

Abstract
Reverse genetics consists in the modification of the activity of a target gene to analyse the phenotypic consequences. Four main approaches are used toward this goal and will be explained in this review. Two of them are centered on genome alterations. Mutations produced by random chemical or insertional mutagenesis can be screened, to recover only mutants in a specific gene of interest. Alternatively, these alterations may be specifically targeted on a gene of interest by homologous recombination. The two other approaches are centered on mRNA. RNA interference is a powerful method to reduce the level of gene products, while morpholino antisense oligonucleotides alter mRNA metabolism or translation. Some model species, such as Drosophila, are amenable to most of these approaches, whereas other model species are restricted to one of them. For example, in mice and yeasts gene targeting by homologous recombination is prevalent, while in Xenopus and zebrafish morpholino oligonucleotides are mainly used. Genome-wide collections of mutants or inactivated models obtained in several species by these approaches have been made and will help decipher gene functions in the post-genome era.
Keywords: Animals; Animals, Genetically Modified; Gene Targeting; Genome; Mice; Models, Genetic; Oligonucleotides, Antisense; genetics; RNA Interference; Recombination, Genetic; Zebrafish
Keywords: Mutation, Gene targeting, phenotype, knock-down, knock-out
Introduction
Genetics relies on mutants. In conventional or “forward” genetics, naturally occurring or artificially induced mutants are first identified on the basis of their specific appearances or properties - their phenotypes. Then they are characterized by addressing several questions such as: (i), How is the mutant phenotype transmitted throughout generations? (ii), Is the mutant phenotype due to a mutation in only one or several genes? (iii), What is the phenotype of the progeny of two different mutants? Ultimately, the gene(s) that confer(s) the phenotype of interest is (are) identified. Forward genetics, which essentially goes from the phenotype to the gene, has been an active field of research for more than one-hundred years, and huge collections of spontaneous or induced mutants in several species have accumulated throughout the 20th century. Even now, trying to identify the genes whose mutations are causal in phenotypes such as complex human diseases remains a challenging goal, although the recent technology breakthrough of massively parallel sequencing associated to the capture of the mapped region should strongly lower the barriers to identify such mutations.
“Reverse” genetics has emerged more recently in the beginning of the 1980s, with the development of molecular tools that allowed considering an alternative genetic process. In reverse genetics, the starting point is the gene. A “reverse geneticist” aims to specifically modify a gene, or its expression, and characterize the phenotypic consequences of this modification. Since the late 1990s, tens of eukaryotic genomes have been sequenced. Genome-wide sequencing revealed a large number of genes whose functions are unknown and cannot be predicted. Assigning functions to these genes can be achieved through several convergent approaches: identifying the subcellular localisations of gene products and the molecular complexes to which they belong, but also describing the phenotypes associated with their manipulation. High-throughput reverse genetics approaches are therefore major tools in the post-genomic era. In the present review, we will present different strategies aimed at specifically manipulating genes or gene expression in eukaryotes. Table 1 summarizes the strategies that can be used in several model species.
Table 1. Overview of reverse genetics approaches in common laboratory species.
The prevalence of every approach in the indicated model species is marked by - (no published report) to +++ (Genome-wide inactivations reported). See main text for references.
| Yeast | A.thaliana | C. elegans | Fruit fly | Zebrafish | Xenopus | Mouse | |
|---|---|---|---|---|---|---|---|
| Insertion mutagenesis | − | +++ Genome-wide collection of T-DNA mutants |
+++ Genome-wide collection of Mos1 mutants in progress |
+++ Genome-wide collection of P element and piggyBac mutants |
+ Proofs-of-principle for retrovirus and transposon-mediated mutagenesis |
+/− | +++ Genome-wide collection of gene-trap ES cells in progress |
| Random chemical mutation and screening | − (used in forward, not reverse genetics) |
+++ Genome-wide collection of TILLING mutants, service |
+ Middle-scale collection of TILLING mutants |
++ Service for the research community |
++ Service for the research community |
+/− Proof-of-principle |
− |
| Homologous recombination | +++ Genome-wide collection of KO and KI mutants |
− | +/− Few examples |
+ Several examples, but no systematic approach |
− | − | +++ Genome-wide collection of cKO ES cells in progress |
| Inserted element mediated gene engineering | − | − | ++ MosTIC, MosSCI, MosDEL |
+ Transposon-controlled deletions |
− | − | − |
| RNAi | − | ++ | +++ Genome-wide “feeding library” |
+++ Genome-wide UAS-driven library |
− | − | +/− Few examples |
| Morpholino | − | − | − | − | ++ Middle-scale screening |
++ Middle-scale screening |
1. Methods derived from forward genetics: random modifications of the genome and screening
A strategy to obtain stable mutants of a gene of interest is by random mutagenesis and screening of the progeny. Mutagenesis is achieved by either the insertion of a piece of DNA (to induce insertion mutations) or by a chemical compound (to induce deletions or point mutations). A gene screening is then performed in the progeny to identify, among hundreds of others, the individuals for which mutations occurred by chance in the gene of interest. Although related to forward genetics, this strategy differs from it at the screening step.
1.1. Insertional mutagenesis
Exogenous pieces of DNA, such as transposons* or retroviruses, can randomly insert in a genome. If the insertion takes place within a gene, this can be mutagenic in several ways. However, this strategy generally leads to an incomplete loss of gene function and most insertion mutants correspond to hypomorphic alleles* but not null alleles*. Indeed, insertion within the promoter is likely to modify, but seldom completely inhibit, the expression of the gene product. In the same way, if the RNA encoded by the inserted DNA is flanked by an upstream acceptor splice site and a downstream polyadenylation site, it behaves like a 3′ terminal exon. Hence, insertion within an intron results in the synthesis of a truncated mRNA and a truncated protein. This gene-trap* approach also generally leads to an incomplete gene inactivation because a reduced amount of wild-type mRNA is still likely to be synthesized from the mutated gene (by skipping the gene-trap cassette). Finally, insertion within the coding sequence that disrupts the open reading frame results in the synthesis of a truncated protein with no or decreased activity.
Several strategies exist to obtain deletion mutants (generally null) from existing insertion mutants. If the mutant phenotype is a consequence of the insertion of a transposon, this one can be “mobilized” (excised) by expressing an appropriate transposase, resulting in restoration of the wild-type phenotype. However, mobilization of the transposon may also result in the deletion of the genomic regions initially flanking the transposon by a process referred to as imprecise mobilization. Hence, deletion mutants may be isolated from screening the progeny of individuals where mobilization occurred. Alternative strategies to obtain deletion mutant from insertional mutants rely on homologous recombination in Caenorhabditis elegans (See paragraph 2.3), and FLP recombinase in flies (Parks et al., 2004).
Large collections of insertion mutants were generated in several model species. Regions flanking the insertion sites were characterized by systematic sequencing (Figure 1A). Hence, a researcher who needs a mutant of a given gene would first query a database of insertion sites to try to find the gene. If no such mutant is available, screening new mutants can be considered. This is achieved by PCR using primers localized in the gene of interest and in the inserted DNA. An amplimere is produced only if the exogenous DNA is inserted within the gene (Figure 1B). To gain efficiency, DNAs from pools are analysed, and only members of positive pools are individually analysed (Krysan et al., 1999).
