | −0.190162108 |
| TCT | Codon usage frequency | 0.194907502 | 0.185575651 |
| TAT | Codon usage frequency | −0.188268811 | −0.173452245 |
| AAC | Codon usage frequency | 0.143590251 | 0.176587498 |
| GO:0006878 | GO (GO:0006878_cellular copper ion homeostasis) | 0.134957407 | 0.131972094 |
| V55 | Normalized Van Der Waals volume | −0.19022717 | −0.191407228 |
Figure 2. The numbers of each kind of features in optimal feature sets.

The numbers of each kind of features for (A) the optimal 37-feature set of rich condition, (B) the optimal 86-feature set of starvation condition.
Discussion
In this study, we have developed a new computational method to predict the translation rate by integrating various sequence-derived features and functional features. In rigorous jackknife cross-validation test, the predictor can achieve an overall prediction accuracy of 68.8% and 70.0% in rich and starvation conditions, respectively. We concluded that the following features are correlated with translation rate: codon usage frequency, some GO enrichment scores, protein features (such as amino acids composition, polarity, normalized Van Der Waals volume), number of RNA binding proteins known to bind its mRNA product, coding sequence length, protein abundance, and 5′UTR free energy. The following elucidations on these features confirmed their informative and importance in understanding the translation rate and translation mechanism in large-scale.
Codon usage frequency
It has been reported by several studies that codon bias is the major factor for translation efficiency [31], [41]. In this study, we analyzed the relationship between the codon usage frequencies of ORFs and their translation rate. Our analysis not only confirmed the strong correlation between codon usage frequencies and translation efficiency, but also showed that more usage of which codon will result in high translation efficiency. It was found that the ORFs with higher frequencies of the following codons (AAC, TCT, ACC, TCC, GCC, GCT, CCA) tend to have higher rate of protein synthesis; on the other hand, higher frequency of the codons (ATA, CGA, TGC, GTA, GGA, CTT, AGG, CGG, TAT) relates to lower translation efficiency.
Gene Ontology (GO) enrichment scores
We also analyzed 4148 Gene Ontology (GO) enrichment score features based on the STRING network context [17]. Interestingly, our analysis indicates that ORFs with different functions or subcellular locations will have different translation rate. The translation differences among different function groups have been mentioned before [42]. According to our analysis, in starvation condition, ORFs with cellular response function tend to have higher translation rate probably to improve the survival in this extreme condition. In starvation, high translation rate correlated with GO groups related to ‘cellular response’ (e.g. GO:0034605 - cellular response to heat, GO:0009409 - response to cold, GO:0009266 - response to temperature stimulus). An interesting contrast is the fact that the GO groups ‘GO:0005737 – cytoplasm’ and ‘GO:0001950 - plasma membrane’ are enriched with genes with high translation rate while the GO group ‘GO:0005634 – nucleus’ is enriched with genes with low translation rate. A possible explanation for this result is that in starvation condition in order to survive proteins in membrane and cytoplasm over-express, and genes related to DNA duplication (replication in the nucleus) under-express.
Protein features
In our study, the protein features such as amino acids composition, polarity, normalized Van Der Waals volume were correlated with translation rate. The correlation between amino acid composition and protein abundance level has been reported [43] and it is possible that the amino acid composition may influence translation. The reason for the importance of protein features in translation efficiency prediction maybe that these features are strongly related to its function. And the translation difference among different function groups was mentioned in Ghaemmaghami's work [42].
Other features
There are additional features that are useful for translation rate prediction. ‘Number of RNA binding proteins known to bind its mRNA product’, ‘Coding sequence length’, ‘Protein abundance’ and ‘5′UTR free energy’ are examples of such features. Firstly, there are a number of RNA binding proteins known to influence mRNA translation rate by bind its mRNA. For instance, RNA-binding proteins HuR and PTB promote the translation of Hypoxia-Inducible Factor 1α [44]. Cytochrome c mRNA translation is controlled by TIA-1 and HuR [45]. Furthermore, the correlation between protein abundance and the level of gene expression has been intensively studied (mainly on yeast). It was suggested that the relatively weak correlation between protein and mRNA abundance is due to different rates of translation and protein degradation [46]. Here we found that the ORFs with higher protein abundance tend to have higher translation rate. Thus, it is possible that the relatively weak correlation between the mRNA levels and protein abundance can be partially explained by the fact that translation rate is an important determinant of protein abundance that can't be estimated from mRNA levels. The last factor is 5′UTR free energy. It supports that previous studies that suggested that base-pairing potentials analysis of 5′UTR in various prokaryotes indicated that 5′UTR free energy is important for translation initiation [47].
