

Abstract
Phosphorylation of the cyclin-dependent kinase inhibitor p27 by upstream mitogenic signaling pathways regulates its stability, localization and biological function. In human cancers, loss of the antiproliferative action of p27 can arise through reduced protein levels and/or cytoplasmic mislocalization leading to increased cell proliferation and/or cell migration, respectively. Reduced p27 expression levels and p27 mislocalization have potential prognostic and therapeutic implications in various types of human cancers. This review highlights mechanisms of functional deregulation of p27 by oncogenic signaling that provide an important molecular rationale for pathway targeting in cancer treatment.
Keywords: p27, cell cycle, cell migration, oncogenic signal transduction, targeted therapies, prognostic biomarkers
Part I – Background
p27 function and subcellular localization
Cell cycle progression is driven by cyclins and their associated cyclin-dependent kinases (CDKs). p27 was discovered as an inhibitor of cyclin E-CDK2 (1–3), but has since been shown to play dual roles to both promote and inhibit cell cycle progression. In G0, p27 translation and protein stability are maximal as it binds and inactivates nuclear cyclin E-CDK2. In early G1, p27 also promotes assembly and nuclear import of D-type cyclin-CDKS (4, 5). The progressive decrease of p27 in G1 permits cyclin E-CDK2 and cyclin A-CDK2 to activate the G1-S transition (6). Mice lacking p27 display multi-organ hyperplasia, increased body size and susceptibility to carcinogen-induced tumors, suggesting that p27 acts as a tumor suppressor to control both tissue expansion and cell proliferation (7–9).
p27 is regulated at transcriptional, translational and post-translational levels(10). p27 mRNA levels usually show little cell cycle periodicity, but may be repressed in a PI3K/AKT-dependent manner (11). In normal cells, p27 protein levels are largely controlled by ubiquitin-dependent proteolysis (6). In early G1, mitogens promote p27 phosphorylation at serine 10 (S10) to facilitate nuclear export (12, 13); this simultaneously relieves cyclin E-CDK2 inhibition and permits KPC -mediated proteolysis of cytoplasmic p27 (14). Tyrosine phosphorylation of p27 by BCR-ABL (Y74, Y88, Y89) or Src family kinases (Y88 and Y74) impairs the CDK2 inhibitory action of p27. This facilitates subsequent cyclin E-CDK2-dependent T187 phosphorylation of p27, that, in turn, allows it to be targeted by SCFSKP2 for degradation (see Figure 1) (15, 16). Tyrosine phosphorylation of p27 is also required for the catalytic activation of p27-cyclin D1-CDK4 complexes (17, 18).
Figure 1. p27 plays multi-faceted roles in the regulation of cell proliferation and cell migration.
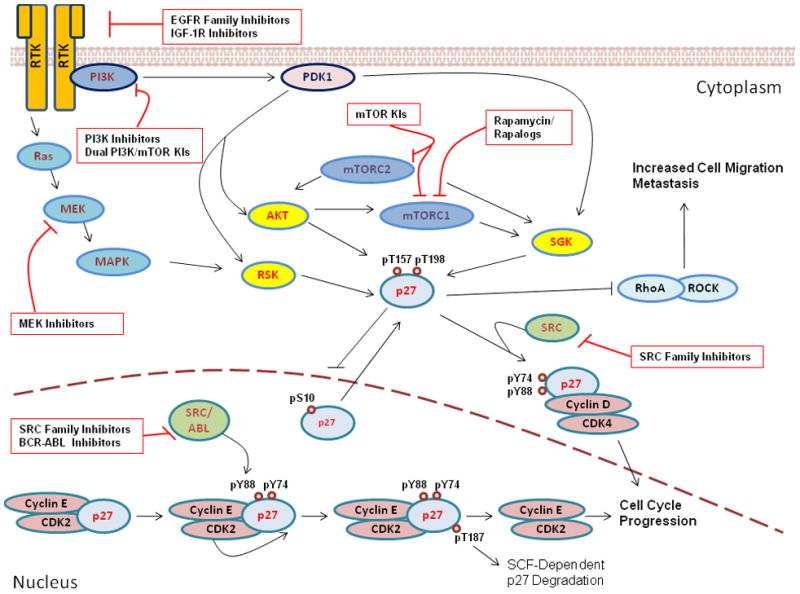
This schematic representation depicts p27 regulation and function as it relates to the protein’s subcellular localization. While in the nucleus, p27 binds to and inhibits cyclin E-CDK2, preventing G1-S transit. Following SRC/ABL-mediated tyrosine phosphorylation and cyclin E-CDK2-mediated T187 phosphorylation, nuclear p27 is targeted for SCFSKP2-dependent degradation, allowing cell cycle progression. S10 phosphorylation promotes nuclear export; while in the cytoplasm p27 may play several roles. Oncogenic signal transduction via PI3K/PDK1 or Ras/MAPK results in the hyperactivation of several AGC family kinases (including AKT, SGK, and RSK), that all mediate C-terminal phosphorylation of p27 at T157 and T198. These phosphorylation events cooperate to sequester and stabilize p27 within the cytoplasm, where it promotes cell proliferation (via cyclin D-CDK4 assembly) and cell motility (via inhibition of RhoA/ROCK signaling). Potential sites of targeted therapeutic intervention are highlighted in the figure.
