

Abstract
The Trans-Proteomic Pipeline (TPP) is a suite of software tools for the analysis of tandem mass spectrometry datasets. The tools encompass most of the steps in a proteomic data analysis workflow in a single, integrated software system. Specifically, the TPP supports all steps from spectrometer output file conversion to protein-level statistical validation, including quantification by stable isotope ratios. We describe here the full workflow of the TPP and the tools therein, along with an example on a sample dataset, demonstrating that the set up and use of the tools is straightforward and well supported and does not require specialized informatics resources or knowledge.
Introduction
Tandem mass spectrometry (MS/MS) has enabled the identification of large numbers of proteins in biological samples in a high throughput manner[1], in an approach termed shotgun proteomics. Peptides from digested proteins are chromatographically separated and then fragmented in the mass spectrometer, yielding fragment ion spectra that can be used to deduce the corresponding peptide sequences. These in turn allow the identification of proteins in the sample. In addition to the protein identities, quantitative information can also be obtained through a variety of stable isotope and isobaric tagging peptide labeling techniques[2]. Tandem mass spectrometry has therefore become the most commonly used method in proteomics today.
There are a wide array of workflows, mass spectrometers, and data analysis tools available. There are typically multiple steps in the data analysis process, with a variety of techniques and options in the software tools used for each step[3]. A typical modern workflow for analysis of MS/MS data that includes validation and quantification can be quite complex, and is described by Deutsch et al.[4]: conversion of raw vendor files to an open (non-proprietary) format that all tools can access downstream; identification of spectra with a sequence or spectrum library search engine; statistical validation of the putative identifications, quantification of results if a quantitative labeling strategy was used; and finally protein inference and interpretation.
Many tools are available from different groups and can be strung together in custom ways, but there are only a few suites of tools that aim to provide a single environment for performing all or most steps in the workflow, including The OpenMS Proteomics Pipeline (TOPP)[5], MaxQuant[6], and the Trans-Proteomic Pipeline[7] (TPP). The TPP, developed at the Seattle Proteome Center (SPC), is the oldest and most comprehensive fully open-source suite of software tools that facilitates and standardizes the analysis of LC-MS/MS data. The TPP includes software tools for MS data representation, MS data visualization, peptide identification and validation, quantification, and protein inference. Here we provide a review of the available TPP tools along with a tutorial demonstrating how easy the analysis and validation of a dataset with the TPP can be.
Online tutorial
An important aspect of the effectiveness of a set of tools is their usability. A tool that implements a clever algorithm but is very difficult for the average researcher to use, or is not linked or linkable to tools performing upstream or downstream steps, is of only modest value. Similarly, a tool for which the underlying algorithm is not described is difficult to assess. Significant effort was therefore devoted to making the TPP and all of its tools easy to use and well integrated into a robust system. To illustrate this, we have developed a short tutorial of 10 steps to accompany this article that demonstrates the use of the tools from installation to final analysis of a sample dataset. The tutorial may be found at http://tools.proteomecenter.org/wiki/index.php?title=TPP_Demo2009.
TPP analysis overview
The full workflow of a typical MS/MS proteomics dataset through the TPP is summarized in Figure 1. Raw mass spectrometer output files are first converted to an open format such as mzXML[8] or mzML[9]. These files are run through one or more search engines such as X!Tandem[10], Mascot[11], SEQUEST[12], or SpectraST[13], and the results are converted to the pepXML[7] file format. The Pep3D tool[14] can be used to visually inspect each MS run to assess the quality of the chromatography. PeptideProphet[15] is then used to validate the search engine results and to model correct vs. incorrect peptide-spectrum matches (PSMs). The datasets can then be validated at the peptide-identification level with the new iProphet tool[16]. Finally, protein-level validation and protein inference are performed with ProteinProphet[17]. Within this workflow, the quantification analysis tools XPRESS[18], ASAPRatio[19], or Libra[20] may be used with data that derive from isotopically or isobarically labeled samples. The final output is a protXML[7] file that includes all the protein- and peptide-level information, including the finally assigned probabilities for all peptides and proteins. This process will be described in more detail in the following subsections, along with references to the aforementioned tutorial.
Figure 1.