Figure 1. Screening for random mutations.
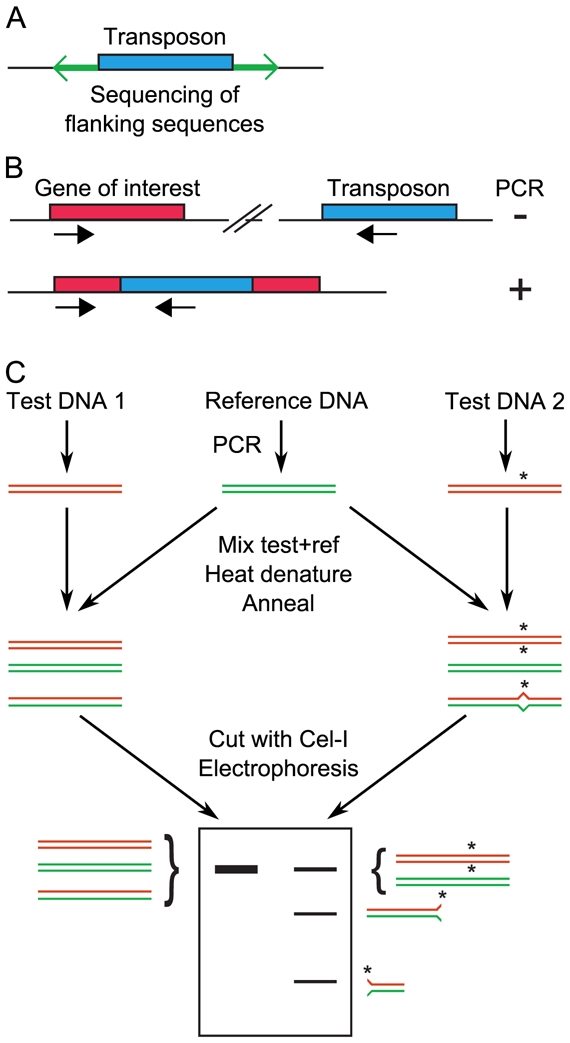
A, Large collections of insertion mutants are generated. For each mutant, the regions flanking the transposon are sequenced, to identify the site of insertion. B, After transposon insertional mutagenesis, screening is achieved by PCR using one primer that hybridizes in the transposon and another that hybridizes in the gene of interest. A PCR product will be obtained only if the transposon is inserted by chance in the gene of interest. To increase throughput, mutants are first tested in pools, then individually for mutants that belong to positive pools. C, TILLING. Regions of interest are simultaneously PCR-amplified from a reference (wild-type) individual and several mutants. Mutant and wild-type amplimeres are mixed together, denatured and reannealed. If a mutation was present in the amplified region of the mutant, then a heteroduplex with one mismatch forms. This heteroduplex is detected by the increased electrophoretic mobility after cleavage by the endonuclease Cel-I.
In the plant Arabidopsis thaliana (A. thaliana), a collection of Agrobacterium transferred DNA (T-DNA*) mediated insertion mutants was described. Mutations were found in more than two-thirds of the genes (Alonso et al., 2003). Together with other independent collections (Rosso et al., 2003) there exist T-DNA mutants in a very large majority of A. thaliana genes.
In C. elegans, initial collections used the Tc1 transposon (Zwaal et al., 1993). A drawback of this endogenous transposon is that it is present in multiple copies in every strain. Nowadays, a large collection of insertion mutants is being generated with the heterologous transposon Mos1 (from fly origin), that is uniquely inserted in every strain (Bazopoulou and Tavernarakis, 2009; Duverger et al., 2007; Granger et al., 2004).
In zebrafish, insertional mutagenesis is achieved with two types of vectors, retroviruses and transposons [reviewed in (Sivasubbu et al., 2007)]. For example, a collection of 525 mutants (390 genes) was generated using a retroviral vector (Amsterdam et al., 2004). Several transposon-mediated gene-trap and enhancer-trap* approaches were also published (Sivasubbu et al., 2007). In principle, inserting a transposon within a gene might result in insertional mutagenesis of the gene. Indeed, 2 lines (out of 14) that were generated using an engineered (“gene-breaking”) transposon showed mutant phenotypes (Sivasubbu et al., 2006) and it was also the case for 2 lines (out of 73) that were generated using the Tol2 transposon (Nagayoshi et al., 2008). The recent identification of mutants of interest after gene-breaking transposon mutagenesis and phenotypic screening (Petzold et al., 2009) suggests that large-scale screening following insertional mutagenesis can be considered and that large collections will ultimately be available. Insertional mutagenesis was also described in Xenopus (Yergeau and Mead, 2009) but, again, no large collection has been developed yet.
Collections of insertion mutants were also developed in flies and mice (Table 1). In Drosophila, the Berkeley Drosophila Genome Project generated a collection of P element (a transposon) insertion strains. Independent strains corresponding to about one-thousand of different genes were reported in the initial article (Spradling et al., 1999), and 5000 genes 5 years later (Bellen et al., 2004). Other collections were generated with alternative transposons, like piggyBac (Thibault et al., 2004). In mice, a collection of 100,000 independent gene-trap ES cells (Embryonic stem cells) was generated, and it is expected that such a collection will eventually cover all the mouse genes (Collins et al., 2007b; Nord et al., 2006). Engineered ES cells are stored frozen, and are the basis to obtain mutant mice within some months by injecting these totipotent cells into a host embryo at the blastula stage that is reimplanted in pseudogestant females ES cells may then contribute to all the organs of the developing embryo including germ cells. The resulting mouse is named a “chimera” because its cells originate from either the host embryo or the ES cells. If germ cells derived from the ES cells participate in fertilization they will transmit their genotype to the next generation and consequently, a mouse bearing homozygous alleles from the ES cells can be readily obtained from the in vitro cultured cells in only two generations [reviewed in (Capecchi, 2005)].
1.2. Random chemical modifications of the genome and screening
Chemical or physical treatments that generate point mutations or deletions are an alternative to transposons to induce random mutations of a genome. Deletions are relatively easy to screen by PCR using several primers hybridizing to a gene of interest. Mutants are evidenced by a shorter PCR product (if the deletion is flanked by the primers) or an absence of PCR product (if the region hybridizing with a primer is deleted) (Gengyo-Ando and Mitani, 2000; Jansen et al., 1997). In C. elegans, throughput can be increased by performing worm culture, DNA preparation and PCR in microtiter plates. Furthermore, pools of DNA, rather than individual DNAs, are screened for the appearance of shorter PCR products, so that roughly 4000 genomes are examined in a single 96-well plate (Liu et al., 1999). A North-american consortium (http://celeganskoconsortium.omrf.org/) and a Japanese consortium (http://www.shigen.nig.ac.jp/c.elegans/index.jsp) are generating deletion alleles for the C. elegans community.