Finally, p27 localization appears to be finely tuned during G1 progression. In early G1, as p27 translation falls, nuclear export of p27 may be needed not only to relieve CDK2 inhibition, but also to promote p27-D type-cyclin-CDK assembly. Transient cytoplasmic retention of newly synthesized p27 is also facilitated in early G1 by PI3K effectors that phosphorylate p27 on T157 and/or T198 to both impair p27 import and promote cyclin D1-CDK4-p27 complex assembly (4).
Oncogenic signaling pathways that disrupt p27 function
In contrast to other tumor suppressors, p27 mutation or deletion is rare in human cancers. Deregulated receptor tyrosine kinases (RTKs) activate Src/BCR-ABL and Ras/MEK/MAPK, or PI3K/AKT signaling which contribute to oncogenesis by inducing p27 loss or mislocalization, respectively (4, 6).
Cell cycle inhibition is a nuclear p27 function. In many cancers, not only is p27 reduced in the nucleus, but tumors also exhibit different degrees of cytoplasmic p27 mislocalization (19). Cytoplasmic p27 appears to acquire a cell cycle-independent oncogenic function to promote cancer cell invasion. TAT-p27 protein transduction was shown to increase Rac-dependent cell motility (20). p27−/− mouse embryonic fibroblasts (MEFs) show reduced motility compared to wild type MEFs that is rescued by re-expression of wild type p27 or mutant p27 that cannot bind cyclins and CDKs (p27CK-) (21). Thus, the pro-motility effects of p27 appear to be independent of its cyclin-CDK regulatory functions. In the cytoplasm, p27 binds RhoA to inhibit RhoA-ROCK mediated actomyosin stabilization (21). Moreover, knock-in of p27CK- increases progenitor cell self-renewal and lung tumor development (22). In malignant lines, overexpression of cytoplasmic p27 increases tumor metastasis in murine models (23, 24) and promotes glioma cell invasion (25). Thus, while nuclear p27 inhibits CDK2 to restrain cell cycle, excessive cytoplasmic p27 can have a pro-oncogenic effect to promote cancer invasion and metastasis.
PI3K is oncogenically activated in many human cancers (26) and not only regulates cell survival, but also modulates cyclin D1, CDK2, p21 and p27 levels and action (27). AKT stabilizes cyclin D1 by inactivating GSK-3β (28). At least three PI3K effectors (AKT, SGK and RSK) contribute to T157 and T198 phosphorylation of p27 (27, 29–31), which impairs import of monomeric p27 and increases p27-cyclin D-CDK4 assembly (17). Phosphorylation at T198 stabilizes p27 (32, 33), increases its cytoplasmic localization and mediates p27-RhoA binding and a p27-dependent increase in cell motility (34). Importantly, PI3K/AKT activation is highly correlated with cytoplasmic p27 localization in breast, renal and thyroid cancers and in certain leukemias (29, 35–37).
The mammalian target of rapamycin (mTOR) integrates nutrient sensing and mitogenic signals to regulate protein synthesis, cell growth, proliferation and motility (38). mTOR signaling is linked to that of PI3K (see Figure 1). AKT-mediated inactivation of the tuberous sclerosis complex (TSC1 and TSC2 proteins) activates mTOR complex 1 (mTORC1, mTOR/Raptor) to upregulate protein biosynthesis required for cell proliferation (38). AKT requires two activating phosphorylations, one via PI3K/PDK1, the second via mTORC2 (mTOR/Rictor) (38). Both mTOR complexes may activate SGK (39, 40). Thus, mTOR-mediated AKT and SGK activation promote p27 phosphorylation at T157/T198, impairing p27 nuclear import and driving cellular proliferation and migration.
