

Abstract
We studied the pH-dependence of ribosome catalyzed peptidyl transfer from fMet-tRNAfMet to the aa-tRNAs Phe-tRNAPhe, Ala-tRNAAla, Gly-tRNAGly, Pro-tRNAPro, Asn-tRNAAsn, and Ile-tRNAIle, selected to cover a large range of intrinsic pKa-values for the α-amino group of their amino acids. The peptidyl transfer rates were different at pH 7.5 and displayed different pH-dependence, quantified as the pH-value, 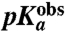 , at which the rate was half maximal. The
, at which the rate was half maximal. The 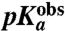 -values were downshifted relative to the intrinsic pKa-value of aa-tRNAs in bulk solution. Gly-tRNAGly had the smallest downshift, while Ile-tRNAIle and Ala-tRNAAla had the largest downshifts. These downshifts correlate strongly with molecular dynamics (MD) estimates of the downshifts in pKa-values of these aa-tRNAs upon A-site binding. Our data show the chemistry of peptide bond formation to be rate limiting for peptidyl transfer at pH 7.5 in the Gly and Pro cases and indicate rate limiting chemistry for all six aa-tRNAs.
-values were downshifted relative to the intrinsic pKa-value of aa-tRNAs in bulk solution. Gly-tRNAGly had the smallest downshift, while Ile-tRNAIle and Ala-tRNAAla had the largest downshifts. These downshifts correlate strongly with molecular dynamics (MD) estimates of the downshifts in pKa-values of these aa-tRNAs upon A-site binding. Our data show the chemistry of peptide bond formation to be rate limiting for peptidyl transfer at pH 7.5 in the Gly and Pro cases and indicate rate limiting chemistry for all six aa-tRNAs.
Keywords: ribosome, kinetics, rate limiting step, accommodation, molecular dynamics
The ribosome promotes protein elongation by transfer of the nascent peptide chain from P-site peptidyl-tRNA to A-site aminoacyl-tRNA (aa-tRNA) (Fig. 1) and translocation of messenger RNA (mRNA) and tRNAs. Peptide bond formation is initiated by a nucleophilic attack of the α-amino group of the amino acid, ester linked to the A-site tRNA, on the ester carbonyl carbon of the peptide chain linked to the 3′-oxygen of the P-site tRNA (Fig. 2). Biochemical data (1–3), crystallographic data (4–6), and molecular dynamics simulations (7, 8) have shown that the 2′OH group of A76 of the P-site tRNA greatly accelerates peptide bond formation by providing a shuttle of the proton from the attacking α-amino group to the leaving 3′O of the deacylated P-site tRNA. The rate of ribosomal peptidyl transfer is further accelerated by a network of H-bonds involving water molecules and conserved bases in the peptidyl transferase center (PTC) of the ribosome (4, 8, 9).
Fig. 1.

Schematic representation of steps leading to ribosomal peptidyl transfer. Here, τGTP and τdip represent the average times to complete GTP hydrolysis and peptide bond formation, respectively, starting from free ternary complexes and initiated ribosomes. By subtracting τGTP from τdip, the average time for all steps subsequent to GTP hydrolysis, τpep, is obtained: τpep = τdip - τGTP. In this scheme we have for simplicity assumed that release of EF-Tu from the ribosome precedes accommodation, so that τpep includes the time, 1/ktu. If release of EF-Tu·GDP from the ribosome occurs in parallel with accommodation (16), then 1/ktu would not be included in τpep (See also Data Evaluation in SI Text).
Fig. 2.

The proposed mechanism of peptidyl transfer (4, 7, 8, 39). A nucleophilic attack of the α-amino group of the A-site aa-tRNA (blue, here Phe-tRNAPhe) on the ester carbonyl carbon of the P-site peptidyl-tRNA (red) results in a six-membered transition state (40).
Peptidyl transfer requires that the α-amino group of the amino acid on the A-site tRNA is in charge neutral rather than in protonated 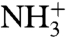 form (Fig. 2) (10). At 25 °C the pKa-values of the α-amino groups of amino acids in bulk water range from 8.8 (Asn) to 10.6 (Pro) units (11). The pKa-values of the aa-tRNAs, approximated by the pKa-values of the corresponding amino acid methyl and ethyl esters (12–14), are downshifted in relation to those of the amino acids by two units, with 20 °C values ranging from 6.8 (Asn) to 8.6 (Pro) (Table 1). The rate of ribosomal peptidyl transfer might therefore be expected to vary differently with pH in the physiological range 6–8 for different A-site bound aa-tRNAs. The sensitivity of peptide bond formation to pH-variation in the 6–8 range has so far only been observed for aa-tRNA analogues but not for native aa-tRNAs (15). It has been suggested that the lack of pH sensitivity for aa-tRNAs is that their accommodation in the A site is so slow that the expected pH-sensitivity of the chemistry of peptidyl transfer is completely masked (16, 17). This view (18–21) was recently challenged in a study (22), based on an Escherichia coli system for protein synthesis optimized for speed and accuracy. Here the activation enthalpy and entropy for peptidyl transfer from initiator tRNA to a native aa-tRNA were similar to the corresponding activation parameters for peptidyl transfer to puromycin, lacking a rate limiting accommodation step (23). From this similarity it was argued that peptidyl transfer to native aa-tRNA and to puromycin may both be rate-limited by the chemistry of peptide bond formation (22). Indeed, incorporation rates in translation of N-alkyl aa-tRNAs (including natural Pro-tRNAs) vs. Phe- and Ala-tRNAs differ in a manner that correlates strongly with α-amino group reactivity (24–26).
form (Fig. 2) (10). At 25 °C the pKa-values of the α-amino groups of amino acids in bulk water range from 8.8 (Asn) to 10.6 (Pro) units (11). The pKa-values of the aa-tRNAs, approximated by the pKa-values of the corresponding amino acid methyl and ethyl esters (12–14), are downshifted in relation to those of the amino acids by two units, with 20 °C values ranging from 6.8 (Asn) to 8.6 (Pro) (Table 1). The rate of ribosomal peptidyl transfer might therefore be expected to vary differently with pH in the physiological range 6–8 for different A-site bound aa-tRNAs. The sensitivity of peptide bond formation to pH-variation in the 6–8 range has so far only been observed for aa-tRNA analogues but not for native aa-tRNAs (15). It has been suggested that the lack of pH sensitivity for aa-tRNAs is that their accommodation in the A site is so slow that the expected pH-sensitivity of the chemistry of peptidyl transfer is completely masked (16, 17). This view (18–21) was recently challenged in a study (22), based on an Escherichia coli system for protein synthesis optimized for speed and accuracy. Here the activation enthalpy and entropy for peptidyl transfer from initiator tRNA to a native aa-tRNA were similar to the corresponding activation parameters for peptidyl transfer to puromycin, lacking a rate limiting accommodation step (23). From this similarity it was argued that peptidyl transfer to native aa-tRNA and to puromycin may both be rate-limited by the chemistry of peptide bond formation (22). Indeed, incorporation rates in translation of N-alkyl aa-tRNAs (including natural Pro-tRNAs) vs. Phe- and Ala-tRNAs differ in a manner that correlates strongly with α-amino group reactivity (24–26).