To specifically generate and identify point mutations within one gene of interest, a method named TILLING (Targeting induced local lesions in genomes) was developed (McCallum et al., 2000). Regions of interest are simultaneously PCR-amplified from a reference (non-mutated) DNA and several test DNAs. Test amplimeres are individually mixed with the reference amplimere, denatured and annealed. If a point mutation was present within a test DNA, then duplexes with one mismatch will form. Initially, these duplexes were detected because they presented altered retention times in denaturing HPLC (McCallum et al., 2000). To gain both throughput and accuracy, an alternative method of screening was proposed that relies on the endonuclease Cel-I that specifically cleaves duplexes with one mismatch (Colbert et al., 2001). After electrophoresis, test DNAs with one mutation within the amplified region are therefore characterized by the appearance of shorter fragments (cut by Cel-I, Figure 1C) the size of which is a first indication of the site of the mutation. Throughput is increased by pooling test DNAs, before individually analysing every DNA from a positive pool. Recently, the screening step of tomato mutants was made by PCR amplification of a gene of interest from a large collection of mutated DNAs and massively parallel sequencing (Rigola et al., 2009), and similar deep sequencing might facilitate screening for point mutations in virtually any organism.
Collections of TILLING mutants were developed in A. thaliana (Till et al., 2003), C. elegans (Gilchrist et al., 2006), zebrafish (Sood et al., 2006; Wienholds et al., 2003), and Drosophila melanogaster (Cooper et al., 2008; Winkler et al., 2005). In Xenopus tropicalis, a proof-of-principle for TILLING was published (Goda et al., 2006). To our knowledge, no TILLING has been reported in mice. A service of TILLING was developed for the scientific community in A. thaliana, fly (http://tilling.fhcrc.org/) and zebrafish (http://www.sanger.ac.uk/Projects/D_rerio/mutres).
1.3. Limitations of random mutagenesis
As seen above, mutations by insertion of a piece of exogenous DNA results in more or less severe alleles. To obtain a mutant with desired characteristics, it may therefore be required to screen a huge number of individuals with no guaranty that a null mutant will eventually be obtained. In addition transposons also display non completely-random insertion profiles because of preference for integration sites. It results that some loci are hot spots of insertions while others may be refractory to integration. Another limitation of insertion mutagenesis in mice is that the mutants are constitutive*, which is much less powerful than the conditional* mutants that will be described below (Paragraph 2.5). In addition, a recent article reported unexpected gene alterations in one of the mouse ES lines, questioning the capacity of the gene-trap approach to easily obtain mutated mice (Vermeire et al., 2009).
The TILLING approach requires a high amount of mutagen. The reason for this is that a low amount of mutagen would result in a low probability of obtaining mutants of the gene of interest and this would imply a too heavy screening. However, massive mutagenesis causes concomitant mutations all over the genome in addition to the gene of interest. Hence, once an individual with a mutation in a gene of interest is isolated, it has to be outcrossed* several times to remove the accompanying mutations. For example, a high-density collection of 3712 A. thaliana mutants was recently developed. Each line carries a mutation on average every 89 kb, and it would be extremely difficult to recover a strain carrying a single mutation from this collection (Martin et al., 2009).
Finally, a practical issue with large collections of mutants is their storage. Whereas it is quite easy to store plant seeds, or to store frozen animals whenever possible (C. elegans), many animals lack a convenient storage form.
2. Targeted modifications of the genome by homologous recombination
2.1. Strategies and designs of homologous recombination
Homologous recombination (HR), the process that generates crossover exchanges between non-sister chromatids upon meiosis, is one of the main pathways used to repair potentially deleterious DNA double strand breaks (DSB) [reviewed in (O’Driscoll and Jeggo, 2006)]. It also permits gene engineering at specific chromosomal loci. The original procedure in the yeast Saccharomyces cerivisiae (S. cerevisiae) was the specific disruption of a gene by insertion of a selection marker within its encoding region (Hinnen et al., 1978). This generated a null allele of that gene. Today, thousands of genes have been invalidated by this way essentially in yeasts, mice and flies and the resulting cells or organisms are described as knock-out (KO, Figure 2A).
Figure 2. Diversity of gene modifications achieved by homologous recombination.
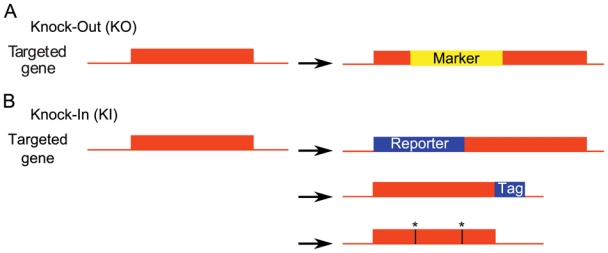
A, Knock-Out (KO). The targeted gene is disrupted by the insertion of a selection marker. B, In Knock-In (KI), a sequence of interest is inserted within the targeted gene. If a reporter gene is inserted at an appropriate place in the targeted gene, this results in a disruption of the targeted gene which generates a null allele, very much like KO (upper panel). Alternatively, the inserted sequence may be a tag (middle panel). The spatio-temporal expression of the tagged protein is the same as that of the wild-type protein in control cells. Finally, the wild-type allele may be replaced by an engineered, mutant allele (stars, lower panel).
HR can also be used to produce more subtle modifications of a gene by substituting an in vitro engineered sequence for the endogenous allele. This approach is referred to as knock-in (KI). A first application of KI strategies is to insert a reporter gene within the endogenous allele. This creates a null allele by interrupting the reading frame of the targeted gene (similar to a KO), and this allows an accurate description of gene expression pattern [e.g. (Kress et al., 2007)] since the expression of the reporter gene is controlled by the promoter of the targeted gene (Figure 2B upper panel).
A second application of KI is to insert a tag-encoding sequence in frame with an endogenous ORF (Figure 2B middle panel). While valid in mice (Zhou et al., 2004), this approach is efficiently used only in yeasts where several tags were fused to proteins on a genomic scale. For example, the subcellular localization of yeast proteins in living cells was systematically determined by GFP tagging and fluorescence microscopy (Huh et al., 2003). Several fused tags correspond to epitopes* that are recognized by commercial monoclonal antibodies, allowing efficient detection and/or purification of the fusion protein. For example, the V5 epitope was fused to ~60% of the ORFs, and the subcellular localization of the corresponding proteins was determined by immunohistochemistry with anti-V5 antibodies (Kumar et al., 2002). A widely used epitope in immunoprecipitations is Flag. Systematic “Flaging” of yeast ORFs, followed by immunoprecipitation with anti-Flag antibodies and identification of co-immunoprecipitated proteins by mass spectroscopy allowed global characterization of yeast protein complexes (Ho et al., 2002). An alternative to Flag epitope to achieve low-background immunoprecipitations is the “Tap-tag” (Tap, tandem affinity purification) (Puig et al., 2001; Rigaut et al., 1999). The Tap-tag consists of two peptides, encoding Calmodulin-binding peptide and Protein A, which permits highly selective isolation of fusion proteins and their partners. As for Flag-epitope tagging, systematic tap-tagging of yeast ORFs allowed the identification of protein complexes (Gavin et al., 2002; Krogan et al., 2006). In addition, thanks to the presence of the Protein A moiety that interacts with immunoglobulins, Tap-tagging also allowed a systematic measurement of protein expression levels in different conditions by Western blotting (Ghaemmaghami et al., 2003).