Oncogenic RTK and Ras trigger MEK/MAPK activation (41) to mediate either p27 loss or inactivation (42–44). In epithelial cells, transfected oncogenic Ras mislocalized p27 to the cytoplasm and increased p27-cyclin D-CDK4/6 complexes (45). This likely reflects constitutive Ras-mediated PI3K activity. Furthermore, Ras-dependent lung tumorigenesis is associated with increased cytoplasmic localization of p27 (46). Constitutively active N-Ras can also mislocalize p27 via the Ral-GEF pathway (47). In fibroblasts, MEK1 overexpression increased p27 degradation and a MEK1 inhibitor increased p27 stability in one study (48) but led to sequestration of p27 in cyclin D-CDK4 complexes in another (44). In breast cancer cells, p27 proteolysis is activated by HER2 and epidermal growth factor receptor in a MEK/MAPK dependent manner (42, 49).
Many human cancers have increased Src levels or activity (50), which would increase tyrosine phosphorylation p27 to promote SCFSKP2-mediated p27 proteolysis. In primary human breast cancers, Src activation is associated with reduced p27 protein (15). Similarly, the Src-family kinase, Lyn, and BCR-ABL phosphorylate p27 at Y88 and this is blocked by the ABL kinase inhibitor drug, imatinib (16). To further complicate matters, BCR-ABL was recently reported to promote AKT-mediated phosphorylation of p27 at T157 in chronic myeloid leukemia progenitors leading to increased cytoplasmic p27 (51). Thus, therapeutic interruption of BCR-ABL may not only increase p27 levels, but also restore its nuclear localization.
Constitutive activation of these different oncogenic pathways may contribute to loss of nuclear p27 and increased cytoplasmic p27, both of which drive tumor growth and progression. Reversal of these effects and restoration of the cell cycle inhibitory action of p27 by targeted inhibition of these oncogenic pathways may contribute importantly to the efficacy of targeted therapies for cancer.
Part II – Clinical-Translational Advances
Loss of nuclear p27 during tumor progression
Given the central roles of p27 in cellular proliferation and migration, it is no surprise that reduced or mislocalized p27, documented by immunohistochemical analysis in primary tumors, is associated with poor clinical outcome in a diverse variety of human malignancies (6). Progressive p27 loss has been observed during the histopathological progression of neoplasia from benign to in situ and invasive cancers of the breast (52, 53). One study showed that, relative to normal breast duct epithelia where 95% of cells show high nuclear p27, pre-malignant atypical ductal hyperplasia shows modest p27 loss (85%), while ductal carcinoma in situ (40%) and invasive cancer (34%) show greater p27 loss (53). In ovarian cancer, lesions of low malignant potential showed intermediate p27 levels compared to high levels in normal epithelia, while highly aggressive cancers showed very low nuclear p27 (54, 55).
Immunohistochemical analysis of p27 for prognostication of human cancers
Reduced nuclear p27 is associated with adverse patient outcome in malignancies of epithelial, hematopoietic, and mesenchymal origin (6). To date, the application of p27 as a clinical prognostic tool has been encumbered by the lack of uniformity in immunohistochemical (IHC) staining protocols. Most IHC studies of p27 have evaluated only nuclear p27 and there is currently no accepted guideline for scoring cutoff values for “low” versus “high.” Despite these limitations, multiple studies demonstrate a correlation between reduced nuclear p27 and poor prognosis. This has been observed in lung (56–58) and prostate cancers (59–63) and more comprehensively in breast cancer, where most analyses show reduced p27 is an independent prognostic indicator of disease relapse or death (52, 64–70).
While far fewer studies have evaluated the prognostic potential of cytoplasmic p27, this is also correlated with adverse outcome in prostate cancer (71), gliomas (72) and high-grade astrocytomas (73). In breast cancer, cytoplasmic p27 staining correlated with AKT activation (29–31) and predicted poor patient prognosis in univariate analysis (29). To date, however, none of these analyses have quantitated both nuclear p27 levels and cytoplasmic p27 mislocalization. An understanding of the potential prognostic and predictive value of p27 (see below) may await this type of study, since reduced nuclear p27 increases proliferation and cytoplasmic p27 would drive tumor cell invasion.