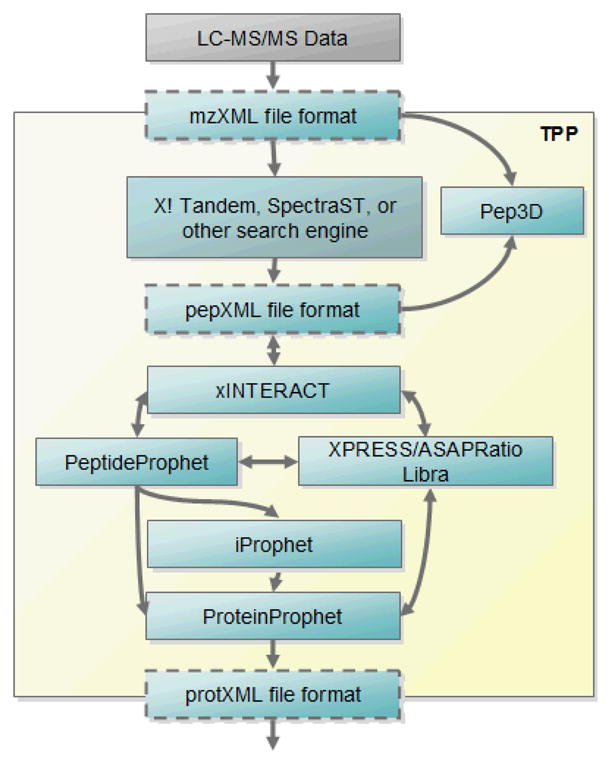
Schematic overview of the TPP workflow. Raw MS/MS data files are first converted to an open XML format such as mzXML or mzML, then analyzed with a search engine, either embedded in the TPP or used externally. Pep3D can allow visualization of the data. The search results, in pepXML format, are processed with tools PeptideProphet for initial spectrum-level validation, iProphet for peptide-level validation, and finally ProteinProphet for protein-level validation and final protein inference. Quantification tools like XPRESS, ASAPRatio, or Libra can be used on labeled data. The final output is protXML, which can be imported into a variety of analysis tools.
Sample dataset
The sample dataset used in this tutorial is derived from a SILAC[21] metabolically labeled yeast (Saccharomyces cerevisiae) whole cell lysate that was digested with trypsin and run on a Thermo Scientific LTQ Orbitrap mass spectrometer as described in detail in the Supplementary Material section. The raw and processed data are available in the PeptideAtlas[22] raw data repository as PAe001337. Downloading of the sample dataset is covered in step 2 of the tutorial, after the installation of the TPP, described immediately hereafter.
Preparation
Considerable effort has been expended to make the TPP easy to install. The TPP can be installed on most major platforms, including Microsoft Windows, UNIX/Linux, and MacOS X. The installation is the easiest on the Windows platform, and since this is the most commonly used environment, we have tailored the tutorial around a session on the Windows platform. However, except for installation, all of the remaining steps will be nearly identical under other platforms. It should be noted that the primary development environment and use of the TPP by the authors is under Linux, and therefore the support for UNIX-like platforms is truly native rather than merely an afterthought.
Step 1 in the tutorial is the preparation for using the TPP on a computer. Here, the installation of the TPP is described as well as launching the graphical user interface. Installation under Windows takes 10–30 minutes, after which the software is ready to use. Installation on Linux or MacOS X platforms does involve some more effort. An installation guide for each of these platforms is available as described in the tutorial.
The TPP is a collection of over 30 tools (Table 1) that can be strung together as a pipeline or run individually as needed. A graphical user interface, Petunia, provides a point-and-click environment from within an ordinary web browser, allowing users to launch tools separately or launch a complete analysis from a single page.
Table 1.
List of tools in the Trans-Proteomic Pipeline distribution.
| Category | Name | Description |
|---|---|---|
| Web interface | Petunia | Graphical user interface for TPP and most other tools in this table. |
| Format converter | ReAdW (1) | Converts Thermo Scientific RAW files to mzXML. |
| Format converter | Trapper (1) | Converts Agilent files to mzXML. |
| Format converter | mzWiff (1) | Converts ABI/Sciex WIFF files to mzXML. |
| Format converter | massWolf (1) | Converts Waters’ MassLynx files to mzXML. |
| Format converter | msconvert (1)(2) | ProteoWizard tool for converting Thermo RAW, Waters RAW, and Bruker FID/YEP/BAF files to mzXML and mzML. |
| Format converter | dta2mzxml | Converts a directory with .dta files into an mzXML file |
| Format converter | MzXML2Search | Converts mzXML files to various search tool input formats. |
| Format conversion support | readmzXML | mzXML parser based on RAMP. |
| Format conversion support | RAMP | mzXML data parser. |
| Data format validation | ValidateXML | Checks mzXML, pepXML, and protXML files for proper format. |
| Data visualization | plot-msms.cgi | Spectrum viewer |
| Data visualization | Pep3D | 2D graphical display of MS/MS runs for quality control. |
| Sequence search tool | X!Tandem (2) | Common, open-source search tool. Augmented with K-score algorithm, optimized to work with PeptideProphet. |
| Sequence search tool | ProbID | Search tool developed at SPC. |
| Sequence search support | decoyFASTA | Creates reverse databases for decoy searching |
| Data visualization | comet-fastadb.cgi | Sequence viewer |
| Spectral library search tool | SpectraST | Identifies MS/MS spectra by comparing against a spectral library. |
| Spectral library support | Lib2HTML | Converts a spectral library to HTML format |
| Format conversion | Out2XML | Converts SEQUEST results to pepXML. |
| Format conversion | Mascot2XML | Converts Mascot results to pepXML. |
| Format conversion | Tandem2XML | Converts X!Tandem results to pepXML. |
| Statistical modeling | PeptideProphet | Provides statistically rigorous probabilities for search identifications. |
| Statistical modeling: internal tool | RefreshParser | Maps peptides in a pepXML file to a protein sequence database different from the search database. Needed for spectral library searching. |
| Statistical modeling | iProphet | Further refines PeptideProphet probabilities. Can combine results from multiple searches. |
| Statistical modeling | ProteinProphet | Provides statistically rigorous probabilities for protein identifications. |
| Statistical modeling: post processing | calctppstats.pl | Summarizes TPP results for an experiment. Only available via command line, not via Petunia. |
| Web interface | PepXMLViewer | Viewer for pepXML files. |
| Web interface | ProtXMLViewer | Viewer for protXML files. |