A third application of KI strategies is to replace the wild-type targeted gene by a mutated or orthologous allele* (Figure 2B lower panel). This is a way to study the basic consequence of a gene mutation described in human. For example, the study of two cancer associated Polymerase δ mutations in the yeast model showed that these mutations are lethal through a catastrophic increase in genome instability (Daee et al., 2009). KI strategies are also promising in the study of evolution. For instance, exchanging the PRX1 gene regulatory region between bat and mice identified a region of this gene responsible for limb length (Cretekos et al., 2008). Another example is given by the mouse Foxp2 gene, whose humanization affected cortico-basal ganglia functions so that the generated mice harbored qualitatively different ultrasonic vocalizations that are in agreement with the potential involvement of this gene in the evolution of speech in human (Enard et al., 2009).
HR-mediated gene engineering is achieved via targeting vectors. Since HR is a low frequency event, the targeting vectors include a (positive) marker aimed at selecting or rapidly identifying the cells or organisms that underwent HR. Generally, selection markers complement an auxotrophic mutation* in yeasts, confer a resistance to a specific antibiotic in the murine cells where HR is achieved (see paragraph 2.5), or confer an easily recognizable phenotype in flies or worms.
Initially, targeted insertions in S. cerevisiae were performed with a circular DNA targeting vector. Such a vector is fully integrated at the homologous locus by a single HR event leading to a duplication of the targeted gene (Figure 3A). A subsequent intrachromosomal recombination between the two copies of the targeted gene (similar to that shown in Figure 3D) can lead either to the recovery of the initial sequence of the targeted gene or to the substitution by the exogenous sequence. Thereafter, it was shown in yeasts that linearization of the circular DNA vector within the exogenous target gene increases strongly the integration process by enhancing the recombination frequencies (Orr-Weaver and Szostak, 1983). Such linearized DNA molecules were designed as “ends-in” (Hastings et al., 1993) because the ends point toward each other when aligned with the endogenous targeted sequence (Figure 3B). Like circular DNA, ends-in molecules lead to the duplication of the gene at the targeted locus. However, gene inactivation can be achieved with the ends-in design, if the targeting gene is designed to be mutated in both the 5′ and 3′ regions. In that case, both duplicated genes are also 5′ and 3′ mutated and inactive.
Figure 3. Designs for gene targeting by homologous recombination.
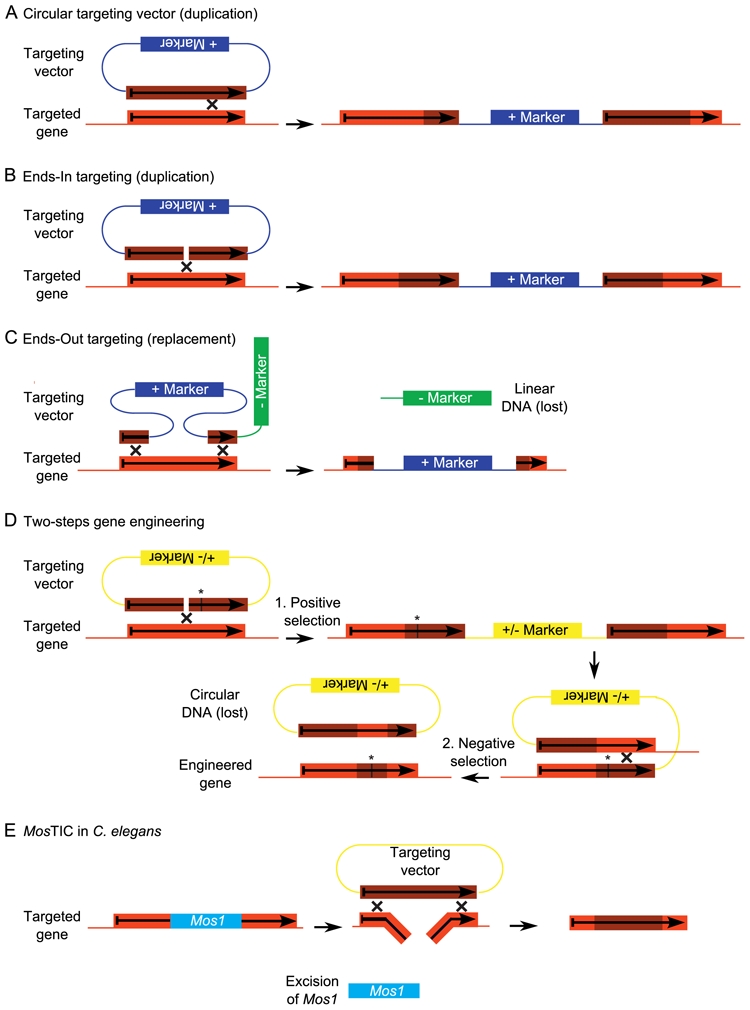
Recombination (cross) occurs at any place within the region of homology designated by an orange box. Throughout the figure, light and dark orange colours represent the targeted and the targeting gene respectively. A, With a circular targeting vector, recombination results in a duplication of the gene. The two copies are most often separated by a positive selection marker, which was used to select recombinant individuals. Importantly, if the targeting gene is both 5′ and 3′ truncated, both copies will be either 5′ or 3′ truncated. B, In ends-in, there is a double-strand break within the targeting gene. Homologous recombination preferably takes place at this double-strand break. As for targeting with a circular targeting vector (A), ends-in design yields duplicated genes often separated by a positive selection marker. C, In ends-out, the double-strand breaks flank the targeting gene to generate two recombination events, leading to the disruption of the targeted gene and its replacement by the positive marker. In mice, a negative selection marker is used to counter-select the cells where the targeting gene was inserted in the genome by non-homologous recombination. If the targeting gene is inserted randomly by non-homologous recombination rather than at the appropriate locus by HR, then the negative selection marker is also inserted. D, Subtle gene engineering in yeast cells, like the introduction of point mutations (indicated here by a star), is achieved in two steps. It requires a marker that can be either positively or negatively selected, depending on the culture medium. The first step is the duplication of the targeted gene by ends-in design. The targeting gene contains the appropriate point mutation. The yeast cells that encountered this duplication are positively selected by the selection marker. The second step is a recombination between the two duplicated genes. The yeast cells that did not encounter this recombination are counter-selected by using the selection marker. The consequence of this recombination is that yeast cells retain only one gene. Depending on where the recombination occurred relative to the point mutation, the allele of this gene is either wild-type or mutant (as shown here). E, MosTIC in Caenorhabditis elegans. The excision of the Mos1 transposon generates a DSB in the genome (targeted gene). This DSB is repaired by gene conversion using the targeting plasmid.