Table 1.
pH dependence of kpep for different aminoacyl-tRNAs at 20 °C
| Aminoacyl-tRNA |
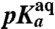 * *
|
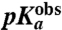 |
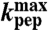 (s-1) (s-1) |
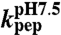 (s-1) † (s-1) †
|
| Asn-tRNAAsn | 6.8 | 5.9 ± 0.2 | 11 ± 1 | 11 ± 1 |
| Phe-tRNAPhe | 7.2 | 6.1 ± 0.1 | 28 ± 1 | 27 ± 1 |
| Ile-tRNAIle | 7.8 | 6.1 ± 0.2 | 22 ± 1 | 21 ± 1 |
| Ala-tRNAAla | 7.9 | 6.3 ± 0.1 | 25 ± 2 | 24 ± 1 |
| Gly-tRNAGly | 7.8 | 7.36 ± 0.04 | 13 ± 1 | 7.6 ± 0.2 |
| Pro-tRNAPro | 8.6 | 7.8 ± 0.2 | 17 ± 5 | 6.0 ± 0.5 |
*pKa of the α-NH2 group of aa-tRNA in bulk water (see Materials and Methods).
†kpep, calculated at pH 7.5 from fits of experimental data (Fig. 3 B–G).
Here, we studied the pH-dependence of the rate of peptidyl transfer from P-site bound initiator tRNA to six different aa-tRNAs entering the ribosome in ternary complex with EF-Tu and GTP. The aa-tRNAs were selected to cover a large range of pKa-values for their α-amino groups (Table 1, left). We used the linear interaction energy (LIE) method (27, 28) to estimate the shift between the pKa-values of these aa-tRNAs in bulk water and in A site.
We conclude that the chemistry of peptidyl transfer limits the rate of peptide transfer to Gly-tRNAGly and Pro-tRNAPro at physiological pH, and that a high pKa-value of Pro-tRNAPro contributes to its small rate of peptidyl transfer (24). This result shows that the chemistry of peptide bond formation and its pH-dependence are relevant for the rate of protein elongation in the living cell and opens a window for kinetic studies of peptide bond formation on the ribosome with native tRNA substrates. The strong correlation that we observe between the molecular dynamics (MD) simulated and the kinetically estimated, apparent pKa-shifts for the six aa-tRNAs suggests that the chemistry of peptide bond formation could, surprisingly, be rate limiting for peptidyl transfer to all six aa-tRNAs at all pH-values.
Results
pH-Dependence of the Rate of Ribosomal Peptidyl Transfer.
The pKa-values of the α-amino groups of aa-tRNAs in bulk solution, approximated by the pKa-values of the corresponding methyl and ethyl esters (12–14), vary greatly with the amino acid side groups. We have selected six aa-tRNAs (Table 1, column 1) with bulk solution pKa-values in the 6.8 (Asn-tRNAAsn) to 8.6 (Pro-tRNAPro) pH unit interval (Table 1, column 2). The pH-dependence of the rate of ribosome catalyzed peptidyl transfer from initiator tRNA to the six aa-tRNAs was studied with quench-flow techniques.
Ternary complex, consisting of EF-Tu, [3H]GTP and one of the six aa-tRNAs, was rapidly mixed in a quench-flow instrument with 70S ribosomes, containing fMet-tRNAfMet ([3H]Met or [35S]Met) in the P site, with an empty A site programmed with either one of the codons AAC (Asn), UUU (Phe), AUC (Ile), GCA (Ala), GGC (Gly), and CCC (Pro). Each one of these codons was read by a single isoaccepting tRNA, charged with amino acid in bulk tRNA or in partially purified form (tRNAPhe). After mixing, the time dependent extents of GTP hydrolysis, quantified as the ratio [3H]GDP/([3H]GDP + [3H]GTP), and dipeptide formation, quantified as ratios [3H]Met-aa/([3H]Met-aa + [3H]Met) or [35S]Met-aa/([35S]Met-aa + [35S]Met), were monitored at 20 °C at pH-values in the 6–8 range. GDP was separated from GTP by TLC or HPLC and fMet from fMet-aa by RP-HPLC.
For measurement precision, GTP hydrolysis and dipeptide formation were always monitored in the very same experiment. The average times for GTP hydrolysis (τGTP, see SI Text: Eq S2) and dipeptide formation (τdip, see SI Text: Eq S1) and their standard errors were estimated by fitting Eqs. S4 (single exponential kinetics, SI Text) and S5 (double exponential kinetics, SI Text) to experimental data. The average time for peptidyl transfer (τpep) was estimated as the difference 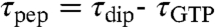 , illustrated schematically in Fig. 1 and graphically as the shaded area in Fig. 3A and Fig. S1 (See Data Evaluation in SI Text). In all cases τpep decreased significantly when pH increased.
, illustrated schematically in Fig. 1 and graphically as the shaded area in Fig. 3A and Fig. S1 (See Data Evaluation in SI Text). In all cases τpep decreased significantly when pH increased.
Fig. 3.

pH dependence of peptidyl transfer. (A) The normalized amount of GDP and fMetPhe dipeptide formed as a function of time when EF-Tu·Phe-tRNAPhe·[3H]GTP ternary complexes react at pH 7.4 and 20 °C with f[35S]Met-tRNAfMet initiated 70S ribosomes displaying UUU (Phe) codon in A site. The shaded area represents the mean time, τpep, for all reaction steps subsequent to GTP hydrolysis up to and including peptidyl transfer. (B–G) The compounded rate constant, kpep, of all reaction steps subsequent to GTP hydrolysis up to and including peptidyl transfer for different aa-tRNAs at different pH. kpep is calculated as the inverse of τpep estimated from experiments as shown in A. The curves represent nonlinear fitting of the data to an equation where a single reacting proton is being titrated (Eq S12 in SI Text). Error bars represent standard deviation of the data points calculated as weighted averages from two or three independent experiments (See Data Evaluation in SI Text). (H) The decimal logarithm of the compounded rate constant kpep for different aa-tRNAs plotted vs. pH. For Gly-tRNAGly and Pro-tRNAPro at low pH, log 10(kpep) is linear in pH with slope one, indicating titration of one proton in a reaction-essential group.