Targeting critical signal transduction cascades upstream of p27
As reviewed above, diverse oncogenic signaling cascades regulate p27 proteolysis, subcellular localization, and function (see Figure 1) (4). The informed use of targeted therapies to attenuate deregulated signaling holds tremendous promise for cancer therapy. Several molecular targeting drugs impact p27 by inhibiting upstream signaling. In chronic myelogenous leukemia driven by the BCR-ABL kinase, reduced p27 levels result from its constitutive tyrosine phosphorylation and accelerated proteolysis (16). Treatment of CML cells with imatinib restores p27 levels and inhibits proliferation (16). In many other pre-clinical models, pathways driving p27 proteolysis were reversed by targeted therapies: in Her2-overexpressing breast cancer, p27 proteolysis is attenuated by treatment with trastuzumab (74); the same is true of EGFR-overexpressing lung cancers treated with gefitinib (75). In a recent study, treatment of breast cancer cells with the EGFR/ErbB-2 inhibitor lapatinib increased p27 and reduced tumor proliferation (76). In melanoma models, MEK inhibition increased p27 to induce growth arrest (77, 78). Similar results were obtained using a MEK inhibitor in human pancreatic cancer lines driven by activated Ras (79).
The placement of p27 downstream of various AGC kinases (SGK, RSK, AKT) highlights the tremendous potential for PI3K/mTOR inhibitors to restore normal p27 function. Hyperactivation of PI3K/mTOR signaling, through C-terminal phosphorylation of p27 at T157 and T198, promotes cytoplasmic p27 mislocalization, increased invasiveness, and may underlie progression in a variety of cancers (4). Early clinical trials with rapamycin and its analogs (rapalogs) met with limited success. This may be due, in part, to incomplete blockade of mTORC1 by rapamycin (80). Furthermore, inhibition of mTORC1 turns on feedback loops leading to PI3K activation (81, 82). This feedback PI3K/AKT activation would promote cytoplasmic p27 sequestration and p27-dependent tumor cell migration and metastasis. Pure mTOR catalytic site inhibitors or dual PI3K/mTOR kinase inhibitors have shown great potential in pre-clinical models and early clinical trials (83). These new compounds inhibit both mTORC1 and mTORC2, effectively attenuating activation of AGC kinases involved in growth, evasion of apoptosis, cell cycle and motility (84).
Joint targeting of ER and oncogenic Src signaling
In estrogen-responsive tumors, estrogen binding to the estrogen receptor (ER) promotes p27 proteolysis and cyclin E-CDK2 activation, driving cell cycle progression (85, 86). In sensitive ER positive breast cancers, ER blocking drugs (tamoxifen and fulvestrant) and aromatase inhibitors oppose estrogen signaling and stabilize p27 to inhibit proliferation and growth. Both pre-clinical studies (85, 87, 88) and analysis of p27 in breast cancers of patients in clinical trials indicate that high nuclear p27 may predict responsiveness to hormonal therapy (67, 70).
Unfortunately, both de novo and acquired resistance limit the efficacy of hormonal therapies. Oncogenic activation of EGFR family receptors and IFG-1R may underlie antiestrogen resistance, potentially via the Src kinase. Src activation by liganded estrogen receptor or by oncogenic receptor tyrosine kinases in breast cancer (89) would facilitate p27 degradation (16, 90) and oppose growth arrest by antiestrogens. Recent pre-clinical studies suggest that combined targeting of both estrogen:ER and Src may prevent the emergence of resistance to antiestrogens (87, 88, 91). Combined use of an aromatase inhibitor (anastrozole) with a Src inhibitor (saracatinib) in vitro resulted in higher p27 levels and greater G1 arrest, reduced xenograft tumor growth in vivo and delayed emergence of drug resistance compared to that observed with either drug alone (87). Similar results were obtained using combined saracatinib and fulvestrant (88). Currently, clinical trials are underway to test the potential efficacy of combined Src and aromatase inhibition in post-menopausal ER positive breast cancer (M. Pegram and JMS, unpublished). Src activation and/or reduced p27 levels may correlate with de-novo lack of responsiveness to ER blockade or aromatase inhibition and a post-treatment increase in nuclear p27 may predict response to combined therapy as observed in pre-clinical models. There appears to be great potential for targeting both Src and ER and a number of Src inhibitor drugs are either approved for clinical use or in clinical development. Furthermore, our knowledge of pathways that disrupt p27 function provides a strong rationale for testing other potential therapeutic combinations. For example, combined ER/MEK blockade or ER/mTOR blockade may both be of clinical value since they would restore p27 function and check tumor proliferation.
p27 as predictor of therapeutic efficacy
Prognostic studies of tumor biomarkers are valuable as they assist in disease stratification and prompt molecular mechanistic studies that shed light upon the pathways driving tumor progression. However, the most valuable biomarkers are those that reliably indicate response to treatment, acting as predictive markers. In certain contexts, p27 may serve just this purpose. For example, in stage III/IV ovarian cancers, reduced p27 predicted improved patient response to adjuvant platinum-based therapy after surgery (92). This likely reflects the efficacy of cell cycle-active chemotherapies in tumors with high proliferation due to low p27. Other studies demonstrated the opposite result, with reduced p27 associated with impaired therapeutic response (55, 93). These latter results may reflect the genetic plasticity of rapidly growing tumors giving rise to chemo-resistant clones.
