

Abstract
Purpose
In this study, we describe the first retinal histopathologic findings in a patient with Susac’s Syndrome (SS).
Design
Observational case report.
Participant
A 51 year-old Caucasian woman diagnosed with Susac’s syndrome.
Methods
Eyes from a 51 year-old Caucasian woman diagnosed with Susac’s syndrome were obtained at autopsy. One retina was dissected and processed for adenosine diphosphatase (ADPase) flat-embedding. Selected areas were further processed for transmission electron microscopy (TEM).
Main outcome measure
Histopathologic examination using ADPase flat-embedding technique.
Results
There were vaso-occlusive changes in retinal periphery resulting in small areas of capillary dropout. Cross sections demonstrated serous filled spaces between the retinal blood vessels and the internal limiting membrane. Lumens adjacent to these spaces appeared compressed and sometimes closed, but without thrombosis. Decreased ADPase activity in some peripheral blood vessels suggested endothelial cell dysfunction and vaso-occlusion. In the optic nerve head, numerous corpora amylacea were observed in the vicinity of capillaries with thickened walls and narrow lumens. TEM demonstrated thickened and amorphous vascular basal lamina and open endothelial cell junctions in some retinal blood vessels.
Conclusions
The serous deposits with compression of retinal vessel lumens observed histologically probably represent the “string of pearls” described clinically in SS. Chronic extension of these serous deposits along the vessel wall are possibly the cause of retinal arterial wall plaques (RAWPs) as described by Gass and others. In the optic nerve head, corpora amylacea are probably a result of microinfarcts due to optic nerve head capillary angiopathy. Accumulation of amorphous material in the basal lamina, loss of viable endothelial cells and capillary dropout suggests that SS may be an endotheliopathy.
Keywords: blood vessels, endothelial cells, permeability, retina, Susac’s syndrome, vaso-occlusion
INTRODUCTION
Susac’s syndrome (SS) consists of the clinical triad of encephalopathy, branch retinal artery occlusions, and hearing loss.1 It is now believed to be an autoimmune disorder that usually effects young women, but is also seen in men [1/3] and the age of onset extends from 7 to 72. The clinical course is usually but not always self-limited, fluctuating and monophasic. It can last from 6 months to 18 years2 and recovery can occur in some patients with little or no apparent residual disease.3 Other patients, however, can be profoundly impaired with cognitive deficits and hearing loss. Surprisingly, vision loss is rarely a serious symptomatic complication. While ophthalmic examinations and fluorescein retinal angiographic studies have been useful in documenting the ocular features that occur in patients with Susac’s syndrome, little is known regarding the pathology of this disease. In this study, we examined the retina and optic nerve head from a patient who was diagnosed with Susac’s syndrome and subsequently died during the course of her disease.
MATERIALS AND METHODS
Clinical History
A 51-year old Caucasian woman with a past history of hypertension, Graves disease, migraine headaches and uterine fibroid developed an enigmatic encephalopathy that was associated with malignant migraine headaches and an inability to stand or walk. Hallucinations and bilateral hearing loss ensued. Initial magnetic resonance imaging (MRI) showed only diffusion weighted abnormalities consistent with an acute microvascular etiology, but an MRI eleven days later showed pathognomonic corpus callosum lesions seen in SS. Lumbar puncture revealed elevated protein. Audiograms confirmed low frequency neurosensory hearing loss.
Ophthalmic examination five days after the second MRI found visual acuity of 20/50 in the right eye and 20/20 in the left eye. Dilated fundus examination showed cotton wool spots in the periphery and retinal and optic nerve head telangiectasis in the right fundus. Fluorescein angiography of the right eye showed focal areas of irregular retinal artery caliber and leakage from small vessels.