We have defined a “compounded” rate constant, kpep, for all steps after GTP hydrolysis up to and including peptide bond formation (see Fig. 1 for details) as the inverse of τpep, i.e., kpep = 1/τpep. Values of kpep estimated from quench-flow data at different pH-values are shown in Figs. 3 B–G.
In the Pro and Gly cases, plots of log 10kpep vs. pH are linear at low pH with positive slopes close to one (Fig. 3H). At further pH-increase, log 10kpep asymptotically approaches its maximal value, 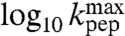 . For these two aa-tRNAs the variation of kpep with pH was due to the titration of a reaction-essential group with a single proton. This group, we propose, is the α-amino group of tRNA-bound Gly or Pro in the ribosomal A site (Fig. 2). By inference, we suggest that the variation of kpep with pH for all six aa-tRNAs (Fig. 3) is due to single proton titration on the α-amino group, i.e., (see SI Text):
. For these two aa-tRNAs the variation of kpep with pH was due to the titration of a reaction-essential group with a single proton. This group, we propose, is the α-amino group of tRNA-bound Gly or Pro in the ribosomal A site (Fig. 2). By inference, we suggest that the variation of kpep with pH for all six aa-tRNAs (Fig. 3) is due to single proton titration on the α-amino group, i.e., (see SI Text):
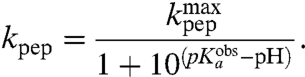 |
[1] |
Here 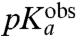 is operationally defined as the pH-value at which
is operationally defined as the pH-value at which 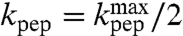 . Estimates of
. Estimates of 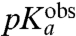 and
and 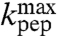 for all six aa-tRNAs are shown in Table 1. The largest
for all six aa-tRNAs are shown in Table 1. The largest 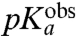 -values are seen for Pro (
-values are seen for Pro (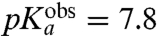 ) and Gly (
) and Gly (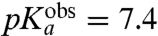 ), and the smallest for Asn (
), and the smallest for Asn (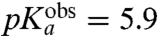 ), while intermediate
), while intermediate 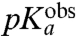 -values are seen for Phe (
-values are seen for Phe (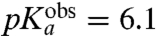 ), Ile (
), Ile (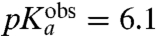 ), and Ala (
), and Ala (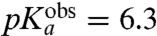 ) (Table 1 and Fig. 3). The
) (Table 1 and Fig. 3). The 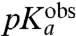 -values are downshifted in relation to the pKa-values for aa-tRNAs in bulk solution,
-values are downshifted in relation to the pKa-values for aa-tRNAs in bulk solution, 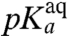 , by about one unit in the Asn, Phe, and Pro cases, 1.5 unit in the Ile and Ala cases, and 0.4 unit in the Gly case (Table 1, columns 2 and 3).
, by about one unit in the Asn, Phe, and Pro cases, 1.5 unit in the Ile and Ala cases, and 0.4 unit in the Gly case (Table 1, columns 2 and 3).
The 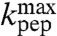 -values were largest for the fast aa-tRNAs cognate to Phe (
-values were largest for the fast aa-tRNAs cognate to Phe (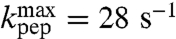 ), Ile (
), Ile (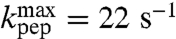 ), and Ala (
), and Ala (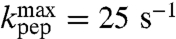 ), smallest for the slow aa-tRNAs cognate to Asn (
), smallest for the slow aa-tRNAs cognate to Asn (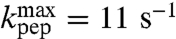 ) and Gly (
) and Gly (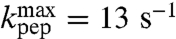 ), while the classification of Pro-tRNAPro as fast or slow was uncertain due to the larger error in the estimate of
), while the classification of Pro-tRNAPro as fast or slow was uncertain due to the larger error in the estimate of 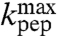 in this case (Table 1, column 4).
in this case (Table 1, column 4).
MD Simulated pKa-Values of A-Site Bound Aminoacyl-tRNAs.
In order to examine the relation between the 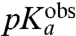 -values (Table 1, column 3) and the physical process of ionizing the α-amino group in the ribosomal A site, we carried out MD simulations of the CCA end of the six different aa-tRNAs in both the neutral and protonated form in bulk water as well as on the ribosome with a dipeptide bound to the P-site tRNA (7). We used the LIE method to estimate the pKa-shifts,
-values (Table 1, column 3) and the physical process of ionizing the α-amino group in the ribosomal A site, we carried out MD simulations of the CCA end of the six different aa-tRNAs in both the neutral and protonated form in bulk water as well as on the ribosome with a dipeptide bound to the P-site tRNA (7). We used the LIE method to estimate the pKa-shifts, 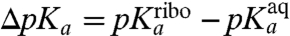 , of the ribosome-bound aa-tRNAs relative to their bulk solution values in terms of electrostatic and nonpolar interaction energy contributions to the neutral and ionized forms of their α-amino groups. In this model, the slope
, of the ribosome-bound aa-tRNAs relative to their bulk solution values in terms of electrostatic and nonpolar interaction energy contributions to the neutral and ionized forms of their α-amino groups. In this model, the slope 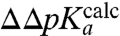 of the straight line that defines ΔpKa from these standard energies is given by (see SI Text):
of the straight line that defines ΔpKa from these standard energies is given by (see SI Text):
| [2] |
Here, ΔΔ〈Uel〉 = Δ〈Uel〉ribo - Δ〈Uel〉aq and ΔΔ〈Uvdw〉 = Δ〈Uvdw〉ribo - Δ〈Uvdw〉aq, where Δ〈Uel〉ribo and Δ〈Uel〉aq are the differences in electrostatic energy between the ionized and neutral forms of ribosome-bound and free aa-tRNA, respectively, while Δ〈Uvdw〉ribo and Δ〈Uvdw〉aq are the corresponding differences for the van der Waals energy. The parameter β is a scaling constant that relates electrostatic potential energy differences to the corresponding free energies and it includes the standard linear response factor of 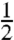 as well as all contributions to the effective dielectric constant that are not represented by the microscopic simulation model of the PTC. α = 0.18 is a standard scaling factor relating van der Waals potential energy differences, Δ〈Uvdw〉, to nonpolar free energy contributions [see, e.g., (29)] and RT log 10 = 1.34 at 20 °C.
as well as all contributions to the effective dielectric constant that are not represented by the microscopic simulation model of the PTC. α = 0.18 is a standard scaling factor relating van der Waals potential energy differences, Δ〈Uvdw〉, to nonpolar free energy contributions [see, e.g., (29)] and RT log 10 = 1.34 at 20 °C.