One can envision several scenarios in which aberrant p27 staining in a pre-treatment biopsy might guide treatment decisions. Cancers with low nuclear p27 due to enhanced proteolytic degradation may be ideal candidates for Src or MEK inhibitors. In responsive tumors, clinical benefit may correlate with restoration of nuclear p27 staining and reduced tumor proliferation in post-treatment biopsies. Alternately, a pre-treatment biopsy showing increased cytoplasmic p27 (with or without low nuclear levels), may herald intratumoral PI3K/mTOR activation and identify tumors most likely to respond to inhibitors of this pathway. PI3K/mTOR inhibitor drugs should shift p27 from cytoplasmic to nuclear localization. A post-treatment increase in nuclear p27 levels (MEK or Src inhibitors) or a shift of p27 from cytoplasm to nucleus (PI3K/mTOR or pure mTOR inhibitors) may prove to be valuable predictors of response to these novel agents. Evaluation of p27 by IHC in pre-treatment tumor samples and re-assessment of p27 levels and localization in a post-treatment biopsy may prove useful in the initial clinical trials of these new agents. Successful attenuation of PI3K/mTOR signaling with the resulting shift from cytoplasmic to nuclear p27 may translate into substantial clinical benefit, due to reduced cellular invasiveness and metastatic potential, while restoration of nuclear p27 action would inhibit tumor proliferation. Establishing reliable predictors of therapeutic response in real-time will dramatically enhance the clinician’s ability to diagnose and treat formerly intractable cancers.
Acknowledgments
The Doris Duke Charitable Foundation supports J.M.S. and S.A.W. NCI grant R01CA105118 supports J.M.S. and D.Z. The authors wish to thank Sandra Williams for technical assistance with the manuscript. We also thank lab members for useful discussions.
Reference List
- 1.Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–9. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- 2.Polyak K, Kato JY, Solomon MJ, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 3.Slingerland JM, Hengst L, Pan CH, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol Cell Biol. 1994;14:3683–94. doi: 10.1128/mcb.14.6.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larrea MD, Wander SA, Slingerland JM. p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle. 2009;8:3455–61. doi: 10.4161/cc.8.21.9789. [DOI] [PubMed] [Google Scholar]
- 5.LaBaer J, Garrett MD, Stevenson LF, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–62. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 6.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–67. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 7.Kiyokawa H, Kineman RD, Manova-Todorova KO, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell. 1996;85:721–32. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 8.Nakayama K, Ishida N, Shirane M, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–20. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 9.Fero ML, Rivkin M, Tasch M, et al. A syndrome of multi-organ hyperplasia with features of gigantism, tumorigenesis and female sterility in p27Kip1-deficient mice. Cell. 1996;85:733–44. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 10.Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–4. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- 11.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 12.Boehm M, Yoshimoto T, Crook MF, et al. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connor MK, Kotchetkov R, Cariou S, et al. CRM1/Ran-Mediated Nuclear Export of p27(Kip1) Involves a Nuclear Export Signal and Links p27 Export and Proteolysis. Mol Biol Cell. 2003;14:201–13. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamura T, Hara T, Matsumoto M, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6:1229–35. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 15.Chu I, Sun J, Arnaout A, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128:281–94. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimmler M, Wang Y, Mund T, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–80. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 17.Larrea MD, Liang J, Da ST, et al. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol. 2008;28:6462–72. doi: 10.1128/MCB.02300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James M, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2007;28:498–510. doi: 10.1128/MCB.02171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000;183:10–7. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 20.Nagahara H, Vocero-Akbani AM, Synder EL, et al. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–52. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 21.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–76. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Besson A, Hwang HC, Cicero S, et al. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev. 2007;21:1731–46. doi: 10.1101/gad.1556607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18:851–5. doi: 10.1101/gad.1205304. [DOI] [PubMed] [Google Scholar]