High-dose glucocorticoids, intravenous immunoglobulin G, and then cyclophosphamide were started one week after the initial MRI but the encephalopathy worsened. Electroencephalography showed left-sided rhythmic slow wave activity without spike activity. Repeat MRI indicated new microinfarcts involving gray and white matter, while some of the older microinfarcts had resolved. One month after the onset of her symptoms, the patient developed tachycardia and a saddle pulmonary embolism. She was intubated but status deteriorated rapidly. She was resuscitated multiple times, given tissue plasminogen activator (tPA) twice, and had thrombectomy by interventional radiology. Upon reperfusion, the patient developed flash pulmonary edema, hypotensive acidosis, coagulopathy, thrombocytopenia, and acute anemia. She received transfusions of packed red blood cells, fresh frozen plasma and platelets, was again resuscitated with the addition of epinephrine, atropine, amiodarone, and cardioversion as appropriate but expired after 12 hours despite heroic efforts. Her autopsy was performed 14 hours after death.
Histology
Eyes were obtained at autopsy as approved by the Johns Hopkins Joint Commission on Clinical Investigations. The left eye was processed for standard paraffin histology and the right eye was dissected, the retina teased from the choroid and incubated for adenosine diphosphatase (ADPase) activity and then flat embedded in glycol methacrylate (JB4, Polysciences) as described previously.4 Briefly, the retina from the right eye was fixed overnight in 2% paraformaldehyde in 0.l M cacodylate buffer at 4°C, washed and incubated for ADPase activity. After washing, the retina was examined microscopically. Tissue containing angiopathy from the right eye was designated for embedment in JB-4 for light microscopy or epon for transmission electron microscopy (TEM). TEM tissue was processed and embedded after the initial fixation and ADPase incubation using previously published techniques.5 For light microscopy, tissue was serially sectioned with a total of 790 2.5-mm thick cross sections collected. Sections were stained using either periodic acid Schiff’s (PAS) and hematoxylin, hematoxylin and eosin, toluidine blue/basic fuchsin, or thionin. For TEM, ultrathin sections were collected and stained with 2% uranyl acetate.
RESULTS
Examination of the ADPase-incubated right retina revealed arterial pruning and occlusions (loss of ADPase activity) in the inferotemporal periphery resulting in focal areas of capillary dropout (Figure 1, available at http://aaojournal.org). The vasculature of the posterior pole was unremarkable. Serial JB-4 sections taken through regions with angiopathy (Figure 2) revealed histopathologic changes in both arteries (Figure 2B–D) and veins (Figure 2E–G), despite their normal appearance in the flat perspective. Along the course of an artery (Figure 2B–D), spaces were observed between the internal limiting membrane and the outer vessel wall (Figure 2B–C). The spaces, which were common but intermittent along the course of the vessel, appeared to be filled with proteinaceous serum-like material (eosinophilic) and were confined to the region just anterior to the blood vessel. Thin cell processes often traversed the spaces (Figure 2E–F), which were reminiscent of astrocytic processes. The lumen at these sites was extremely compressed. Where the spaces were absent, the vessel wall appeared thickened with an amorphous eosinophilic material (Figure 2D and G) that was not strongly PAS positive (Figure 2B). Veins within the same region also demonstrated similar pathologic changes with thickened walls (Figure 2E–F); however, their lumens were more severely compressed than were the arteries at sites adjacent to serous spaces. This occasionally resulted in almost complete closure of the luminal space (Figure 2E–F). Serous fluid was also found creating spaces in the internal limiting membrane near degenerating capillaries in the nerve fiber layer (Figure 3, available at http://aaojournal.org). Sections taken through an arterial occlusion (loss of ADPase activity)(Figure 2H) revealed that the vessel consisted primarily of a collapsed collagenous tube with a few viable smooth muscle cells. No evidence of a thrombus was observed within the lumen at the occluded segment. The distal portion of the vessel, which had endothelial cells, may have survived due to retrograde blood flow.
Figure 1.

Adenosine diphosphatase (ADPase) labeled inferotemporal retina showing an arterial occlusion (arrow), arteriolar pruning, and areas of capillary dropout (asterisk). The outlined areas indicate the regions examined in cross section using light (large box) and electron microscopy (small box). Scale bar = 1mm
Figure 2.