An alternative design of the linear DNA molecules in which the ends point outwards when hybridized with the targeted sequence was named “ends-out”. It results in the disruption of the target gene concomitantly with the insertion of the selection marker through a double HR event (Figure 3C). In yeasts, more subtle modifications, like point mutations, can be introduced by a two-steps procedure (Figure 3D). This requires a marker that can be successively positively then negatively selected. For example, yeasts containing the URA3 marker are positively selected in the absence of uracil in the culture medium. However, the same gene product metabolizes 5-fluoroorotic acid into 5-fluorouracil, a toxic compound. Consequently, when the culture medium contains both uracil and 5-fluoroorotic acid, only yeasts devoid of the URA3 marker are able to grow (Boeke et al., 1984).
2.2. Systematic gene disruption in yeast
Gene disruption can be carried out directly with PCR engineered ends-out molecules that consist generally of a selection marker flanked by two short DNA sequences homologous to the DNA target. About 40 nucleotides are sufficient in S. cerevisiae for an efficient HR. This approach was efficient enough to systematically invalidate the ~6200 annotated open reading frames, by substituting the targeted genes from the start to stop codon with a kanMX4 cassette that confers resistance to the drug G418 (Giaever et al., 2002; Winzeler et al., 1999). Analysis of the resulting strains revealed that ~71 % of the genes are not essential for growth on rich glucose medium. The cassettes used to generate the deletion collection contain two distinct 20 nucleotide sequences on both sides that serve as unique “molecular bar codes” to identify and quantify each KO strain in a mixed population. These features allowed a simultaneous analysis of the growth of the mutant collection under different culture conditions (Giaever et al., 2002).
Because yeasts can grow as haploid or diploid cells, they are a unique model to study synthetic-lethal relations between genes. Two mutations are synthetically lethal if cells with either of the single mutations are viable while cells with both mutations are not. Synthetic-lethal phenotypes are diagnostic of a functional interaction between the products of two genes. Combinations of synthetic-lethal mutations can give information about the products needed to fulfill a cellular process. Strategies to seek synthetic-lethal interactions at the genome scale have been developed [reviewed in (Dixon et al., 2009)]. Two deletion collections are needed, in which genes are inactivated by the insertion of cassettes conferring resistance to two different antibiotics. After crossing yeasts from these two collections, the viability of the progeny resistant simultaneously to the two antibiotics (hence, disrupted for the two genes) is measured. In S. cerivisiae, 4000 genetic interactions were tested. Thirty-four of them were synthetic-lethal. By extrapolating to the whole genome, this suggests that 200,000 synthetic-lethal interactions may exist (Tong et al., 2004). Similar conclusions were drawn in Schizosaccharomyces pombe (S. pombe) (Roguev et al., 2008).
2.3. Gene targeting by homologous recombination in Caenorhabditis elegans
In multicellular organisms, targeted modification of the genome by HR is especially useful for the geneticist if it is achieved in germ cells or very early embryos, so that successive matings lead to mutant organisms. In the nematode Caenorhabditis elegans (C. elegans), introduction of linear DNA into meiotic oocyte nuclei results in a low rate of HR events within the targeted gene (Berezikov et al., 2004). HR in C. elegans is more widely associated with transposon-mediated insertional mutagenesis (Table 1). The rationale is that transposon excision generates DSBs that are a site for HR. To target a mutation into a gene of interest, the first step is to get a strain with a transposon inserted within that gene. Then, plasmids containing the targeting gene (with the appropriate modifications) are introduced into these worms by gonadal microinjection. When the transposon is excised, the DSB is potentially repaired by HR using the targeting gene as a template. Consequently, the modifications of the targeting gene are introduced into the genome, a process named gene conversion. As compared with HR in yeast cells or mice, an interesting difference is that the DSB that is required for HR is localised within the targeted gene (due to transposon excision) and not the targeting gene (Figure 3E).
A proof-of-principle for this approach was first brought with endogenous transposons (Barrett et al., 2004). However, in that case transposon excision is a very rare event in germ cells and “mutator” strains (with increased rates of transposition, but also increased rates of background spontaneous mutations) are required. Furthermore, hundreds of copies of the transposons are present in the genome and the frequency of excision of a particular transposon is low. Alternatively, a collection of Mos1 transposon uniquely inserted strains is available (See Paragraph 1.1). Transposon excision is achieved by a transposase whose expression is controlled by an inducible promoter. Hence, transposition is locus-specific and user-controlled. A procedure relying on controlled excision of Mos1 transposon and HR with a targeting transgene was named MosTIC (Mos1 excision-induced transgene instructed gene conversion) (Robert and Bessereau, 2007). Two derivatives of MosTIC were named MosSCI (Mos1-mediated single-copy insertion) and MosDEL (Mos1-mediated deletion). In MosSCI, excision of Mos1 transposon allows single-copy insertion of a transgene (Frokjaer-Jensen et al., 2008). MosDEL is used to generate large (up to 25 kb) targeted deletions (Frokjaer-Jensen et al., 2010). Accurate gene engineering is now possible for most C. elegans genes thanks to these recent approaches and to a large library of Mos1 insertion mutants (Bazopoulou and Tavernarakis, 2009).
2.4. Gene targeting by homologous recombination in Drosophila
As explained above, HR is much more efficiently achieved with linear than with circular DNA molecules. In Drosophila, generating linear DNA in the nuclei of early embryos was challenging. This difficulty was solved a decade ago (Rong and Golic, 2000) by the combination of a site-specific recombinase (SSR) and a rare-cutting endonuclease. SSRs are a family of enzymes that are able to recognize sites on the genome with a high specificity and to ligate two different sites after cleavage. SSR target sites are directional and depending on their relative orientations, SSR activity can have two different consequences. If the sites are in opposite orientations, SSR causes the inversion of the DNA between the sites while sites in the same orientation cause its excision and circularization, leading to the genomic deletion of the sequence located between the sites [Figure 4A; reviewed in (Birling et al., 2009; Feil, 2007)]. The most frequently used SSRs and their sites are listed in Figure 4B. Among them, the yeast FLP recombinase catalyzes recombinations between two FRT (FLP Recombination Targets) sites.
Figure 4. Site-Specific Recombinases (SSRs).