Taken together, these sequence-derived and functional features are significantly-related to mRNA translation. Therefore, our prediction model might become a high throughput tool for annotating the translation rate of mRNAs. As a preliminary predictor of translation rate, the current model can only give the high or low categories of translation rate. When more in-depth understanding of translation is accumulated, the regression model might be tried to construct a more practical predictor which can directly estimate the translation rate.
Supporting Information
The features for translation rate prediction in rich condition.
(XLS)
The features for translation rate prediction in starvation condition.
(XLS)
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the Knowledge Innovation Program of Chinese Academy of Sciences (KSCX1-YW-R-74), Systems Biology Research Foundation of Shanghai University, Shanghai Science and Technology Committee (09DZ227180), Shanghai Science and Technology Committee (09DZ227180), the National Basic Research Program of China (2011CB510102, 2011CB510101). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Washburn MP, Koller A, Oshiro G, Ulaszek RR, Plouffe D, et al. Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2003;100:3107–3112. doi: 10.1073/pnas.0634629100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3:960–969. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Nie L, Wu G, Zhang W. Correlation between mRNA and protein abundance in Desulfovibrio vulgaris: a multiple regression to identify sources of variations. Biochem Biophys Res Commun. 2006;339:603–610. doi: 10.1016/j.bbrc.2005.11.055. [DOI] [PubMed] [Google Scholar]
- 6.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 7.Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilchrist MA, Wagner A. A model of protein translation including codon bias, nonsense errors, and ribosome recycling. J Theor Biol. 2006;239:417–434. doi: 10.1016/j.jtbi.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann JE, Lodish HF. A kinetic model of protein synthesis. Application to hemoglobin synthesis and translational control. J Biol Chem. 1979;254:11927–11937. [PubMed] [Google Scholar]
- 10.Liljenstrom H, von Heijne G. Translation rate modification by preferential codon usage: intragenic position effects. J Theor Biol. 1987;124:43–55. doi: 10.1016/s0022-5193(87)80251-5. [DOI] [PubMed] [Google Scholar]
- 11.Varenne S, Buc J, Lloubes R, Lazdunski C. Translation is a non-uniform process. Effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J Mol Biol. 1984;180:549–576. doi: 10.1016/0022-2836(84)90027-5. [DOI] [PubMed] [Google Scholar]
- 12.Le Quesne JP, Spriggs KA, Bushell M, Willis AE. Dysregulation of protein synthesis and disease. J Pathol. 2010;220:140–151. doi: 10.1002/path.2627. [DOI] [PubMed] [Google Scholar]
- 13.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dwight SS, Balakrishnan R, Christie KR, Costanzo MC, Dolinski K, et al. Saccharomyces genome database: underlying principles and organisation. Brief Bioinform. 2004;5:9–22. doi: 10.1093/bib/5.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charif D, Thioulouse J, Lobry JR, Perriere G. Online synonymous codon usage analyses with the ade4 and seqinR packages. Bioinformatics. 2005;21:545–547. doi: 10.1093/bioinformatics/bti037. [DOI] [PubMed] [Google Scholar]
- 16.Sharp PM, Tuohy TM, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, Pascual-Montano A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 2007;8:R3. doi: 10.1186/gb-2007-8-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS: Structure, Function, and Genetics (Erratum: ibid, 2001, Vol44, 60) 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 20.Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Current Proteomics. 2009;6:262–274. [Google Scholar]
- 21.Chou KC. A novel approach to predicting protein structural classes in a (20-1)-D amino acid composition space. Proteins: Structure, Function & Genetics. 1995;21:319–344. doi: 10.1002/prot.340210406. [DOI] [PubMed] [Google Scholar]
- 22.Chou KC, Zhang CT. Predicting protein folding types by distance functions that make allowances for amino acid interactions. Journal of Biological Chemistry. 1994;269:22014–22020. [PubMed] [Google Scholar]
- 23.Dubchak I, Muchnik I, Mayor C, Dralyuk I, Kim SH. Recognition of a protein fold in the context of the Structural Classification of Proteins (SCOP) classification. Proteins: Structure, Function, and Genetics. 1999;35:401–407. [PubMed] [Google Scholar]