The 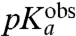 -values, operationally defined from the pH-dependence of the rate, kpep, of peptidyl transfer on the ribosome as described in the previous section, are all downshifted in relation to the experimentally estimated pKa-values,
-values, operationally defined from the pH-dependence of the rate, kpep, of peptidyl transfer on the ribosome as described in the previous section, are all downshifted in relation to the experimentally estimated pKa-values, 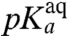 , of the α-amino groups of aa-tRNAs in bulk solution (Table 1). If the chemistry of peptidyl transfer, rather than a preceding step, is rate limiting for kpep (Fig. 1), one would expect a strong correlation between pKa-shifts calculated according to Eq. 2 above and the experimentally determined
, of the α-amino groups of aa-tRNAs in bulk solution (Table 1). If the chemistry of peptidyl transfer, rather than a preceding step, is rate limiting for kpep (Fig. 1), one would expect a strong correlation between pKa-shifts calculated according to Eq. 2 above and the experimentally determined 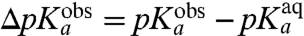 values (Table 1). If, in contrast, a step preceding the chemistry of peptidyl transfer is rate limiting for kpep, no such correlation would be expected a priori.
values (Table 1). If, in contrast, a step preceding the chemistry of peptidyl transfer is rate limiting for kpep, no such correlation would be expected a priori.
We plotted the empirical 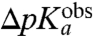 -values for the six different aa-tRNAs vs. the MD-estimated
-values for the six different aa-tRNAs vs. the MD-estimated 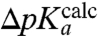 -values for the six tRNAs with the unknown dielectric scaling constant, β, as a fitted parameter (Fig. 4). The figure reveals a remarkably strong, positive correlation between the
-values for the six tRNAs with the unknown dielectric scaling constant, β, as a fitted parameter (Fig. 4). The figure reveals a remarkably strong, positive correlation between the 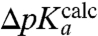 - and
- and 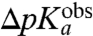 -values.
-values.
Fig. 4.

Observed pKa shifts vs. MD-simulated pKa shifts. Experimentally observed shifts in pKa of the α-amino group of aa-tRNAs on the ribosome relative to aa-tRNAs in bulk water, 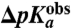 , plotted vs. MD-simulated shifts,
, plotted vs. MD-simulated shifts, 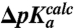 , in the pKa-value of aa-tRNAs as they move from bulk water to ribosomal A site.
, in the pKa-value of aa-tRNAs as they move from bulk water to ribosomal A site.
Discussion
We have studied the pH-dependence at 20 °C of the rate constant, kpep, for fMet transfer from initiator tRNA in the P site to each one of six aa-tRNAs cognate to Asn, Phe, Ile, Ala, Gly, or Pro in the A site of the ribosome. kpep defined as the inverse of the average time, τpep, for aa-tRNA to receive fMet after hydrolysis of GTP on EF-Tu (Fig. 1), displays distinct pH-dependence for all six tRNAs (Fig. 3 B–G). The pH-dependence of the peptidyl transfer reaction, quantified as the pH-value, 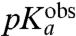 , at which kpep is half its maximal value at high pH,
, at which kpep is half its maximal value at high pH, 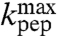 , varies among the six aa-tRNAs by almost two pH units (Table 1, column 3). There is, at the same time, a positive correlation between the pKa-value of the α-amino group of an aa-tRNA in bulk water (Table 1, column 2),
, varies among the six aa-tRNAs by almost two pH units (Table 1, column 3). There is, at the same time, a positive correlation between the pKa-value of the α-amino group of an aa-tRNA in bulk water (Table 1, column 2), 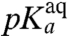 , and its
, and its 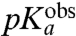 -value, suggesting that the pH-dependence of kpep reflects inhibition of peptide bond formation due to protonation of the α-amino group of the A-site aa-tRNA. Furthermore, plots of log 10kpep vs. pH in the Pro and Gly cases give straight lines with slopes very close to one in the low pH range (Fig. 3H), suggesting titration of a single proton for peptidyl transfer to Gly-tRNAGly and Pro-tRNAPro (11). From this result, we propose that titration of the α-amino groups of Gly and Pro is the sole explanation for the pH-dependence of the rate of peptidyl transfer to these aa-tRNAs (Fig. 2). Because also the pH-dependence of kpep of the other four tRNAs is accounted for by single proton titrations of their α-amino groups, we suggest that the pH-dependence of kpep has the same origin for all six tRNAs. It is, we suggest, not likely that this variegated spectrum of
-value, suggesting that the pH-dependence of kpep reflects inhibition of peptide bond formation due to protonation of the α-amino group of the A-site aa-tRNA. Furthermore, plots of log 10kpep vs. pH in the Pro and Gly cases give straight lines with slopes very close to one in the low pH range (Fig. 3H), suggesting titration of a single proton for peptidyl transfer to Gly-tRNAGly and Pro-tRNAPro (11). From this result, we propose that titration of the α-amino groups of Gly and Pro is the sole explanation for the pH-dependence of the rate of peptidyl transfer to these aa-tRNAs (Fig. 2). Because also the pH-dependence of kpep of the other four tRNAs is accounted for by single proton titrations of their α-amino groups, we suggest that the pH-dependence of kpep has the same origin for all six tRNAs. It is, we suggest, not likely that this variegated spectrum of 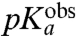 -values and its correlation with the spectrum of
-values and its correlation with the spectrum of 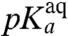 -values associated with its aa-tRNAs is caused by proton titration on any other functional group in the ribosome or by different tRNA-bodies. Yet another hypothesis, by which the rate of accommodation of aa-tRNA in the A site is severely inhibited by protonation of its amino group, so far lacks a clear mechanistic interpretation.
-values associated with its aa-tRNAs is caused by proton titration on any other functional group in the ribosome or by different tRNA-bodies. Yet another hypothesis, by which the rate of accommodation of aa-tRNA in the A site is severely inhibited by protonation of its amino group, so far lacks a clear mechanistic interpretation.