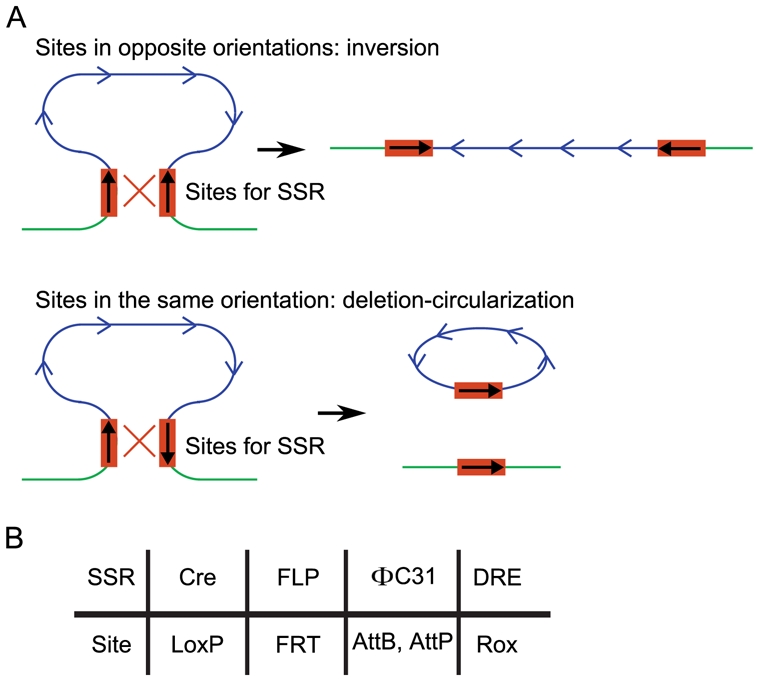
A, If two sites of a SSR are in relative proximity on the DNA in opposite orientations, SSR-mediated recombination between these two sites results in an inversion of the DNA fragment between the two sites (upper panel). If the two sites are in the same orientation, SSR-mediated recombination between these two sites results in an excision of the DNA fragment between the two sites, and the appearance of a circular DNA corresponding to this excised fragment (lower panel). B, SSRs frequently used in gene targeting, and their corresponding sites.
In the pioneer article (Rong and Golic, 2000), the targeting construct consisted of the yellow+ (y+) gene, flanked by two FRT sites in the same orientation. The y+ gene also contained a site for the rare-cutting endonuclease I-SceI. Transgenic flies for this construct were obtained by P-element transgenesis. They were crossed with Drosophila conditionally expressing the FLP recombinase and the I-SceI endonuclease. In early embryos, FLP-mediated recombination between the two FRT sites yielded an intact genomic DNA with one FRT site, and a circular DNA that consisted of the y+ gene and one FRT site (owing to the mechanism shown in Figure 4A, lower panel). The circular DNA was next linearized within the y+ gene (due to I-SceI cleavage). This was expected to drive “ends-in” targeting of the endogenous y locus. This targeting was actually observed at a rate of one for some hundreds of progeny.
In that article, the consequence of HR was to duplicate a gene, which is still of limited interest for reverse geneticists. Following articles next demonstrated that it was possible to inactivate a gene or obtain a specific allele by HR in Drosophila both with the ends-in and ends-out designs (Gong and Golic, 2003; Xie and Golic, 2004). Successful HR is revealed in the progeny by an easily recognizable phenotype associated with a phenotypic marker in the targeting construct. HR is a rare event, but screening hundreds of flies to find one that encountered such an event is possible thanks to this marker gene.
2.5. Gene targeting by homologous recombination in mice
In 2007 Mario R. Capecchi, Sir Martin J. Evans and Oliver Smithies received the Nobel Prize for their discoveries on the principles that allowed the introduction of modifications into mice genes by using ES cells [review on gene targeting in mice, (Capecchi, 2005)]. Gene targeting (KO or KI) by HR is performed in ES cells before their injection into host embryos. ES cells are transfected with the targeting construct and the cells that encountered recombination are selected on the basis of their resistance to an antibiotic conferred by the selection marker. In contrast to yeast in which HR is the major DSB repair pathway, the non homologous end-joining pathway based on the ligation of the two ends of a DSB is the prevalent mechanism to repair a broken chromosome in mice (O’Driscoll and Jeggo, 2006). Consequently, illegitimate recombinations (insertion of the targeting sequence away from the targeted locus) are highly frequent in mouse cells. To prevent them, a second (negative) selection marker is required in mice. This marker is excised only upon HR, and not upon non homologous end-joining (Figure 3C).
As promising as they appear to be, conventional (constitutive) KO and KI suffer from a number of limitations in mice. If a gene disruption is lethal at a given age or developmental stage, it is impossible to analyse the phenotypic consequences of that disruption in older mice. Furthermore, if a protein is ubiquitously (or at least widely) expressed, the phenotype caused by the gene invalidation may result from an abrogated gene function in virtually any cell type or tissue and it is often difficult to identify in which cell types the inactivation initiates the mutant phenotype. A solution was brought by the use of SSRs. The Cre recombinase, from bacteriophage P1, is the most widely used SSR in mice because, in contrast to the wild-type yeast FLP recombinase, it is stable at 37°C. The Cre recombinase catalyzes a recombination between two loxP sites (Figure 4B). The “conditional KO” (cKO) strategy consists first in the insertion of two loxP sites oriented in the same direction in two different introns of the targeted gene. This insertion is achieved as above by HR in ES cells and injection of the modified cells in host embryos to obtain chimeras and finally genetically modified mice. The “floxed” (flanked by two loxP sites) allele is generally associated with a wild-type phenotype, since it only differs from the wild-type allele by two short loxP sites (34 nucleotides) located in introns. However, in tissues where the Cre recombinase is expressed, it causes the deletion of the genomic DNA between the two loxP sites, including the exons. If the loxP sites were localised in adequate introns, this deletion creates a null allele for the target gene (Collins et al., 2007a; Collins et al., 2007b).
Generally, floxed mice are crossed with mice expressing the Cre recombinase specifically in certain cell types or tissues to achieve tissue-specific inactivation in the progeny. These Cre-expressing mice can be obtained by transgenesis. In that case, the transgene consists of the Cre gene controlled by a promoter expected to drive tissue-specific expression. Alternatively, Cre-expressing mice can be obtained by HR in ES cells, by substituting the coding sequence of a given gene (displaying an interesting expression pattern) with the Cre coding sequence. Hundreds of mouse strains that express the Cre recombinase in a large variety of cell types have been generated to date. These strains are generally made available to the scientific research community (http://www.emmanet.org/; http://www.informatics.jax.org/recombinase.shtml). An International Mouse Knockout Consortium coordinates North-American and European efforts to systematically inactivate mouse genes. The goal is to obtain thousands of ES cells with conditional (floxed) targeted mutations. These recombined cells are stored frozen before being injected in a host embryo to finally obtain mutant mice (Collins et al., 2007a; Collins et al., 2007b).
Engineered Cre recombinases were also described. The most common one is a fusion protein between the Cre protein and a modified form of the estrogen receptor ligand binding domain (Indra et al., 1999). This mutated estrogen receptor is unable to bind endogenous estrogens, but efficiently binds, and is activated by, the drug tamoxifen. In control conditions, this fusion protein is sequestered in the cytoplasm, through the interaction of the ligand binding domain with HSP90. Upon treatment of mice with tamoxifen, this interaction is released and the Cre recombinase is translocated into nuclei, where recombination occurs. This system provides a means to inactivate a given gene in a given tissue at any time.
2.6. Limitations of gene targeting by homologous recombination
HR is probably the best method to achieve gene targeting whenever it is possible. Unfortunately, only few model organisms are amenable to this approach: yeasts, mice, flies and C. elegans (Table 1). Consequently, other strategies for gene inactivation have to be considered in other species.