- 24.Wu FY, Wang SE, Sanders ME, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66:2162–72. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- 25.See WL, Heinberg AR, Holland EC, Resh MD. p27 deficiency is associated with migration defects in PDGF-expressing gliomas in vivo. Cell Cycle. 2010:9. doi: 10.4161/cc.9.8.11259. [DOI] [PubMed] [Google Scholar]
- 26.van der Heijden MS, Bernards R. Inhibition of the PI3K pathway: hope we can believe in? Clin Cancer Res. 2010;16:3094–9. doi: 10.1158/1078-0432.CCR-09-3004. [DOI] [PubMed] [Google Scholar]
- 27.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–45. [PubMed] [Google Scholar]
- 28.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–72. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 29.Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 30.Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–52. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 31.Viglietto G, Motti ML, Bruni P, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–44. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 32.Liang J, Shao SH, Xu Z, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 33.Kossatz U, Vervoorts J, Nickeleit I, et al. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. 2006;25:5159–70. doi: 10.1038/sj.emboj.7601388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larrea MD, Hong F, Wander SA, et al. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci USA. 2009;106:9268–73. doi: 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Jonasch E, Alexander A, et al. Cytoplasmic sequestration of p27 via AKT phosphorylation in renal cell carcinoma. Clin Cancer Res. 2009;15:81–90. doi: 10.1158/1078-0432.CCR-08-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motti ML, Califano D, Troncone G, et al. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol. 2005;166:737–49. doi: 10.1016/S0002-9440(10)62295-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min YH, Cheong JW, Kim JY, et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004;64:5225–31. doi: 10.1158/0008-5472.CAN-04-0174. [DOI] [PubMed] [Google Scholar]
- 38.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 39.Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–11. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–85. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 41.Sebolt-Leopold JS. Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin Cancer Res. 2008;14:3651–6. doi: 10.1158/1078-0432.CCR-08-0333. [DOI] [PubMed] [Google Scholar]
- 42.Yang HY, Zhou BP, Hung MC, Lee MH. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J Biol Chem. 2000;275:24735–9. doi: 10.1074/jbc.C000147200. [DOI] [PubMed] [Google Scholar]
- 43.Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276:40888–95. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- 44.Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci USA. 1998;95:1091–6. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Sun Y, Ehrlich M, et al. Disruption of TGF-beta growth inhibition by oncogenic ras is linked to p27Kip1 mislocalization. Oncogene. 2000;19:5926–35. doi: 10.1038/sj.onc.1203991. [DOI] [PubMed] [Google Scholar]
- 46.Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20:47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kfir S, Ehrlich M, Goldshmid A, Liu X, Kloog Y, Henis YI. Pathway- and expression level-dependent effects of oncogenic N-Ras: p27(Kip1) mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol Cell Biol. 2005;25:8239–50. doi: 10.1128/MCB.25.18.8239-8250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delmas C, Manenti S, Boudjelal A, Peyssonnaux C, Eychene A, Darbon JM. The p42/p44 mitogen-activated protein kinase activation triggers p27Kip1 degradation independently of CDK2/cyclin E in NIH 3T3 cells. J Biol Chem. 2001;276:34958–65. doi: 10.1074/jbc.M101714200. [DOI] [PubMed] [Google Scholar]
- 49.Lane HA, Beuvink I, Motoyama AB, Daly JM, Neve RM, Hynes NE. ErbB2 potentiates breast tumor proliferation through modulation of p27(Kip1)-Cdk2 complex formation: receptor overexpression does not determine growth dependency. Mol Cell Biol. 2000;20:3210–23. doi: 10.1128/mcb.20.9.3210-3223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010;16:3526–32. doi: 10.1158/1078-0432.CCR-09-1834. [DOI] [PubMed] [Google Scholar]