A distinct pH-dependence of the rate of peptidyl transfer to native aa-tRNAs in the ribosomal A site has not been observed before (15). The absence of pH-dependence has been rationalized as due to rate limiting accommodation of aa-tRNA in the A site, masking the (expected) pH-dependence of the rate constant, kpt (see Fig. 1), for the chemistry of peptide bond formation (15). In fact, the pH-dependence of the peptidyl transfer reaction has previously been observed only for small aa-tRNA-analogs, like puromycin, C-puromycin, CC-puromycin (23, 30, 31), or puromycin analogs (32). For these, the pH-dependence is complex, with the effect of two protons on the rate of peptidyl transfer to puromycin at 37 °C (23, 30) as well as at 20 °C (31). The effect of two protons or one proton was observed for C-puromycin at 37 °C (30) or 20 °C (31), respectively. Furthermore, the rate of peptidyl transfer to CC-puromycin displayed a qualitatively different and much weaker pH-dependence (30).
In contrast, the present findings show that the rate constant for the chemistry of peptidyl transfer, kpt in Fig. 1, can be directly assessed in kinetic experiments with native aa-tRNAs performed at pH-values below the 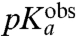 -values reported in Table 1: in these experimentally accessible pH-regimes the chemistry of peptidyl transfer is slower than the accommodation of aa-tRNA in the A site. An interesting question is now whether the chemistry of peptidyl transfer is slower than preceding steps on the pathway from GTP hydrolysis only at low pH-values. Or, alternatively, if also the maximal rate constant for peptide bond formation (
-values reported in Table 1: in these experimentally accessible pH-regimes the chemistry of peptidyl transfer is slower than the accommodation of aa-tRNA in the A site. An interesting question is now whether the chemistry of peptidyl transfer is slower than preceding steps on the pathway from GTP hydrolysis only at low pH-values. Or, alternatively, if also the maximal rate constant for peptide bond formation (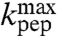 ) in the high pH range is rate limited by the chemistry of peptidyl transfer for some or all aa-tRNAs. The answer to this question depends on the cause of the varying downshifts of the kinetically determined
) in the high pH range is rate limited by the chemistry of peptidyl transfer for some or all aa-tRNAs. The answer to this question depends on the cause of the varying downshifts of the kinetically determined 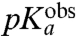 -values in relation to the
-values in relation to the 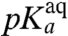 -values for aa-tRNAs in bulk water (Compare columns 2 and 3 in Table 1).
-values for aa-tRNAs in bulk water (Compare columns 2 and 3 in Table 1).
In our kinetic analysis based on average times, 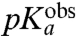 is determined by the pKa-value,
is determined by the pKa-value, 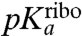 , of the α-amino group of a ribosome-bound aa-tRNA through (SI Text):
, of the α-amino group of a ribosome-bound aa-tRNA through (SI Text):
| [3] |
Here, 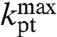 is the high pH-limit of the rate constant, kpt, for the chemistry of peptidyl transfer, i.e., when the α-amino group is neutral (Fig. 2) and
is the high pH-limit of the rate constant, kpt, for the chemistry of peptidyl transfer, i.e., when the α-amino group is neutral (Fig. 2) and 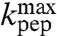 is the corresponding high pH-limit of the compounded rate constant for all steps leading to peptidyl transfer after GTP hydrolysis on EF-Tu (Fig. 1). Eq. 3 shows that when the chemistry of peptide bond formation,
is the corresponding high pH-limit of the compounded rate constant for all steps leading to peptidyl transfer after GTP hydrolysis on EF-Tu (Fig. 1). Eq. 3 shows that when the chemistry of peptide bond formation, 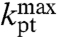 , is rate limiting for
, is rate limiting for 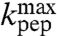 , i.e.,
, i.e., 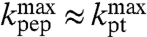 so that
so that 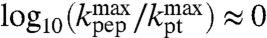 , then
, then 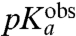 is approximated by
is approximated by 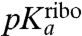 . When, in contrast, a step prior to the chemistry of peptide bond formation is rate limiting for
. When, in contrast, a step prior to the chemistry of peptide bond formation is rate limiting for 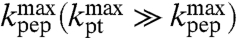 then
then 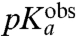 is much smaller than
is much smaller than 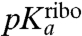 . In general, the downshifts,
. In general, the downshifts, 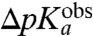 , of the
, of the 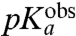 -values relative to the
-values relative to the 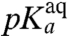 -values for the amino groups of aa-tRNAs in bulk solution (Table 1) are given by:
-values for the amino groups of aa-tRNAs in bulk solution (Table 1) are given by:
| [4] |
This expression shows that if a putative downshift, ΔpKa, of the pKa-value of the amino group of an aa-tRNA as it moves from bulk water to the A site of the ribosome, were known, the ratio 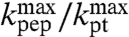 could be determined from
could be determined from 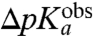 . This ratio could then be used to decide the extent to which the overall peptidyl transfer reaction is rate limited by aa-tRNA accommodation and other steps preceding the chemistry of peptide bond formation. Because ΔpKa cannot be directly measured, we have used MD techniques and the LIE approximation (29) to estimate ΔpKa according to Eq. 2. When the estimated values of ΔpKa,
. This ratio could then be used to decide the extent to which the overall peptidyl transfer reaction is rate limited by aa-tRNA accommodation and other steps preceding the chemistry of peptide bond formation. Because ΔpKa cannot be directly measured, we have used MD techniques and the LIE approximation (29) to estimate ΔpKa according to Eq. 2. When the estimated values of ΔpKa, 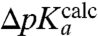 , are plotted vs. the
, are plotted vs. the 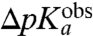 -values, a strong correlation is seen (Fig. 4). The major part of the correlation stems from the electrostatic energy contribution, ΔΔ〈Uel〉 (Fig. S2), while the nonpolar contribution ΔΔ〈Uvdw〉 mainly affects the pKa-shift of Phe-tRNAPhe. The structural explanation for the different observed pKa-shifts is straightforward, as judged from the average MD structures (Fig. 5). That is, water molecules are generally occluded from the volume surrounding the protonated amino group in the PTC, due to the position of the amino acid side chain in the A site. This water occlusion leads to pKa downshifts because desolvation destabilizes the
-values, a strong correlation is seen (Fig. 4). The major part of the correlation stems from the electrostatic energy contribution, ΔΔ〈Uel〉 (Fig. S2), while the nonpolar contribution ΔΔ〈Uvdw〉 mainly affects the pKa-shift of Phe-tRNAPhe. The structural explanation for the different observed pKa-shifts is straightforward, as judged from the average MD structures (Fig. 5). That is, water molecules are generally occluded from the volume surrounding the protonated amino group in the PTC, due to the position of the amino acid side chain in the A site. This water occlusion leads to pKa downshifts because desolvation destabilizes the 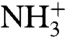 in relation to the NH2 state (Fig. 5A). The only exception is Gly-tRNAGly, for which a water molecule can enter the volume normally occupied by the Cβ atom, thus providing more solvation of the amine and thus explaining why Gly had the smallest pKa-shift (Fig. 5B). In the case of Pro-tRNAPro, on the other hand, because of the cyclic structure of Pro the volume is already more blocked for the side chain in bulk water than for most side chains, so that the desolvation effect in the PTC is not as pronounced and this leads to a smaller pKa-shift than for most side chains. Also, Asn-tRNAAsn is somewhat special because the side chain amide carbonyl group can form a favorable interaction (H-bond) with its protonated N terminus, thus somewhat attenuating the desolvation effect. The remaining three aa-tRNAs (Phe, Ala, and Ile) have the largest downshifts because of the lack of compensating interactions with the amino group.