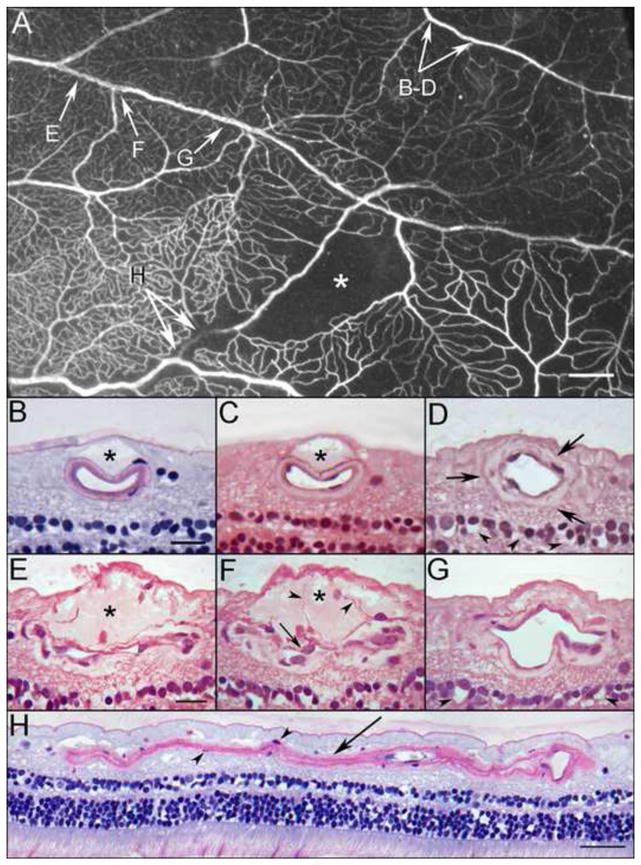
Area of ADPase labeled retina embedded in JB-4 polymer showing an arteriolar occlusion (arrow), capillary dropout (asterisk) and a ribbon-like appearance of a vein (paired arrows). This region was serially sectioned and the arrows correspond to areas shown in panels A–H below.
Panels B and C demonstrate a dome-shaped serous–like material located internal to the artery just below the internal limiting membrane (asterisks). These bleb-like features were common but intermittent along the course of this artery. Even when these bleb-like structures were absent, the wall of the artery appeared thickened with an amorphous material (arrows) as shown in “D”. The spaces in the inner nuclear layer (arrowheads) probably represent a postmortem artifact. Cross sections in panels E–F taken through a portion of the venous segment demonstrate a serous–like material located internal to the vessel just below the internal limiting membrane (asterisks in E–F). The lumen in some areas appears to be compressed or possibly collapsed. The wall of the vein is thick and contains an amorphous material. Cell processes extend into the site (arrowheads in F) and endothelial cell nuclei appear quite large with pale basophilic staining (arrow in F). A more distal segment of the vein is less affected (G), however, the wall is abnormally thickened. The spaces in the inner nuclear layer (arrowheads) probably represent a postmortem artifact. An old arteriolar occlusion in longitudinal section (H). Some smooth muscle cells remain within the collapsed basement membrane matrix (arrowheads in H). Scale bar = 1mm in A, 20 microns in B–G, 40 microns in H [B and H. Periodic acid Schiff’s (PAS) and hematoxylin; C. hematoxylin and eosin; D–G. toluidine blue/basic fuchsin]
Figure 3.

Sections showing serous-like exudate that extends along the internal limiting membrane creating a bleb-like structure (asterisks). In “B”, a degenerating capillary is shown (arrow). The spaces in the inner nuclear layer in “B” (white arrowheads) probably represent a postmortem artifact. Occasional inflammatory cells are found within the bleb (arrowheads in A and B). Scale bar = 20 microns (A. toluidine blue/basic fuchsin; B. thionin)
Examination of blood vessel ultrastructure revealed diffuse thickening of the basal lamina with amorphous material (Figure 4, available at http://aaojournal.org). Notable also was the loss of the basal lamina that would usually extend to surround the adjacent exposed pericyte (Figure 4B, available at http://aaojournal.org). The endothelial cell cytoplasm was often vacuolated and contained electron dense particles, which were the product of the ADPase histochemical reaction. Occasional open junctions were seen between some endothelial cells (Figure 4E, available at http://aaojournal.org). However, it must be mentioned that the tissue was fixed initially for the ADPase technique (2% paraformaldehyde) and not for TEM, and the pieces were excised from the retina after ADPase processing, so some of the ultrastructural changes may represent artifacts of tissue processing.