| Command line interface | xinteract | Runs TPP statistical modeling tools from command line. |
| Spectrum processing | QualScore | Finds high quality spectra among those unassigned by search. |
| Quantification | XPRESS | Quantitative analysis for isotopic labeling (simple tool). |
| Quantification | ASAPRatio | Quantitative analysis for isotopic labeling (more sophisticated tool). |
| Quantification | Libra | Quantitative analysis for multi-channel isobaric labeling. |
| SRM | MaRiMba | Creates SRM transitions lists from a set of spectra. |
requires vendor DLLs
third-party software included in TPP
Conversion to an open format
Each instrument vendor has a different proprietary file format for its mass spectrometer output files; in fact, some vendors have several formats depending on the instrument type. In order to support all kinds of data from all mass spectrometers, the TPP allows the conversion of the data into either of two open, vendor-neutral formats: mzXML or mzML. The mzXML format was developed by our group. Recently, this format has been supplanted by the mzML format, developed by the HUPO Proteomics Standards Initiative (PSI). Both mzXML and mzML are currently supported by TPP.
The TPP tools for conversion of file formats include msconvert[23] (as well as the older ReAdW tool) for conversion of Thermo Scientific RAW files, mzWiff for conversion of ABI/Sciex WIFF files, Trapper for conversion of Agilent files, and massWolf for conversion of Waters’ MassLynx files. Bruker provides a freely downloadable tool, CompassXport, for conversion of their formats to mzXML. All of these conversion tools can only work on a Windows computer with vendor libraries installed. However, some vendors are starting to agree to allow the distribution of these libraries free of charge. See http://tools.proteomecenter.org/wiki/index.php?title=Formats:mzXML for the current status of conversion tools.
To perform the conversion, follow the detailed items in step 3 of the tutorial, which demonstrates how the vendor RAW files for the sample dataset are transformed into mzML through Petunia using msconvert. If your computer does not have the appropriate Thermo Scientific libraries installed, the tutorial shows how to obtain pre-converted mzML data files for use within the tutorial.
Search with a sequence search engine
The main step in processing MS/MS data is a search with a spectrum interpretation algorithm that attempts to match the spectra with possible peptide sequences extracted from a given list of protein sequences. This is most commonly done with one of the many available sequence search engines. It is a common misconception that the TPP only supports SEQUEST search results as it did at the very beginning. In reality the TPP now supports X!Tandem, Mascot, SEQUEST, OMSSA[24], Phenyx[25], and ProbID[26]. Only X!Tandem, an open source search engine, is bundled with the TPP; other engines must be installed separately.
In the tutorial step 4 we demonstrate the search of the sample dataset with the bundled X!Tandem. For better results, the X!Tandem packaged with TPP was augmented with a pluggable scoring mechanism[27] called K-score, which has been optimized to work well with PeptideProphet.
Search with a spectral library search engine
An alternative or complementary approach to sequence searching is spectral library searching. In this approach, spectra are compared against a library of previously identified spectra rather than against hypothetical spectra predicted from a sequence database. Spectral library searching is shown to be much faster and more capable of identifying low-quality spectra than sequence search engines[13]. This is mainly because spectral searching benefits from a smaller search space (fewer candidates to choose from) and the use of real reference spectra as opposed to theoretical ones predicted, often simplistically, by sequence search engines. The obvious limitation is that only peptides corresponding to spectra in the library can be identified; previously undetected peptides will be missed. Therefore, spectral searching is best seen as a complementary approach to sequence searching, and the best results are often obtained from combining results from multiple search engines, as described below.
In step 5 of the tutorial document, we demonstrate how to download the yeast ion trap spectrum library version 2 created by the National Institute for Standards and Technology (NIST) and search the sample dataset using the SpectraST[13] spectral library search engine. SpectraST is part of the TPP and every TPP installation already comes with SpectraST.
Validating search results with PeptideProphet
Every search engine returns one or more best interpretations (peptide-spectrum matches; PSMs) of each input MS/MS spectrum along with scores that quantify the quality of the matches. Some engines provide a normalized score (e.g. an E-value) that indicates the likelihood that the PSM is an incorrect, random event, while other search engines provide raw scores without absolute significance. It is thus challenging to directly compare the results of different search engines. The PeptideProphet algorithm[15] was developed to separate all of the resulting PSM scores into a population of correct and incorrect PSMs using an expectation maximization algorithm. PeptideProphet assigns a probability of correctness to each PSM and calculates a set of global false discovery rates[28] as a function of the probability cutoff. In step 6 of the tutorial, this statistically rigorous validation of both the X!Tandem and SpectraST search results is demonstrated.