in relation to the NH2 state (Fig. 5A). The only exception is Gly-tRNAGly, for which a water molecule can enter the volume normally occupied by the Cβ atom, thus providing more solvation of the amine and thus explaining why Gly had the smallest pKa-shift (Fig. 5B). In the case of Pro-tRNAPro, on the other hand, because of the cyclic structure of Pro the volume is already more blocked for the side chain in bulk water than for most side chains, so that the desolvation effect in the PTC is not as pronounced and this leads to a smaller pKa-shift than for most side chains. Also, Asn-tRNAAsn is somewhat special because the side chain amide carbonyl group can form a favorable interaction (H-bond) with its protonated N terminus, thus somewhat attenuating the desolvation effect. The remaining three aa-tRNAs (Phe, Ala, and Ile) have the largest downshifts because of the lack of compensating interactions with the amino group.
Fig. 5.

Snapshots from MD simulations with Gly-Phe-tRNAPhe in P site and Phe-tRNAPhe (A) or Gly - tRNAGly (B) in A site. The red spheres represent water molecules.
Although the absolute value calibration of the MD calculations depended on a fitting of the unknown dielectric scaling constant, β, in Eq. 2 above, a straightforward interpretation of the observed downshifts, 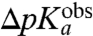 , is that they mainly reflect amino acid specific downshifts, ΔpKa, in the pKa-values of aa-tRNAs as they become ribosome bound. This interpretation is in line with the fact that already the unnormalized, β-independent electrostatic potential energy differences, ΔΔ〈Uel〉, correlate significantly with the observed downshifts (Fig. S2). The correspondence of
, is that they mainly reflect amino acid specific downshifts, ΔpKa, in the pKa-values of aa-tRNAs as they become ribosome bound. This interpretation is in line with the fact that already the unnormalized, β-independent electrostatic potential energy differences, ΔΔ〈Uel〉, correlate significantly with the observed downshifts (Fig. S2). The correspondence of 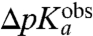 to ΔpKa would imply that
to ΔpKa would imply that 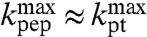 , meaning that the chemistry of peptidyl transfer would dominate the overall rate of peptidyl transfer for all six aa-tRNAs also at high pH-values.
, meaning that the chemistry of peptidyl transfer would dominate the overall rate of peptidyl transfer for all six aa-tRNAs also at high pH-values.
However, this conclusion is controversial, because it is generally assumed that accommodation of native aa-tRNAs in the A site is rate limiting for peptidyl transfer (18–21). Evidence for rate limiting A-site accommodation of aa-tRNA was originally based on stopped-flow experiments, showing that rapid mixing of EF-Tu·GTP in complex with fluorescence-labeled Phe-tRNAPhe with ribosomes containing fMet-tRNAfMet or deacylated tRNAfMet in the P site leads to the same fast fluorescence change, interpreted as GTPase activation of EF-Tu, followed by the same slow fluorescence change, interpreted as Phe-tRNAPhe accommodation in the A site. From the equivalence of the rate of the slow fluorescence change and the overall rate of peptide bond formation (kdip in Fig. 1), accommodation was concluded to be rate limiting for kdip (16, 17). In those (16, 17) and similar (15) experiments the kdip-values were much smaller than kdip-values reported by us (22), suggesting that a rate limiting accommodation step had been removed in our optimized buffer system, thereby allowing for direct kinetic studies of the chemistry of peptide bond formation. Very recent quench-flow experiments (33) now confirm the previously reported (22) large maximal values of kdip at saturating ternary complex concentration. The authors (33) also performed stopped-flow experiments using fluorescence-labeled Phe-tRNAPhe at a single ribosome concentration in excess over ternary complex and found a kdip value identical to a fluorescence relaxation rate about five times smaller than the maximal value of kdip at saturating concentration of ternary complexes. The slow fluorescence relaxation was ascribed to accommodation and the authors argued that its similarity with kdip proves accommodation limited peptidyl transfer (33). However, the accommodation rate constant must always be larger than the maximal value of kdip. Therefore, the observations in (33) do not support the assignment of the slow fluorescence change to the aa-tRNA accommodation step. Further experiments will be needed to define the exact contributions of aa-tRNA accommodation and the chemistry of peptide bond formation to the overall kinetics of peptidyl transfer.
Conclusions
We have found that ribosomal peptidyl transfer to Gly-tRNAGly and Pro-tRNAPro at pH 7.5 is rate limited by the chemistry of peptidyl transfer, rather than by a preceding step following GTP hydrolysis on EF-Tu. A straightforward interpretation of the results of our MD simulations, revealing amino acid specific downshifts of the pKa-values of aa-tRNAs upon A-site binding, is that the chemistry of peptide bond formation is rate limiting in a pH-independent manner for all six of the studied aa-tRNAs. Resolution of the apparent contradiction between this strong conclusion from the present work and the fluorescence based conclusion that peptidyl transfer to Phe-tRNAPhe is accommodation limited at physiological pH will require more detailed kinetic experiments. In any case, our results demonstrate that careful choices of pH in the 6–8 unit range open kinetic windows for direct study of the chemistry of peptide bond formation for six aa-tRNAs in an approach that is extendable to a much larger set of aa-tRNAs. Such kinetic windows are essential for characterization of the phenotypic effects of mutations in ribosomal RNA and protein with implications for cell physiology and molecular evolution.
Materials and Methods
Reagents.