- 51.Chu S, McDonald T, Bhatia R. Role of BCR-ABL-Y177-mediated p27kip1 phosphorylation and cytoplasmic localization in enhanced proliferation of chronic myeloid leukemia progenitors. Leukemia. 2010 doi: 10.1038/leu.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Catzavelos C, Bhattacharya N, Ung YC, et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. 1997:227–30. doi: 10.1038/nm0297-227. [DOI] [PubMed] [Google Scholar]
- 53.Han S, Park K, Kim HY, Lee MS, Kim HJ, Kim YD. Reduced expression of p27Kip1 protein is associated with poor clinical outcome of breast cancer patients treated with systemic chemotherapy and is linked to cell proliferation and differentiation. Breast Cancer Res Treat. 1999;55:161–7. doi: 10.1023/a:1006258222233. [DOI] [PubMed] [Google Scholar]
- 54.Sui L, Dong Y, Ohno M, et al. Implication of malignancy and prognosis of p27(kip1), Cyclin E, and Cdk2 expression in epithelial ovarian tumors. Gynecol Oncol. 2001;83:56–63. doi: 10.1006/gyno.2001.6308. [DOI] [PubMed] [Google Scholar]
- 55.Korkolopoulou P, Vassilopoulos I, Konstantinidou AE, et al. The combined evaluation of p27Kip1 and Ki-67 expression provides independent information on overall survival of ovarian carcinoma patients. Gynecol Oncol. 2002;85:404–14. doi: 10.1006/gyno.2002.6627. [DOI] [PubMed] [Google Scholar]
- 56.Yatabe Y, Nakamura S, et al. p27Kip1 in human lung cancers: differential changes in small cell and non-small cell carcinomas. Cancer Res. 1998;58:1042–7. [PubMed] [Google Scholar]
- 57.Tsukamoto S, Sugio K, Sakada T, Ushijima C, Yamazaki K, Sugimachi K. Reduced expression of cell-cycle regulator p27(Kip1) correlates with a shortened survival in non-small cell lung cancer. Lung Cancer. 2001;34:83–90. doi: 10.1016/s0169-5002(01)00216-1. [DOI] [PubMed] [Google Scholar]
- 58.Hirabayashi H, Ohta M, Tanaka H, et al. Prognostic significance of p27KIP1 expression in resected non-small cell lung cancers: analysis in combination with expressions of p16INK4A, pRB, and p53. J Surg Oncol. 2002;81:177–84. doi: 10.1002/jso.10176. [DOI] [PubMed] [Google Scholar]
- 59.Vis AN, Noordzij MA, Fitoz K, Wildhagen MF, Schroder FH, van der Kwast TH. Prognostic value of cell cycle proteins p27(kip1) and MIB-1, and the cell adhesion protein CD44s in surgically treated patients with prostate cancer. J Urol. 2000;164:2156–61. [PubMed] [Google Scholar]
- 60.Tsihlias J, Kapusta LR, DeBoer G, et al. Loss of cyclin-dependent kinase inhibitor p27Kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res. 1998;58:542–8. [PubMed] [Google Scholar]
- 61.Yang RM, Naitoh J, Murphy M, et al. Low p27 expression predicts poor disease-free survival in patients with prostate cancer. J Urol. 1998;159:941–5. [PubMed] [Google Scholar]
- 62.Cote RJ, Shi Y, Groshen S, et al. Association of p27Kip1 levels with recurrence and survival in patients with stage C prostate carcinoma. J Natl Cancer Inst. 1998;90:916–20. doi: 10.1093/jnci/90.12.916. [DOI] [PubMed] [Google Scholar]
- 63.Kuczyk MA, Bokemeyer C, Hartmann J, et al. Predictive value of altered p27Kip1 and p21WAF/Cip1 protein expression for the clinical prognosis of patients with localized prostate cancer. Oncol Rep. 2001;8:1401–7. doi: 10.3892/or.8.6.1401. [DOI] [PubMed] [Google Scholar]
- 64.Porter PL, Malone KE, Heagerty PJ, et al. Expression of cell cycle regulators p27kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med. 1997;3:222–5. doi: 10.1038/nm0297-222. [DOI] [PubMed] [Google Scholar]
- 65.Tan P, Cady B, Wanner M, et al. The cell cycle inhibitor p27 is an independent prognostic marker in small (T1a,b) invasive breast carcinomas. Cancer Res. 1997;57:1259–63. [PubMed] [Google Scholar]
- 66.Han S, Park K, Bae BN, et al. Prognostic implication of cyclin E expression and its relationship with cyclin D1 and p27Kip1 expression on tissue microarrays of node negative breast cancer. J Surg Oncol. 2003;83:241–7. doi: 10.1002/jso.10268. [DOI] [PubMed] [Google Scholar]
- 67.Porter PL, Barlow WE, Yeh IT, et al. p27(Kip1) and cyclin E expression and breast cancer survival after treatment with adjuvant chemotherapy. J Natl Cancer Inst. 2006;98:1723–31. doi: 10.1093/jnci/djj467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu J, hen ZZ, u J. Prognostic role of p27Kip1 and apoptosis in human breast cancer. Br J Cancer. 1999;79:1572–8. doi: 10.1038/sj.bjc.6690250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nohara T, Ryo T, Iwamoto S, Gon G, Tanigawa N. Expression of cell-cycle regulator p27 is correlated to the prognosis and ER expression in breast carcinoma patients. Oncology. 2001;60:94–100. doi: 10.1159/000055303. [DOI] [PubMed] [Google Scholar]