3. Targeted gene silencing by double stranded RNAs
3.1. RNA interference in Caenorhabditis elegans…
In 2006, Andrew Fire and Craig Mello were awarded a Nobel Prize for their discovery (Fire et al., 1998) that in C. elegans, exogenous double-stranded RNAs (dsRNA) could specifically and efficiently repress genes showing strong sequence identity to the dsRNA. This property was named RNA interference (RNAi). A similar phenomenon had been previously discovered in plants and named co-suppression (Napoli et al., 1990). Since, extensive efforts have been made to understand the genetic and biochemical bases of RNAi. It was shown that dsRNA-mediated gene silencing occurs both at transcriptional (transcriptional repression) and post-transcriptional (mRNA degradation and translation inhibition) levels. RNAi shares several features with endogenous miRNA (microRNA)-mediated gene silencing. The mechanisms of RNAi are beyond the scope of this review, and the reader may refer to several reviews (Carthew and Sontheimer, 2009; Ghildiyal and Zamore, 2009; Liu and Paroo, 2010). Briefly, small interfering RNAs (siRNAs) are the triggers of RNAi. They enter the cell by direct transfection, or they originate from a longer precursor (short hairpin RNA or long double-stranded RNA). This precursor arises from transfection or in vivo transcription from a transgene (Figure 5A). siRNAs associated with RISC (RNA-induced silencing complex) cause the rapid degradation of cellular mRNAs containing a region perfectly complementary to the siRNA, or repress the translation of cellular mRNAs with partial complementarity, resulting in either case in gene silencing (Figure 5B). siRNAs showing strong sequence identity with a gene promoter (untranscribed region) also repress transcription by epigenetic mechanisms, but this property is only seldom used in reverse genetics.
Figure 5. RNA interference.
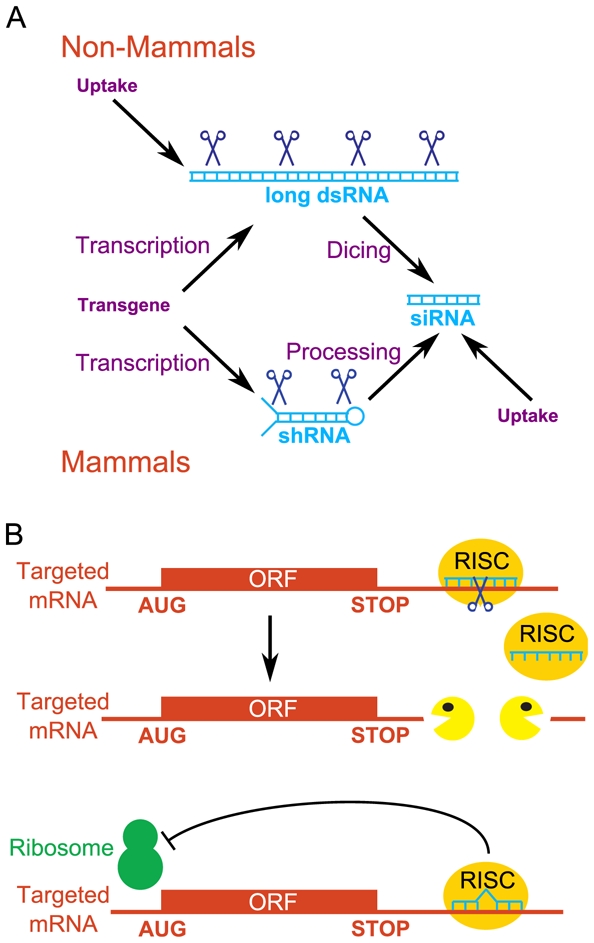
A, Origin of siRNAs (small interfering RNAs). siRNAs are small double-stranded RNA molecules and are the triggers of RNAi. In cultured mammalian cells, they are directly transfected. In mammals in vivo (or in mammalian cultured cells whenever a stable expression of the siRNA is required), they are produced in cells by the maturation of a precursor (shRNA, short hairpin RNA) that is transcribed from a transgene. In non-mammalian species, siRNAs arise from long double-stranded RNAs (dsRNAs) by dicing. These dsRNAs are directly introduced into the cells (C. elegans, cultured Drosophila cells) or transcribed from a transgene (Drosophila in vivo). B, Mechanisms of action of siRNAs. One strand of the initially double-stranded siRNA associates with RISC (RNA-induced silencing complex), and the RISC-RNA complex scans cellular mRNAs. In general, mRNAs with a perfect complementarity to the RNAi are rapidly degraded endonucleolytically then exonucleolytically (upper panel) whereas mRNAs with a partial complementarity are translationnally repressed (lower panel). In either case, siRNAs repress gene expression.
Geneticists rapidly realized that RNAi was a highly powerful strategy for reverse genetics analyses (Table 1). Introducing a dsRNA into cells can potentially silence or at least knock-down* virtually any target gene. In C. elegans, this dsRNA may be introduced by direct injection into the gonad or, even easier, by soaking the worms in a dsRNA solution or feeding them with bacteria expressing dsRNA (Fire et al., 1998; Tabara et al., 1998; Timmons and Fire, 1998). Systematic, genome-wide screenings were published using either of these methods (Gonczy et al., 2000; Kamath et al., 2003), giving for the first time an access to the phenotypes associated with silencing of a vast majority of genes in a multicellular organism. The “feeding library” (Kamath et al., 2003) is now widely used by C. elegans researchers. The easy uptake of dsRNA in C. elegans also allowed some systematic analysis of genetic interactions to be undertaken. Tens of different null mutants were grown on thousands of different dsRNA-expressing bacteria, so as to examine the phenotypes associated with double genetic inactivation. This led to the discovery of several hundreds of genetic interactions (Byrne et al., 2007; Lehner et al., 2006) [reviewed in (Dixon et al., 2009)].
3.2. ...And in other organisms
In Drosophila, RNAi is most of ten achieved via a transgene containing an inverted repeat sequence (Figure 5A). Transcription of this transgene produces a long RNA with self-complementarity. It is processed into a long dsRNA that is finally diced in several siRNAs against the targeted gene. RNAi was combined with the Gal4-UAS-mediated control of gene expression in Drosophila to achieve cell-specific inactivation of virtually any gene (Dietzl et al., 2007). If a transgene contains an UAS (Upstream Activating Sequence) in its promoter, it is expressed only in the presence of the yeast transcription factor Gal4. Therefore, cell-specific expression of the transgene is achieved in the progeny of a cross between the transgenic fly and the appropriate “driver”, a fly expressing Gal4 in the desired cell type. Since that strategy is highly popular, a large numbers of drivers with several different expression patterns are available for the scientific community. In the above article (Dietzl et al., 2007), a genome-wide collection of UAS-inverted repeats Drosophila was made. Crossing a Drosophila transgenic for an inverted repeat against a gene of interest with a tissue-specific Gal4 driver results in the inactivation of the gene of interest in the cell types expressing Gal4. This and other (http://www.shigen.nig.ac.jp/fly/nigfly/index.jsp) collections are currently widely used by the Drosophila community. Expression of a long dsRNA is also the basis for systematic inactivation of A. thaliana genes by RNAi (http://www.agrikola.org/index.php?o=/agrikola/html/index). The targeting sequences are based on previously described sequence tags covering the genome (Hilson et al., 2004). The RNAi resources (and other Omics resources) in A. thaliana and other plants were recently surveyed (Mochida and Shinozaki, 2010).