70S ribosomes (E. coli strain MRE 600), synthetic mRNAs, initiation factors, elongation factors, and radiolabeled fMet-tRNAfMet were prepared according to ref. 22 and references therein. tRNAs were from Sigma-Aldrich and Chemical Block (Russia). Radioactive compounds were from GE Healthcare and all other chemicals were from Merck or Sigma-Aldrich. All experiments were carried out in polymix buffer [95 mM KCl, 5 mM NH4Cl, 5 mM Mg(OAc)2, 0.5 mM CaCl2, 8 mM putrescine, 1 mM spermidine, 5 mM potassium phosphate and 1 mM dithioerythritol (DTE)] (34).
Initiated 70S Ribosomes and Ternary Complexes.
Initiated 70S ribosomes, carrying fMet-tRNAfMet ([35S]Met or [3H]Met) in P site and displaying either of the codons UUU (Phe), CCC (Pro), AAC (Asn), GGC (Gly), GCA (Ala), or AUC (Ile) in A site, were prepared by incubating 70S ribosomes (80–85% active in dipeptide formation), fMet-tRNAfMet (1.5 times the ribosome concentration), mRNA, IF1, IF3 (all in twice the ribosome concentration), and IF2 (same concentration as ribosomes) for 10 min at 37 °C. Ternary complexes were prepared by first equilibrating EF-Tu alone with [3H]GDP (1∶1 with EF-Tu) for 15 min at 37 °C, and then incubating it for 20 min at 37 °C in a mixture consisting of the amino acid of interest (400 μM), the corresponding aa-tRNA synthetase (2 units/μL) and tRNA (Purified tRNAPhe for fMetPhe dipeptide formation, bulk tRNA in all other cases). In addition, the ribosome mix contained ATP (1 mM) and GTP (1 mM), and the ternary complex mix contained ATP (2 mM) but no extra GTP. Both mixtures contained phosphoenolpyruvate (PEP) (10 mM), pyruvate kinase (PK) (50 μg/mL), and myokinase (MK) (2 μg/mL). For the fMetPhe dipeptide experiments, where purified tRNAPhe was available, the concentration of ribosomes (1 μM final) was less than the concentration of ternary complex (4 μM final), whereas in all other experiments, the concentration of ribosomes (1–2 μM final) was higher than that of ternary complex (0.5–1 μM final).
pH Adjustments.
The two reaction mixtures were prepared in pH adjusted buffer. Before adding the stock ribosomes, pH of the ribosome mixture was checked (using a Hamilton Minitrode) and corrected if necessary by the addition of KOH (0.5 M) or HCl (1 M). After adding the ribosomes, pH of the two mixtures was equalized if necessary by the addition of KOH (0.5 M) or HCl (1 M) to the ternary complex mix.
Rapid Quench-Flow Experiments.
The two mixtures were rapidly mixed in equal volumes in a temperature controlled quench-flow instrument (RQF-3, KinTek Corp.) as described previously (22). The quenched samples were centrifuged for 15 min at 20,800 × g and the [3H]GTP and [3H]GDP in the supernatants were separated and analyzed using either TLC (35) or a MonoQ ion-exchange column (GE Healthcare) with an on-line scint counter (βRAM3; INUS Inc.) (24). The extent of dipeptide formation was analyzed by RP-HPLC according to ref. 36.
pKa in Bulk Water.
The pKa-values of the α-amino group of different aa-tRNAs in solution at 20 °C were extrapolated from measured values of the pKa of free amino acids and amino acid esters. It has previously been shown that the α-amino pKa is very similar for tRNA-bound amino acids and more simple amino acid esters (14). From the temperature dependence of the pKa for several amino acid methyl and ethyl esters (12, 13), the pKa at 20 °C was calculated to between 0.10 and 0.15 units higher than at 25 °C. The pKas of the amino acid methyl and ethyl esters are downshifted between 2.0 and 2.2 units in relation to the corresponding amino acids (12, 13). As neither Asn nor Pro were included in the studies, we have for all six aa-tRNAs estimated the α-amino pKa at 20 °C (Table 1) as a downshift of 2.0 units from the pKa of free amino acids at 25 °C (11).
MD Simulations.
The calculations were based on the crystallographic coordinates of the 50S Haloarcula marismortui ribosome with the bound peptidyl transfer transition state analog RAP, PDB code 1VQP (4). The RAP moiety was converted to an S-enantiomeric tetrahedral intermediate corresponding to CCA-Phe in the A site and CCA-Phe-Gly in the P site where the terminal amino group was kept neutral. The system was prepared using essentially the same protocols as described previously (7, 8) and consisted of all residues with atoms within 20 Å of the P-site carboxyl carbon and was solvated using the program Q (37). All Sr2+ ions were converted to Mg2+ ions and phosphate linkages and ions outside 15 Å were neutralized as described earlier. The system was relaxed to the reactant state with the aa-tRNA with a free amine in the A site and dipeptide-tRNA ester in the P site. Five additional systems were constructed in this way with A-site aa-tRNAs corresponding to the amino acids Ala, Asn, Gly, Ile, and Pro. MD simulations were performed (SI Text) using the program Q (37) at 300 K with the CHARMM22 force field (38).
Supplementary Material
Acknowledgments.
We thank Tony Forster for detailed discussions about the manuscript. This work was supported by grants from the Swedish Research Council (Project and Linné Uppsala RNA Research Center, M.E., J.Å.) and COST (European Coordination of Science and Technology) Action CM0703 on “Systems Chemistry” (P.S.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information on-line at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012612107/-/DCSupplemental.