In calculating PSM probabilities, PeptideProphet can be made to consider not only the search score, but also PSM attributes such as mass deviation, number of enzymatic termini, number of missed cleavages, and retention time. This allows better discrimination between correct and incorrect interpretations and leads to improved probability estimates. PSMs whose attributes conform better to those of the correct population are rewarded, while PSMs whose attributes conform better to those of the incorrect population are penalized. For example, a PSM whose mass deviation (between that measured by the instrument and that predicted if the sequence interpretation is correct) is near the average for other correct identifications (not necessarily 0.0) is rewarded with a higher probability.
When the dataset is from a high accuracy instrument, PeptideProphet can employ a high mass accuracy model which models the deviation in observed mass to the nearest isotopic peak of the putative identification. The overall accuracy is learned from the data and, therefore, the better the accuracy of the input masses, the better the discrimination by mass deviation. If external tools such as DeconMSn[29] are used to improve the input precursor m/z values, PeptideProphet learns this and applies a more discriminating model. An additional recently developed model is the elution time model, which fits for an entire MS run the actual elution time for each PSM to its predicted elution time. In the same way, PSMs that conform well to the predicted model are rewarded and large deviations are penalized. A manuscript describing these features in detail is in preparation.
PeptideProphet does not require a sequence search performed with a target-decoy strategy[30] as input in order to perform its modeling. An ordinary target-only search can significantly decrease computation time over target-decoy strategies. However, if a target-decoy strategy is employed, PeptideProphet can use this information to help refine the models[31], or even model the incorrect population directly to the decoy population[32]. PeptideProphet can also be set to ignore decoy information during modeling so that the decoy PSMs can be used for independent evaluation of the TPP results.
The final result of PeptideProphet analysis is a pepXML format file that reports a probability for every PSM as well as the results of the modeling, including a representation of the receiver operating characteristic (ROC) curves. The pepXML file can be interactively explored with PepXMLViewer application.
Visualizing the chromatography with Pep3D
There are a number of problems with chromatography that can degrade the number of identifiable MS/MS spectra in a run. For example, non-optimal chromatography can result in the sample eluting over a short range of the entire LC gradient. Or polymer contamination can wastefully consume CID scan acquisitions. Most of these problems are not apparent by looking through the search results. However, a visual image derived from all the MS1 spectra can make apparent many chromatography problems. The TPP tool Pep3D[14] can be used to create images from one or more LC runs for quality control and visual inspection. Overlaid on the images are symbols for each MS/MS spectrum acquisition, color coded when the MS/MS spectrum is identified with high confidence. Figure 2 shows the Pep3D output for one of the sample runs. Step 7 in the tutorial demonstrates the use of Pep3D to examine the sample runs.
Figure 2.

Pep3D visualization of one of the runs in the sample dataset. Elution time in minutes is along the X axis, and m/z is along the Y axis. MS1 intensities are rendered in grayscale. Blue and red squares depict the precursor m/z and time of the MS/MS scans. Red squares denote high probability PSMs as derived by PeptideProphet. Blue squares are low probability PSMs. Although some contaminant streaking is seen at low m/z, the wide ramp of elution features, many of them annotated red as high probability, indicates that this is a good quality run.
Further peptide-level validation with iProphet
After validation of a dataset with PeptideProphet, each PSM is assigned a probability of being correct. However, there is still a significant amount of corroborating evidence from other identifications that can be used to further discriminate between correct and incorrect PSMs. The iProphet tool[16] is an additional tool that can improve the probability estimates coming out of PeptideProphet. In addition, it offers the opportunity to combine search results from multiple search engines. In step 8 of the tutorial, iProphet is used to combine the results of the X!Tandem and SpectraST searches of the sample dataset.
When a dataset is searched with two or more search engines, a mostly overlapping yet somewhat different set of spectra are scored highly by PeptideProphet. When a spectrum is scored highly by multiple search engines, it is more likely to be correct (although, of course, multiple search engines can very occasionally agree on the wrong answer). If two or more search engines disagree on the matching peptide sequence, each probability is penalized by the magnitude of the probabilities of the conflicting results.
Besides corroborating evidences from multiple search engines, iProphet also takes into account repeated discoveries of the same peptide ion, or the same peptide sequence with a different charge state or modification, all of which increase confidence in the identification. The iProphet tool creates mixture models for several lines of corroborating evidence (number of sibling search results, number of replicate spectra, number of sibling ions, number of sibling modifications, number of sibling experiments) and adjusts the models for each dataset processed. For each model, the probability of each PSM is either rewarded or penalized based on its similarity to the correct population or incorrect population, respectively. The final result is better discrimination, yet still with accurate probabilities and global false discovery rate (FDR).