Figure 4.

Higher magnification view of the region of retina shown in Figure 1 examined using electron microscopy showing capillary dropout (asterisk) and arterial pruning (arrow). Electron micrograph of a viable capillary (B and C) from the region shown in “A”. The electron dense particles in the endothelial cytoplasm are lead reaction product from the ADPase reaction. The endothelial ultrastructure appears intact with characteristic nucleus, plasmalemma vesicles and cytoplasmic inclusions. A pericyte is seen in the close contact with the basal lamina (B) but the granular appearance of this cell and poorly defined cytoplasmic membrane may represent postmortem changes. The basal lamina is thickened and amorphous (asterisk in “C”). Endothelial cells sometimes were vacuolated (arrow in D) and open cell junctions were occasionally observed (asterisk in E). The majority of the intercellular junctions lacked the distinct zonula occludens (tight junction), which tend to be extensive in healthy retinal vessels. Scale bar = 1mm in A, 2 microns in B, 100nm in C, 500nm in D, 100nm in E.
Sections taken through the optic nerve head (Figure 5C–D, available at http://aaojournal.org) demonstrated numerous PAS positive corpora amylacea. Many were in close proximity to capillaries that had abnormally thickened walls (Figure 5E, available at http://aaojournal.org). Corpora amylacea were also present in the nerve fiber layer of peripapillary retina and occasionally seen adjacent to capillaries in the paramacular nerve fiber layer (Figure 5F, available at http://aaojournal.org).
Figure 5.

Color fundus photograph (A) of the right fundus shows patchy irregular caliber of the inferonasal branch retinal artery and of the small macular arteries and veins. Venous phase fluorescein angiogram (B) of the same field shows better vascular detail and telangiectasis of the optic nerve head. Note patchy abnormal retinal hyperfluorescence superior to the optic nerve corresponding to leaking capillaries. ADPase flat embedded optic nerve head (C) and peripapillary retina of the right eye (scale bar = 300 μm). Section through nerve head (D) showing numerous corpora amylacea (arrows) stained with PAS (scale bar = 100 μm). Higher magnification of corpora amylacea (E) in nerve head (arrow) near a capillary (arrowhead) with an abnormally thickened wall (scale bar = 20 μm). Corpora amylacea (arrow) in the nerve fiber layer (F) of the paramacular retina. Scale bar = 20 μm (D–F. PAS and hematoxylin).
DISCUSSION
This is the first detailed histopathological description of a Susac’s Syndrome retina and optic nerve head. The findings confirm the clinical observations of vascular occlusions and subsequent ghost vessels3 but, in this particular subject, the occlusions were not just in branch arteries but in vessels of all caliber in the vascular hierarchy. The occlusions were present at sites of endothelial cell dysfunction and death in that ADPase activity was reduced or absent in blood vessels, as we have found at sites of occlusion in diabetic and sickle cell retinopathies.4, 6–8 These peripheral blood vessels often lacked viable endothelial cells (EC) but had viable smooth muscle cells, suggesting that the source of the fluid between the internal limiting membrane and superficial blood vessels was due to breakdown of blood retinal barrier (BRB). The EC vacuolation and open junctions further supports this concept but these ultrastructural changes may represent artifacts of tissue processing for TEM. Recent work has shown elevated circulating Factor VIII and the presence of anti-endothelial cell antibodies,3 suggesting that SS is an endotheliopathy, which our study supports.