- 24.Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins-Structure Function and Genetics. 2002;47:228–235. doi: 10.1002/prot.10082. [DOI] [PubMed] [Google Scholar]
- 25.Pollastri G, Baldi P, Fariselli P, Casadio R. Prediction of coordination number and relative solvent accessibility in proteins. Proteins-Structure Function and Genetics. 2002;47:142–153. doi: 10.1002/prot.10069. [DOI] [PubMed] [Google Scholar]
- 26.Chothia C, Finkelstein AV. The classification and origins of protein folding patterns. Annu Rev Biochem. 1990;59:1007–1039. doi: 10.1146/annurev.bi.59.070190.005043. [DOI] [PubMed] [Google Scholar]
- 27.Charton M, Charton BI. The structural dependence of amino acid hydrophobicity parameters. J Theor Biol. 1982;99:629–644. doi: 10.1016/0022-5193(82)90191-6. [DOI] [PubMed] [Google Scholar]
- 28.Fauchere JL, Charton M, Kier LB, Verloop A, Pliska V. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int J Pept Protein Res. 1988;32:269–278. doi: 10.1111/j.1399-3011.1988.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 29.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 30.Huang T, Shi XH, Wang P, He Z, Feng KY, et al. Analysis and prediction of the metabolic stability of proteins based on their sequential features, subcellular locations and interaction networks. PLoS One. 2010;5:e10972. doi: 10.1371/journal.pone.0010972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuller T, Ruppin E, Kupiec M. Properties of untranslated regions of the S. cerevisiae genome. BMC Genomics. 2009;10:391. doi: 10.1186/1471-2164-10-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalem O, Dahan O, Levo M, Martinez MR, Furman I, et al. Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Mol Syst Biol. 2008;4:223. doi: 10.1038/msb.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ringner M, Krogh M. Folding free energies of 5′-UTRs impact post-transcriptional regulation on a genomic scale in yeast. PLoS Comput Biol. 2005;1:e72. doi: 10.1371/journal.pcbi.0010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 37.Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J Bioinform Comput Biol. 2005;3:185–205. doi: 10.1142/s0219720005001004. [DOI] [PubMed] [Google Scholar]
- 38.Huang T, Cui W, Hu L, Feng K, Li YX, et al. Prediction of pharmacological and xenobiotic responses to drugs based on time course gene expression profiles. PLoS One. 2009;4:e8126. doi: 10.1371/journal.pone.0008126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang T, Tu K, Shyr Y, Wei CC, Xie L, et al. The prediction of interferon treatment effects based on time series microarray gene expression profiles. J Transl Med. 2008;6:44. doi: 10.1186/1479-5876-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glass GV, Hopkins KD. Allyn & Bacon; 1995. Statistical Methods in Education and Psychology (3rd edition ed.): [Google Scholar]
- 41.Lithwick G, Margalit H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 2003;13:2665–2673. doi: 10.1101/gr.1485203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 43.Tuller T, Kupiec M, Ruppin E. Determinants of protein abundance and translation efficiency in S. cerevisiae. PLoS Comput Biol. 2007;3:e248. doi: 10.1371/journal.pcbi.0030248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galban S, Kuwano Y, Pullmann R, Jr, Martindale JL, Kim HH, et al. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2008;28:93–107. doi: 10.1128/MCB.00973-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawai T, Lal A, Yang X, Galban S, Mazan-Mamczarz K, et al. Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol Cell Biol. 2006;26:3295–3307. doi: 10.1128/MCB.26.8.3295-3307.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brockmann R, Beyer A, Heinisch JJ, Wilhelm T. Posttranscriptional expression regulation: what determines translation rates? PLoS Comput Biol. 2007;3:e57. doi: 10.1371/journal.pcbi.0030057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osada Y, Saito R, Tomita M. Analysis of base-pairing potentials between 16S rRNA and 5′ UTR for translation initiation in various prokaryotes. Bioinformatics. 1999;15:578–581. doi: 10.1093/bioinformatics/15.7.578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The features for translation rate prediction in rich condition.
(XLS)
The features for translation rate prediction in starvation condition.
(XLS)