Differential labeling quantification analysis
Isotopic labeling is an important technique for adding a quantitative dimension to shotgun proteomics experiments. In techniques such as ICAT[33] or SILAC[21], one or more samples are labeled with heavy isotopes and then run through an instrument together with matched samples consisting of light isotopes (control vs. treatment(s); disease vs. normal, etc.). The relative abundances of two samples can be measured via the ratio of the two extracted ion currents of a heavy-light peptide pair. There are two TPP tools for analysis of such data. XPRESS[18] is the earlier, simpler tool that is still appropriate for some uses. The more recent ASAPRatio[19] is more sophisticated in its measurement of, and aggregation of measurements from, multiple peptide ions from the same peptide, as well as aggregation at the protein level. Step 9 of the tutorial demonstrates the use of ASAPRatio on the sample dataset to derive abundance ratios for the two samples.
Another popular technique is isobaric labeling such as iTRAQ[34] and TMT[35], wherein peptides in up to 8 samples can be labeled with N-terminal isobaric labels, each with a different fragment ion peak near 100 m/z. Measurement of the relative peak intensities of these reporter peaks in the fragment ion spectra yield relative abundances of peptides and hence proteins in the original samples. The TPP tool Libra performs the quantitative analysis of such datasets even though this is not illustrated in the tutorial. Although TPP tools can be used to aid with quantification on label-free MS/MS datasets using extracted ion chromatogram or spectral counting techniques, these workflows are not explicitly supported.
Protein inference and validation with ProteinProphet
The final step of processing data with the TPP entails combining all of the peptide observations into a final list of proteins. This is a complex problem, mainly due to the fact that many related proteins share peptide sequences[17]. The ProteinProphet tool[36] performs this final analysis step.
ProteinProphet first applies a mixture model based on the number of distinct peptides per protein (sibling peptides) to boost the probabilities of peptides with multiple siblings while penalizing peptides without siblings. ProteinProphet then performs a protein inference analysis to create the simplest list of proteins that can explain all the peptide observations. Each protein is assigned a probability of being in the sample. Step 10 in the tutorial demonstrates the analysis of the sample dataset with ProteinProphet.
The final result is a protXML [7] file with a list of all proteins corresponding to the PSMs, along with protein probabilities and global FDRs at different thresholds. Each protein is annotated with the relative abundance ratio, if a quantification technique was used, along with uncertainties. The output can be viewed using the TPP’s protXML viewer, and exported to Excel or other analysis tools in order to address the biological question that prompted the experiment. A few of these tools are described below.
Data formats
In order to support the vision of the TPP as a complete set of tools to analyze shotgun proteomics data from any instrument or vendor, it was necessary to develop common, open (non-proprietary) formats through which the tools can exchange data. Since there were no such formats at the time, we developed and made accessible the mzXML, pepXML, and protXML formats. The mzXML is used for raw spectra, as described above. Search engine results are converted to the pepXML format for downstream analysis. PeptideProphet reads pepXML files from the search engines and creates a combined pepXML file with all the model information. The iProphet tool also reads pepXML and writes out new pepXML with additional model information. The quantification tools write their results into the pepXML files. Finally, ProteinProphet reads one or more pepXML files and writes out a protXML file that is the final end result of TPP processing, including protein identifications and supporting information. None of these formats is an official standard, but all have become de facto standard formats in the community and are used by many software tools unrelated to the TPP, primarily due to the free availability of software that reads and writes these formats.
The TPP team worked with the PSI mass spectrometry standards working group to develop a next-generation format for mass spectrometer output files, mzML[9], that meets not only the needs of the TPP, but also other needs in the community. The PSI proteomics informatics group, also with cooperation from the TPP development team, has developed a new format that can encode downstream informatic analysis of proteomics MS data. The new format, mzIdentML, combines the information encoded in pepXML and protXML and much more into a single file format (except for quantification information, which is expected from a subsequently released mzQuantML format). Although it is likely that the TPP will continue to use pepXML and protXML as internal working formats for the pipeline, in the future the TPP will convert all the final results to mzIndentML once its development is complete.
All of the information required by the Guidelines for Proteomic Data Publication[37, 38] is encoded in the standard file formats mzML, pepXML, or protXML, with the exception of the annotated spectra. Therefore these files can be included with journal submissions to fulfill the requirements. For the annotated spectra, we have developed a program that will generate a PDF document of annotated spectra as required by the guidelines. The functionality is currently in beta testing (beta version available in the source code repository) and will be officially released in version 4.4 of the TPP soon.
Subsequent analysis
The final result of a TPP analysis is a protXML file containing a list of proteins, their probabilities of presence in the sample, supporting peptide information, and possibly quantification results. The results can be browsed with the protXML viewer or exported into a tab-delimited format suitable for Excel. However, many other tools exist for further downstream analysis.