Blood vessels were abnormal in other ways as well. Thickened vascular walls were present as in diabetes except that material appeared serous (eosinophilic) in nature and, like glycated diabetic basement membrane material, was not strongly PAS positive. Also unlike diabetes, the walls of all blood vessels were not thickened. Even in the same blood vessel there were areas with greatly thickened wall and other areas of the same vessel had a normal wall (Figure 2E–G). The patient was reported to have been hypertensive, and thickened walls and arteriolar narrowing are observed in hypertension.9–11 However, the thickening of the wall was not suggestive of hypertension, in that there was never an onion skin-like appearance to the arterial walls. The material in the thickened walls was amorphous (Figure 4, available at http://aaojournal.org) and did not contain remnants of smooth muscle cells or dense bodies as reported in hypertension.12 In addition, the vessels were not as tortuous as reported in hypertension.13
Lumens are also highly constricted in hypertension. However, the lumens in this Susac’s subject were narrow but this appeared to be due to compression of lumens by the abluminal fluid-filled spaces. This was more apparent in veins (Figure 2E–G) than arteries and arterioles (Figure 2B–D), but arteries and arterioles were compressed as well. Edema around blood vessels occurs in hypertension as well,14 but it encircles the blood vessels and is not confined to the space between superficial plexus blood vessels and internal limiting membrane as in this Susac’s syndrome subject.
Although the data suggests that Susac’s syndrome is an endotheliopathy, there is evidence for glial dysfunction as well. The fluid filled spaces were anterior to the blood vessels of the superficial plexus and not observed in deep capillary network. It could be argued that the subject had received large volumes of fluids, which could be the source of the fluid. However, the fluid was confined to the inner aspect of retina and was not present adjacent to all blood vessels, so global fluid overload does not seem likely as the source of the fluid. The fluid was sequestered in a glial domain of retina, anterior to blood vessels where Muller cell foot processes and astrocytes are present. So why was fluid retained in this particular area? In cystoid macular edema, some investigators have suggested that Muller cells cordon off the fluid from breakdown of the BRB by forming cysts in retina while others suggest that Muller cells actually accumulate the fluid intracellularly.15–17 Glial cells and their processes appeared to be present in and around the fluid in some areas (Figure 2E–F).
The large number of corpora amylacea in retina and optic nerve in this 50-year-old subject might also suggest glial dysfunction. These formations are thought to be related to glial dysfunction and occur most frequently with aging.18, 19 Woodward and Tso found that these formations are composed of neuronal components and suggested that they accumulate because of disruption in axonal transport.20 It may be that neural breakdown products accumulate within astrocytes, which then create the deposit. However, a recent study by Meng et al of brain tissue from aged subjects and vascular dementia cases demonstrated immunostaining for the serum proteins thrombospondin-1 and ADAMTS13, in corpora amylacea.21 They suggested that because of the characteristic predilection for corpora amylacea to accumulate near blood vessels, that proteins extravasated from blood via blood brain barrier defects may contribute to the formation of these structures. Apparent barrier defects may have contributed to the corpora amylacea formations in the SS subject reported herein. Alternatively, corpora amylacea might be secondary to other contributory factors in this subject like hypotension and coagulopathy, although there is no connection that we have found between these and corpora amylacea in the literature.
The perivascular fluid-filled spaces observed by histopathology may represent the Gass plaques previously described with fluorescein angiography in SS patients.22 Unfortunately, we do not have an angiogram of the area of the fundus that had several fields of vascular loss that are the focus of most of this manuscript. However, the sites of occlusion were analyzed in serial sections from areas documented in tissue blocks. It is notable that there were no platelet-fibrin thrombi at sites of vascular loss. There were also very few white blood cells apparent anywhere in the tissue that was analyzed and we did not observe the platelet fibrin thrombi reported previously at occluded arterioles.3 This disagrees with one rare report by Do et al on a SS patient with leptomeningitis whose brain biopsy showed areas of capillary hyaline thrombus with perivascular inflammatory cells.23 The initial differential diagnosis in Susac’s patients often includes multiple sclerosis, another autoimmune disease that can result in peripheral retinal vascular occlusions, retinal venous sheathing with fluorescein leakage.24 However, the break down in the BRB in multiple sclerosis is annular and not just anterior to the superficial retinal blood vessels and is always accompanied by periphlebitis 25, 26, 27, whereas the SS subject examined in this manuscript had very few if any leukocytes associated with areas of serum leakage.