- 70.Pohl G, Rudas M, Dietze O, et al. High p27Kip1 expression predicts superior relapse-free and overall survival for premenopausal women with early-stage breast cancer receiving adjuvant treatment with tamoxifen plus goserelin. J Clin Oncol. 2003;21:3594–600. doi: 10.1200/JCO.2003.02.021. [DOI] [PubMed] [Google Scholar]
- 71.Li R, Wheeler TM, Dai H, et al. Biological correlates of p27 compartmental expression in prostate cancer. J Urol. 2006;175:528–32. doi: 10.1016/S0022-5347(05)00151-5. [DOI] [PubMed] [Google Scholar]
- 72.Piva R, Cancelli I, Cavalla P, et al. Proteasome-dependent degradation of p27/kip1 in gliomas. J Neuropathol Exp Neurol. 1999;58:691–6. doi: 10.1097/00005072-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 73.Hidaka T, Hama S, Shrestha P, et al. The combination of low cytoplasmic and high nuclear expression of p27 predicts a better prognosis in high-grade astrocytoma. Anticancer Res. 2009;29:597–603. [PubMed] [Google Scholar]
- 74.Nahta R, Hung MC, Esteva FJ. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004;64:2343–6. doi: 10.1158/0008-5472.can-03-3856. [DOI] [PubMed] [Google Scholar]
- 75.Busse D, Doughty RS, Ramsey TT, et al. Reversible G(1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J Biol Chem. 2000;275:6987–95. doi: 10.1074/jbc.275.10.6987. [DOI] [PubMed] [Google Scholar]
- 76.D’Alessio A, De LA, Maiello MR, et al. Effects of the combined blockade of EGFR and ErbB-2 on signal transduction and regulation of cell cycle regulatory proteins in breast cancer cells. Breast Cancer Res Treat. 2010;123:387–96. doi: 10.1007/s10549-009-0649-x. [DOI] [PubMed] [Google Scholar]
- 77.Kortylewski M, Heinrich PC, Kauffmann ME, Bohm M, Mackiewicz A, Behrmann I. Mitogen-activated protein kinases control p27/Kip1 expression and growth of human melanoma cells. Biochem J. 2001;357:297–303. doi: 10.1042/0264-6021:3570297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lefevre G, Calipel A, Mouriaux F, Hecquet C, Malecaze F, Mascarelli F. Opposite long-term regulation of c-Myc and p27Kip1 through overactivation of Raf-1 and the MEK/ERK module in proliferating human choroidal melanoma cells. Oncogene. 2003;22:8813–22. doi: 10.1038/sj.onc.1207099. [DOI] [PubMed] [Google Scholar]
- 79.Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer Res. 2005;65:4870–80. doi: 10.1158/0008-5472.CAN-04-2848. [DOI] [PubMed] [Google Scholar]
- 80.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harrington LS, Findlay GM, Gray A, et al. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu K, Toral-Barza L, Shi C, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–40. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 84.Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci USA. 2000;97:9042–6. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y, Guggisberg N, Jorda M, et al. Combined Src and Aromatase Inhibition Impairs Human Breast Cancer Growth In vivo and Bypass Pathways Are Activated in AZD0530-Resistant Tumors. Clin Cancer Res. 2009;15:3396–405. doi: 10.1158/1078-0432.CCR-08-3127. [DOI] [PubMed] [Google Scholar]
- 88.Chen Y, Alvarez EA, Azzam D, et al. Combined Src and ER blockade impairs human breast cancer proliferation in vitro and in vivo. Breast Cancer Research & Treatment. 2010 doi: 10.1007/s10549-010-1024-7. In press. [DOI] [PubMed] [Google Scholar]
- 89.Belsches-Jablonski AP, Biscardi JS, Peavy DR, Tice DA, Romney DA, Parsons SJ. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene. 2001;20:1465–75. doi: 10.1038/sj.onc.1204205. [DOI] [PubMed] [Google Scholar]
- 90.Chu I, Arnaout A, Loiseau S, et al. Src promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancer. J Clin Invest. 2007;117:2205–15. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hiscox S, Jordan NJ, Smith C, et al. Dual targeting of Src and ER prevents acquired antihormone resistance in breast cancer cells. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-0058-6. [DOI] [PubMed] [Google Scholar]
- 92.Psyrri A, Bamias A, Yu Z, et al. Subcellular localization and protein levels of cyclin-dependent kinase inhibitor p27 independently predict for survival in epithelial ovarian cancer. Clin Cancer Res. 2005;11:8384–90. doi: 10.1158/1078-0432.CCR-05-1270. [DOI] [PubMed] [Google Scholar]
- 93.Masciullo V, Ferrandina G, Pucci B, et al. p27Kip1 expression is associated with clinical outcome in advanced epithelial ovarian cancer: multivariate analysis. Clin Cancer Res. 2000;6:4816–22. [PubMed] [Google Scholar]