The Protein Information and Property Explorer[39] (PIPE) is a web-based interactive interface developed at the SPC for exploring the significance of protein lists. It includes functionality for mapping to other identifiers (Entrez gene, Unigene, Uniprot, etc.), Gene Ontology (GO) enrichment calculations (to see which biological processes are overrepresented in the list), annotation of the list to specific GO terms, and more. The PIPE can also serve as a data management tool, saving lists with user specified metadata (i.e., description of the data).
The results of TPP processing can be imported into the SBEAMS-Proteomics database, which is part of the SBEAMS (Systems Biology Experiment Analysis Management System) Project[40], a framework for collecting, storing, and accessing data produced by proteomics experiments and other experiment types. Several other database systems such as CPAS[41] and YPED[42] provide a data management layer on top of the embedded TPP tools.
Other tools
In addition to the tools describe herein, many others come bundled with the TPP. The full set available is summarized in Table 1. A few tools of special note are described hereafter.
The xinteract tool is a command-line wrapper program that can be used to run several of the TPP tools at once. It has an extensive set of parameters that can be used to chain together programs such as PeptideProphet, iProphet, and ProteinProphet in one command. This is often quite useful in an environment where the processing of many experiments is scripted or for other automation scenarios.
The QualScore[43] tool takes a PeptideProphet analysis result as input and takes very high probability PSMs and very low probability PSMs as proxies for high quality and low quality spectra, respectively. Then a machine learning technique attempts to discriminate between high quality and low quality spectra based on the attributes of the input training sets. The result is then a population of spectra that share many attributes with the population of highest probably PSMs, but could not be identified. This population is suitable for another round of exhaustive searching with expanded modification parameters or proteins lists.
The MaRiMba tool[44] is a workflow for selecting selected reaction monitoring (SRM; sometimes called multiple reaction monitoring or MRM) transitions for a targeted proteomics assay based on a consensus spectral library as created by SpectraST. MaRiMba creates SRM transition lists from downloaded or custom-built spectral libraries, restricts output to specified proteins or peptides of interest, and filters SRM lists based on user-defined precursor peptide and product ion properties. MaRiMba can also create SRM lists containing isotopically heavy transitions for use with isotopic labeling strategies such as SILAC. MaRiMba outputs the final SRM list to a text file convenient for upload to a mass spectrometer. The TPP itself does not presently provide analysis for acquired SRM data; however, the TIQAM (Targeted Identification for Quantitative Analysis by MRM)[45] and MRMer[46] programs are available and the ATAQS (Automated and Targeted Analysis with Quantitative SRM) pipeline is under development and will be distributed by our group similarly to the TPP.
Support for the tools
Support for the tools is provided via an email discussion list at Google Groups, with a browsable archive. Over 800 users are signed up for the list and many questions are answered by members of the TPP community other than those at the SPC. Members of the TPP team are often found in a booth at major conferences helping to install the TPP on laptops of booth visitors or answering questions. The SPC also hosts a 5-day course twice per year. At each course, 30 students are accepted to attend lectures describing each of the tools, usually taught by the original authors or active maintainers of the software. Students work in pairs on the provided course computers to process sample datasets (including the dataset described in the accompanying on-line tutorial) through the various tools in guided tutorials and independent exercises. All information about the email discussion list, booths, and courses is available on the SPC web site.
Comparisons
As mentioned in the introduction, there are several software packages similar to the TPP that are freely available to the community. OpenMS provides a large number of C++ classes that can be used to facilitate the building of proteomics software tools, and TOPP (built on OpenMS) consists of several small applications that can be put together to create a custom analysis pipeline tailored for a specific problem. Mascot and OMSSA are supported and a tool is available to create a consensus of two searches of the same data. TOPP uses the PSI mzML format as input and then implements additional XML-based formats for internal communication among tools, similar to pepXML and protXML. TOPP is available for all three major operating system platforms and is completely open source, but lacks the more sophisticated modeling algorithms and quantification tools of the TPP.
MaxQuant is for use with high resolution LTQ FT and LTQ Orbitrap instruments from Thermo Fisher. This software uses .RAW files directly and therefore standard formats are not needed or supported. The software works on Windows XP or Vista, is used with the Mascot search engine, and can perform quantitative analysis with the SILAC technique. For experiments that fall within this scope, MaxQuant provides an effective analysis platform. It is freely available as a downloadable binary, but the source code is not open.
Among free software solutions, the TPP provides the most advanced analysis algorithms, and the only ones that are truly able to improve upon the native results for a variety of search engines from datasets derived from a variety of instrument vendors and types. The TPP is not without shortcomings, however. Installation difficulties still provide an immediate barrier to some, although installation is greatly improved over earlier versions. The user interface is sometimes lacking; considerably more effort has gone into developing advanced algorithms than advanced user interfaces. Although the algorithms work well on very large datasets, there are known deficiencies with the data viewers that cause performance problems with large datasets. The large numbers of configurable options can be daunting, although the default values are usually adequate. No features are available to assist with annotation of experiments, e.g. sample information. Finally, converting the raw vendor formats can be problematic for some vendors, and if the raw files cannot be converted to mzML or mzXML, then the TPP cannot be fully applied. Work is underway to address these shortcomings, although solutions will take time.