In summary, this 50-year old Susac’s patient had serous deposits, which appeared to compress retinal vessel lumens. These serous-filled spaces may represent the Gass plaques or retinal arterial wall plaques (RAWPs) reported by others with fluorescein angiography.22 The source of the fluid appears to be serous due to BRB breakdown from death and dysfunction of endothelial cells, although glia appear to be involved in sequestering the fluid between retinal blood vessels and the internal limiting membrane. The contribution of this subject’s end-of-life acute clinical conditions and treatments (t-PA, transfusions, corticosteroids, immunoglobulins, and cyclophosphamide) and prior medical history (Graves’ disease and hypertension) to the histopathological findings can not be excluded, but the fact that these pathological changes were in isolated retinal blood vessels and adjacent areas were normal may suggest local rather than systemic increased vascular permeability and vaso-occlusive events. Finally, in the nerve head, corpora amylacea are probably a result of micro-infarcts due to optic nerve head angiopathy.
Supplementary Material
Acknowledgments
Grant support: NIH grants EY016151 (GL), EY019347 (HY), EY01765 (Wilmer), the Altsheler-Durell Foundation, and an RPB Unrestricted Grant (Wilmer). Gerard Lutty received an RPB senior scientific investigator award in 2008.
Footnotes
The authors have neither financial disclosures nor conflicts of interest concerning the contents of this manuscript.
This article contains online-only material. The following should appear online-only: Figures 1, 3, 4, 5.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Susac JO. Susac’s syndrome: the triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology. 1994;44:591–3. doi: 10.1212/wnl.44.4.591. [DOI] [PubMed] [Google Scholar]
- 2.Petty GW, Matteson EL, Younge BR, et al. Recurrence of Susac syndrome (retinocochleocerebral vasculopathy) after remission of 18 years. Mayo Clin Proc. 2001;76:958–60. doi: 10.4065/76.9.958. [DOI] [PubMed] [Google Scholar]
- 3.Susac JO, Egan RA, Rennebohm RM, Lubow M. Susac’s syndrome: 1975–2005 microangiopathy/autoimmune endotheliopathy. J Neurol Sci. 2007;257:270–2. doi: 10.1016/j.jns.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 4.Lutty GA, McLeod DS. A new technique for visualization of the human retinal vasculature. Arch Ophthalmol. 1992;110:267–76. doi: 10.1001/archopht.1992.01080140123039. [DOI] [PubMed] [Google Scholar]
- 5.Baba T, Grebe R, Hasegawa T, et al. Maturation of the fetal human choriocapillaris. Invest Ophthalmol Vis Sci. 2009;50:3503–11. doi: 10.1167/iovs.08-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lutty GA, McLeod DS. Phosphatase enzyme histochemistry for studying vascular hierarchy, pathology, and endothelial cell dysfunction in retina and choroid. Vision Res. 2005;45:3504–11. doi: 10.1016/j.visres.2005.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLeod D, Goldberg M, Lutty G. Dual perspective analysis of vascular formations in sickle cell retinopathy. Arch Ophthalmol. 1993;111:1234–45. doi: 10.1001/archopht.1993.01090090086026. [DOI] [PubMed] [Google Scholar]
- 8.McLeod DS, Merges C, Fukushima A, et al. Histopathological features of neovascularization in sickle cell retinopathy. Am J Ophthalmol. 1997;124:473–87. doi: 10.1016/s0002-9394(14)70862-1. [DOI] [PubMed] [Google Scholar]
- 9.Klein R, Sharrett AR, Klein BE, et al. Are retinal arteriolar abnormalities related to atherosclerosis? The Atherosclerosis Risk in Communities Study. Arterioscler Thromb Vasc Biol. 2000;20:1644–50. doi: 10.1161/01.atv.20.6.1644. [DOI] [PubMed] [Google Scholar]