References
- 1.Dorner S, Panuschka C, Schmid W, Barta A. Mononucleotide derivatives as ribosomal P-site substrates reveal an important contribution of the 2′-OH to activity. Nucleic Acids Res. 2003;31:6536–6542. doi: 10.1093/nar/gkg842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krayevsky AA, Kukhanova MK. The peptidyltransferase center of ribosomes. Prog Nucleic Acid Re. 1979;23:1–51. doi: 10.1016/s0079-6603(08)60130-0. [DOI] [PubMed] [Google Scholar]
- 3.Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat Struct Mol Biol. 2004;11:1101–1106. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 4.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 6.Hansen JL, Schmeing TM, Moore PB, Steitz TA. Structural insights into peptide bond formation. Proc Natl Acad Sci USA. 2002;99:11670–11675. doi: 10.1073/pnas.172404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trobro S, Åqvist J. Analysis of predictions for the catalytic mechanism of ribosomal peptidyl transfer. Biochemistry. 2006;45:7049–7056. doi: 10.1021/bi0605383. [DOI] [PubMed] [Google Scholar]
- 8.Trobro S, Åqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc Natl Acad Sci USA. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lang K, Erlacher M, Wilson DN, Micura R, Polacek N. The role of 23 S ribosomal RNA residue A2451 in peptide bond synthesis revealed by atomic mutagenesis. Chem Biol. 2008;15:485–492. doi: 10.1016/j.chembiol.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 10.Green R, Lorsch JR. The path to perdition is paved with protons. Cell. 2002;110:665–668. doi: 10.1016/s0092-8674(02)00965-0. [DOI] [PubMed] [Google Scholar]
- 11.Fersht A. Structure and mechanism in protein science : a guide to enzyme catalysis and protein folding. New York: Freeman and Company; 1999. [Google Scholar]
- 12.Hay RW, Morris PJ. Proton ionization constants and kinetics of base hydrolysis of some α-amino-acid esters in aqueous solution. Part II. J Chem Soc B. 1970:1577–1582. [Google Scholar]
- 13.Hay RW, Porter LJ. Proton ionization constants and kinetics of base hydrolysis of some α-amino-acid esters in aqueous solution. J Chem Soc B. 1967:1261–1264. [Google Scholar]
- 14.Wolfenden R. The mechanism of hydrolysis of amino acyl Rna. Biochemistry. 1963;2:1090–1092. doi: 10.1021/bi00905a031. [DOI] [PubMed] [Google Scholar]
- 15.Bieling P, Beringer M, Adio S, Rodnina MV. Peptide bond formation does not involve acid-base catalysis by ribosomal residues. Nat Struct Mol Biol. 2006;13:423–428. doi: 10.1038/nsmb1091. [DOI] [PubMed] [Google Scholar]
- 16.Pape T, Wintermeyer W, Rodnina MV. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. Embo J. 1998;17:7490–7497. doi: 10.1093/emboj/17.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodnina MV, Fricke R, Wintermeyer W. Transient conformational states of aminoacyl-tRNA during ribosome binding catalyzed by elongation factor Tu. Biochemistry. 1994;33:12267–12275. doi: 10.1021/bi00206a033. [DOI] [PubMed] [Google Scholar]
- 18.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FVt. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ledoux S, Uhlenbeck OC. Different aa-tRNAs are selected uniformly on the ribosome. Mol Cell. 2008;31:114–123. doi: 10.1016/j.molcel.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beringer M, Rodnina MV. The ribosomal peptidyl transferase. Mol Cell. 2007;26:311–321. doi: 10.1016/j.molcel.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 21.Zaher HS, Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson M, Bouakaz E, Lovmar M, Ehrenberg M. The kinetics of ribosomal peptidyl transfer revisited. Mol Cell. 2008;30:589–598. doi: 10.1016/j.molcel.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Katunin VI, Muth GW, Strobel SA, Wintermeyer W, Rodnina MV. Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol Cell. 2002;10:339–346. doi: 10.1016/s1097-2765(02)00566-x. [DOI] [PubMed] [Google Scholar]
- 24.Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc Natl Acad Sci USA. 2009;106:50–54. doi: 10.1073/pnas.0809211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watts RE, Forster AC. Chemical models of peptide formation in translation. Biochemistry. 2010;49:2177–2185. doi: 10.1021/bi1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Tan Z, Dickson LG, Nalam MN, Cornish VW. Specificity of translation for N-alkyl amino acids. J Am Chem Soc. 2007;129:11316–11317. doi: 10.1021/ja0734871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marelius J, Hansson T, Åqvist J. Calculation of ligand binding free energies from molecular dynamics simulations. Int J Quantum Chem. 1998;69:77–88. [Google Scholar]
- 28.Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 29.Carlsson J, Boukharta L, Aqvist J. Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to HIV-1 reverse transcriptase. J Med Chem. 2008;51:2648–2656. doi: 10.1021/jm7012198. [DOI] [PubMed] [Google Scholar]
- 30.Beringer M, Rodnina MV. Importance of tRNA interactions with 23 S rRNA for peptide bond formation on the ribosome: studies with substrate analogs. Biol Chem. 2007;388:687–691. doi: 10.1515/BC.2007.077. [DOI] [PubMed] [Google Scholar]
- 31.Brunelle JL, Youngman EM, Sharma D, Green R. The interaction between C75 of tRNA and the A loop of the ribosome stimulates peptidyl transferase activity. RNA. 2006;12:33–39. doi: 10.1261/rna.2256706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okuda K, Seila AC, Strobel SA. Uncovering the enzymatic pKa of the ribosomal peptidyl transferase reaction utilizing a fluorinated puromycin derivative. Biochemistry. 2005;44:6675–6684. doi: 10.1021/bi047419c. [DOI] [PubMed] [Google Scholar]
- 33.Wohlgemuth I, Pohl C, Rodnina MV. Optimization of speed and accuracy of decoding in translation. Embo J. 2010;29:3701–3709. doi: 10.1038/emboj.2010.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jelenc PC, Kurland CG. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc Natl Acad Sci USA. 1979;76:3174–3178. doi: 10.1073/pnas.76.7.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bilgin N, Claesens F, Pahverk H, Ehrenberg M. Kinetic properties of Escherichia coli ribosomes with altered forms of S12. J Mol Biol. 1992;224:1011–1027. doi: 10.1016/0022-2836(92)90466-w. [DOI] [PubMed] [Google Scholar]
- 36.Pavlov MY, Freistroffer DV, MacDougall J, Buckingham RH, Ehrenberg M. Fast recycling of Escherichia coli ribosomes requires both ribosome recycling factor (RRF) and release factor RF3. EMBO J. 1997;16:4134–4141. doi: 10.1093/emboj/16.13.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marelius J, Kolmodin K, Feierberg I, Åqvist J. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J Mol Graph Model. 1998;16:213–225. doi: 10.1016/s1093-3263(98)80006-5. [DOI] [PubMed] [Google Scholar]
- 38.MacKerell AD, Wiorkiewicz-Kuczera J, Karplus M. An all-atom empirical energy function for the simulation of nucleic acids. J Am Chem Soc. 1995;117:11946–11975. [Google Scholar]
- 39.Schmeing TM, Huang KS, Strobel SA, Steitz TA. An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature. 2005;438:520–524. doi: 10.1038/nature04152. [DOI] [PubMed] [Google Scholar]
- 40.Changalov MM, Ivanova GD, Rangelov MA, Acharya P, Acharya S. 2′/3′-O-peptidyl adenosine as a general base catalyst of its own external peptidyl transfer: implications for the ribosome catalytic mechanism. Chembiochem. 2005;6:992–996. doi: 10.1002/cbic.200400349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.