Future outlook for the TPP
The TPP is a complete and working system that forms part of the routine analysis workflow of hundreds of researchers. Yet robust development activity by the TPP team continues. The tools are continually adapted to improvements in wet lab techniques and advances in instrumentation. The iProphet tool has just recently been completed. Adaption of the tools for instruments that can use the novel fragmentation technique electron transfer dissociation (ETD) has recently been completed[47], including support for higher charge states, additional enzymes, charge-state prediction algorithms, ETD-spectrum library building, and improved annotation of c- and z -type ions.
We have preliminary beta support for some additional search engines such as InsPecT[48] and MyriMatch[49] with support for more search engines planned, as well as support for additional quantitative labeling techniques. Work is underway to adapt the TPP to work seamlessly in a cloud computing environment. We envision a system whereby a user can instantiate a preconfigured node on the Amazon Elastic Compute Cloud (EC2) already running the TPP (by paying a modest usage fee to EC2) as well as instantiating a user-selectable number of other nodes to work as search engine slave nodes for processing large jobs.
The TPP will be better integrated with the current proteomics data repositories. From within the TPP interface, users will be able to easily upload their results to one of the repositories such as PeptideAtlas[22], PRIDE[50], Tranche[51], or Peptidome (http://www.ncbi.nlm.nih.gov/projects/peptidome/). Similarly, the interface will be adapted to easily download raw data from Tranche or the other repositories for viewing or reprocessing in the TPP environment. We are also planning a visual dashboard application that provides a quick overview of the results of TPP processing with auto-generated charts and figures, as well as an analysis advisor that can point out common problems in processing, sample preparation, or instrument methods. These and other developments will continue to provide an ever better user experience and a more capable suite of tools to keep up with improving MS/MS proteomics techniques.
Conclusion
We have described the various tools that comprise the TPP, and demonstrated the utility of the TPP via a sample dataset in the accompanying tutorial. The TPP provides both a graphical user interface for ease of use in addition to a discrete set of back-end tools that may be easily combined into custom pipelines for large amounts of data, as is used to enable the PeptideAtlas project. Further, the TPP tools provide an advanced, statistically sound, and continually improving suite that is a huge improvement upon the use of a single search tool with no post-validation. For users who want an intensive hands-on training in the use of these tools, the Seattle Proteome Center offers a biannual course that has trained over 500 members of the community thus far.
The TPP tools are all free and open source, and have become more robust and far easier to use in the past few years. For the Windows platform, there is an easy-to-follow quick installation guide available at the Seattle Proteome Center web site (http://www.proteomecenter.org). The TPP is also compatible with Linux and MacOS X, although the installation for those platforms is somewhat more complex. However, the entire process is sufficiently simple that all users can easily take advantage of the advanced tools in the TPP to analyze their data.
Supplementary Material
Acknowledgments
We are grateful to the many users of and contributors to the TPP project; their feedback, bug reports, and code contributions. This work has been funded in part with federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, under contract No. N01-HV-28179 and grant R44HG004537, and from PM50 GMO76547/Center for Systems Biology.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Bantscheff M, et al. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389(4):1017–31. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 3.Vitek O. Getting Started in Computational Mass Spectrometry–Based Proteomics. PLoS Comput Biol. 2009;5(5):e1000366. doi: 10.1371/journal.pcbi.1000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deutsch EW, Lam H, Aebersold R. Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics. Physiol Genomics. 2008;33(1):18–25. doi: 10.1152/physiolgenomics.00298.2007. [DOI] [PubMed] [Google Scholar]
- 5.Kohlbacher O, et al. TOPP--the OpenMS proteomics pipeline. Bioinformatics. 2007;23(2):e191–7. doi: 10.1093/bioinformatics/btl299. [DOI] [PubMed] [Google Scholar]
- 6.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 7.Keller A, et al. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005 0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pedrioli PG, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22(11):1459–66. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 9.Deutsch E. mzML: a single, unifying data format for mass spectrometer output. Proteomics. 2008;8(14):2776–7. doi: 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- 10.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 11.Perkins DN, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Eng J, McCormack AL, Yates JR. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 13.Lam H, et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics. 2007;7(5):655–67. doi: 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- 14.Li XJ, et al. A tool to visualize and evaluate data obtained by liquid chromatography-electrospray ionization-mass spectrometry. Anal Chem. 2004;76(13):3856–60. doi: 10.1021/ac035375s. [DOI] [PubMed] [Google Scholar]
- 15.Keller A, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 16.Shteynberg D, et al. Postprocessing and validation of tandem mass spectrometry datasets improved by iProphet. in preparation. [Google Scholar]
- 17.Nesvizhskii AI, Aebersold R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Discovery Today. 2004;9:173–181. doi: 10.1016/S1359-6446(03)02978-7. [DOI] [PubMed] [Google Scholar]