- 10.Walsh JB. Hypertensive retinopathy. Description, classification, and prognosis. Ophthalmology. 1982;89:1127–31. [PubMed] [Google Scholar]
- 11.Wong TY, Klein R, Klein BE, et al. Retinal microvascular abnormalities and their relationship with hypertension, cardiovascular disease, and mortality. Surv Ophthalmol. 2001;46:59–80. doi: 10.1016/s0039-6257(01)00234-x. [DOI] [PubMed] [Google Scholar]
- 12.Kimura T, Mizota A, Fujimoto N, Tsuyama Y. Light and electron microscopic studies on human retinal blood vessels of patients with sclerosis and hypertension. Int Ophthalmol. 2005;26:151–8. doi: 10.1007/s10792-007-9033-1. [DOI] [PubMed] [Google Scholar]
- 13.Tso MOM, Jampol LM. Hypertensive retinopathy, choroidopathy, and optic neuropathy of hypertensive ocular disease. A clinical and pathophysiologic approach to classification. In: Laragh JH, Brenner MB, editors. Hypertension: Pathophysiology, Diagnosis, and Management. New York: Raven Press, Ltd; 1990. pp. 433–465. [Google Scholar]
- 14.Spencer WR. An Atlas and Textbook. Chicago: American Academy of Ophthalmology; 1990. Ophthalmic Pathology; pp. 1108–22. [Google Scholar]
- 15.Bringmann A, Reichenbach A, Wiedemann P. Pathomechanisms of cystoid macular edema. Ophthalmic Res. 2004;36:241–9. doi: 10.1159/000081203. [DOI] [PubMed] [Google Scholar]
- 16.Fine BS, Brucker AJ. Macular edema and cystoid macular edema. Am J Ophthalmol. 1981;92:466–81. doi: 10.1016/0002-9394(81)90638-3. [DOI] [PubMed] [Google Scholar]
- 17.Yanoff M, Fine BS, Brucker AJ, Eagle RC., Jr Pathology of human cystoid macular edema. Surv Ophthalmol. 1984;28 (Suppl):505–11. doi: 10.1016/0039-6257(84)90233-9. [DOI] [PubMed] [Google Scholar]
- 18.Keller JN. Age-related neuropathology, cognitive decline, and Alzheimer’s disease. Ageing Res Rev. 2006;5:1–13. doi: 10.1016/j.arr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Mrak RE, Griffin ST, Graham DI. Aging-associated changes in human brain. J Neuropathol Exp Neurol. 1997;56:1269–75. doi: 10.1097/00005072-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Woodfard B, Tso MOM. An ultrastructural study of corpora amylacea of the optic nerve and retina. Am J Ophthalmol. 1980;90:492–502. doi: 10.1016/s0002-9394(14)75018-4. [DOI] [PubMed] [Google Scholar]
- 21.Meng H, Zhang X, Blaivas M, Wang MM. Localization of blood proteins thrombospondin1 and ADAMTS13 to cerebral corpora amylacea. Neuropathology. 2009 doi: 10.1111/j.1440-1789.2009.01024.x. epub April 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egan RA, Ha Nguyen T, Gass JD, et al. Retinal arterial wall plaques in Susac syndrome. Am J Ophthalmol. 2003;135:483–6. doi: 10.1016/s0002-9394(02)02085-8. [DOI] [PubMed] [Google Scholar]
- 23.Do TH, Fisch C, Evoy F. Susac syndrome: report of four cases and review of the literature. AJNR Am J Neuroradiol. 2004;25:382–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Younge BR. Fluorescein angiography and retinal venous sheathing in multiple sclerosis. Can J Ophthalmol. 1976;11:31–6. [PubMed] [Google Scholar]
- 25.Arnold AC, Pepose JS, Hepler RS, Foos RY. Retinal periphlebitis and retinitis in multiple sclerosis. I. Pathologic characteristics. Ophthalmology. 1984;91:255–62. doi: 10.1016/s0161-6420(84)34296-8. [DOI] [PubMed] [Google Scholar]
- 26.Vine AK. Severe periphlebitis, peripheral retinal ischemia, and preretinal neovascularization in patients with multiple sclerosis. Am J Ophthalmol. 1992;113:28–32. doi: 10.1016/s0002-9394(14)75748-4. [DOI] [PubMed] [Google Scholar]
- 27.Kerrison JB, Flynn T, Green WR. Retinal pathologic changes in multiple sclerosis. Retina. 1994;14:445–51. doi: 10.1097/00006982-199414050-00010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.