- 18.Han DK, et al. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19(10):946–51. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li XJ, et al. Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal Chem. 2003;75(23):6648–57. doi: 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- 20.Pedrioli PG, et al. Automated identification of SUMOylation sites using mass spectrometry and SUMmOn pattern recognition software. Nat Methods. 2006;3(7):533–9. doi: 10.1038/nmeth891. [DOI] [PubMed] [Google Scholar]
- 21.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 22.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9(5):429–34. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessner D, et al. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–6. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geer LY, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3(5):958–64. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 25.Colinge J, et al. High-performance peptide identification by tandem mass spectrometry allows reliable automatic data processing in proteomics. Proteomics. 2004;4(7):1977–84. doi: 10.1002/pmic.200300708. [DOI] [PubMed] [Google Scholar]
- 26.Zhang N, Aebersold R, Schwikowski B. ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics. 2002;2(10):1406–12. doi: 10.1002/1615-9861(200210)2:10<1406::AID-PROT1406>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 27.MacLean B, et al. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics. 2006;22(22):2830–2. doi: 10.1093/bioinformatics/btl379. [DOI] [PubMed] [Google Scholar]
- 28.Choi H, Nesvizhskii AI. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J Proteome Res. 2008;7(1):47–50. doi: 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
- 29.Mayampurath AM, et al. DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics. 2008;24(7):1021–3. doi: 10.1093/bioinformatics/btn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 31.Choi H, Nesvizhskii AI. Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. J Proteome Res. 2008;7(1):254–65. doi: 10.1021/pr070542g. [DOI] [PubMed] [Google Scholar]
- 32.Choi H, Ghosh D, Nesvizhskii AI. Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J Proteome Res. 2008;7(1):286–92. doi: 10.1021/pr7006818. [DOI] [PubMed] [Google Scholar]
- 33.von Haller PD, et al. The Application of New Software Tools to Quantitative Protein Profiling Via Isotope-coded Affinity Tag (ICAT) and Tandem Mass Spectrometry: II. Evaluation of Tandem Mass Spectrometry Methodologies for Large-Scale Protein Analysis, and the Application of Statistical Tools for Data Analysis and Interpretation. Mol Cell Proteomics. 2003;2:428–442. doi: 10.1074/mcp.M300041-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Zieske LR. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J Exp Bot. 2006;57(7):1501–8. doi: 10.1093/jxb/erj168. [DOI] [PubMed] [Google Scholar]
- 35.Thompson A, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 36.Nesvizhskii AI, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 37.Bradshaw RA. Revised draft guidelines for proteomic data publication. Mol Cell Proteomics. 2005;4(9):1223–5. [PubMed] [Google Scholar]
- 38.Bradshaw RA, et al. Reporting protein identification data: the next generation of guidelines. Mol Cell Proteomics. 2006;5(5):787–8. doi: 10.1074/mcp.E600005-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Ramos H, Shannon P, Aebersold R. The protein information and property explorer: an easy-to-use, rich-client web application for the management and functional analysis of proteomic data. Bioinformatics. 2008;24(18):2110–1. doi: 10.1093/bioinformatics/btn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marzolf B, et al. SBEAMS-Microarray: database software supporting genomic expression analyses for systems biology. BMC Bioinformatics. 2006;7:286. doi: 10.1186/1471-2105-7-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rauch A, et al. Computational Proteomics Analysis System (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J Proteome Res. 2006;5(1):112–21. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- 42.Shifman MA, et al. YPED: a web-accessible database system for protein expression analysis. J Proteome Res. 2007;6(10):4019–24. doi: 10.1021/pr070325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nesvizhskii AI, Vogelzang M, Aebersold R. Measuring MS/MS spectrum quality using a robust multivariate classifier. Proc. 52th ASMS Conf. Mass Spectrom; Nashville, TN. 2004. [Google Scholar]
- 44.Sherwood C, et al. MaRiMba: a Software Application for Spectral Library-Based MRM Transition List Assembly. J Proteome Res. 2009 doi: 10.1021/pr900010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lange V, et al. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Mol Cell Proteomics. 2008;7(8):1489–500. doi: 10.1074/mcp.M800032-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin DB, et al. MRMer, an interactive open source and cross-platform system for data extraction and visualization of multiple reaction monitoring experiments. Mol Cell Proteomics. 2008;7(11):2270–8. doi: 10.1074/mcp.M700504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deutsch EW, et al. Trans-Proteomic Pipeline supports and improves analysis of electron transfer dissociation datasets. Proteomics. doi: 10.1002/pmic.200900567. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanner S, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77(14):4626–39. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 49.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res. 2007;6(2):654–61. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martens L, et al. PRIDE: the proteomics identifications database. Proteomics. 2005;5(13):3537–45. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 51.Falkner JA, Andrews PC. Tranche: Secure Decentralized Data Storage for the Proteomics Community. Journal of Biomolecular Techniques. 2007;18(1):3. [Google Scholar]
- 52.Klimek J, et al. The standard protein mix database: a diverse data set to assist in the production of improved Peptide and protein identification software tools. J Proteome Res. 2008;7(1):96–103. doi: 10.1021/pr070244j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.