In mammals, long dsRNAs nonspecifically shut off translation by an interferon pathway. Hence, siRNAs, rather than long dsRNAs, are used to avoid this interferon response (Elbashir et al., 2001). In mammalian cultured cells, RNAi can be initiated by the direct transfection of siRNAs or the expression of “short hairpin” RNAs (shRNAs) that are transcribed from an appropriate transgene and processed in siRNAs through the miRNA biogenesis pathway (Figure 5A). In mice, several strategies for conditional expression of shRNA have been published. A potential advantage of these methods over gene engineering by homologous recombination would be that they are easier to set up (they do not require manipulations of ES cells, but zygotic injection of transgenes). In one approach, the expression of the shRNA was controlled by an engineered tetracycline promoter which made its expression dependent on doxycycline administration (Szulc et al., 2006). In an alternative approach, the promoter governing the expression of the shRNA was interrupted by a cassette between two LoxP sites, and was therefore inactive. In cells expressing the Cre recombinase, this cassette was deleted and the shRNA was expressed (Yu and McMahon, 2006). However, to date, there are few articles in which RNAi was used to silence genes in whole mice [recent review (Cazzin and Ring, 2009)].
3.3. Limitations of RNAi approaches
The RNAi approach has several drawbacks. The modifications of gene activity caused by RNAi are only poorly transmitted to the next generation (Alcazar et al., 2008), unless the interfering molecule is produced from a stably inserted transgene. Genes may be incompletely silenced, and the level of gene silencing is sometimes not reproducible. In C. elegans, the procedure to deliver dsRNAs (injection into the gonad, soaking in dsRNA solution or feeding the worms with bacteria expressing dsRNA) influences the silencing efficiency.
The major issue of RNA interference is the occurrence of off-target effects*. siRNAs are able to repress translation of mRNAs sharing only limited sequence conservation (Figure 5B). Hence, any siRNA is likely to silence several genes in addition to the target gene. The sequences of these off-targets are related, but not identical, to the siRNA. Consequently a mutant phenotype associated with the expression of a siRNA may be due to the inactivation of one or several of these unknown off-targets rather than or in addition to the target gene. In mammalian cultured cells, where siRNAs are directly transfected, several controls are therefore strongly recommended: different siRNAs against the same gene product should cause the same phenotype, the interfering RNA must cause a reduction in the level of the targeted gene product, and restoring the normal level of gene product (by introducing a modified transgene whose sequence does not match with siRNAs) should restore a wild-type phenotype (“rescue” experiment).
In other organisms, long dsRNAs diced in several different siRNAs are used. Consequently, each individual siRNA is diluted by several other siRNAs and the off-target effects of each individual siRNA are expected to be reduced as compared with those of unique siRNAs used in mammalian cells. However, the total number of off-targets may also increase with the number of different siRNAs. Indeed, two articles showed that off-target effects exist in cultured Drosophila cells, although different prevalences were reported (DasGupta et al., 2007; Ma et al., 2006). Hence rescue experiments are required to demonstrate the specificity of RNAi. Rescuing transgenes harbour several mutations making them immune to RNAi. These mutations may be silent, altering the nucleotide sequence without modifying the peptide sequence owing to the degenerate genetic code (Schulz et al., 2009). Orthologs of the targeted gene in a highly related species (like Caenorhabditis briggsae for C. elegans, or Drosophila pseudoviridis for D. melanogaster) are also a valuable resource for rescuing transgenes (Kondo et al., 2009; Langer et al., 2010; Sarov et al., 2006).
4. Antisense oligonucleotide-mediated inhibition of mRNA metabolism and translation
4.1. Morpholino antisense oligonucleotides in zebrafish and Xenopus
In antisense approaches, a RNA or a DNA oligonucleotide targets a given mRNA by nucleotide complementarity, and may inhibit its translation via steric interference (antisense RNA) or cause its degradation through an RNase H activity that hydrolyses the RNA moiety of RNA-DNA heteroduplexes (antisense DNA). While these approaches have met some success, they were largely supplanted by RNAi since 1998. Interestingly, the first paper on RNAi in C. elegans indicates that double-stranded RNAs are far more efficient than single-stranded, complementary RNA to silence a given gene (Fire et al., 1998). However, two popular models in developmental biology, zebrafish and Xenopus, have not switched to RNAi for two reasons (Table 1). Firstly, RNAi is inefficient or non-specific in these species [reviewed by (Eisen and Smith, 2008)]. Secondly, “morpholino” antisense oligonucleotides were introduced and proved to be more efficient than conventional DNA or RNA oligonucleotides. Morpholino oligonucleotides are characterized by morpholine rings which replace the riboses or deoxyriboses moieties, and non-ionic phosphorodiamidate links between rings. Although mimicking true nucleotide sequences, they are extremely resistant to nucleases. The mechanism of action of the morpholinos relies on the stability of the duplex formed on the RNA that leads to steric hindrance.
Morpholinos (MOs) are generally microinjected into Xenopus or zebrafish early embryos. Several cytoplasmic bridges connect cells in a zebrafish embryo, so that MO injection into any cell of an embryo up to the 8-cell stage provides ubiquitous delivery (Bill et al., 2009). In contrast, if a Xenopus embryo is injected in one of the two first blastomeres, a phenotype resulting from the MO is detected only on the side of the embryo resulting from the injected cell, the other side being usable as an internal developmental control [e.g. (Gautier-Courteille et al., 2004)]. Three strategies are used to specifically alter gene expression using MOs: translational repression, RNA processing modification or target protection.
4.2. Translational inhibition by Morpholinos
Morpholino-mediated translational inhibition uses antisense MOs directed to the 5′ untranslated region or the sequence spanning the start codon of an mRNA [Figure 6A, reviewed by (Eisen and Smith, 2008)]. While non-demonstrated, it is assumed that they act by interfering with the scanning of the small ribosomal subunit. A morpholino directed against the coding region has essentially no effect, probably because translating ribosomes are able to strip the morpholino off the mRNA. This feature is important since it reduces (but does not eliminate, see below) the off-target effects. Middle-scale phenotypic screens using translation-blocking MOs were reported both in zebrafish and Xenopus tropicalis (X. tropicalis). In zebrafish, 150 genes encoding secreted proteins and 61 genes orthologous to human genes expressed in hematopoietic stem cells were targeted. Distinct phenotypes were observed for 18 and 14 of them respectively (Eckfeldt et al., 2005; Pickart et al., 2006). X. tropicalis, rather than Xenopus laevis (X. laevis) was used for a phenotypic screen of MO-mediated developmental defects, essentially because it is diploid (whereas X. laevis is pseudotetraploid) and several genomic resources were developed. Two-hundreds-and-two genes were targeted, and almost 70% of them yielded developmental phenotypes (Rana et al., 2006).
Figure 6. Three designs of morpholinos in reverse genetics.
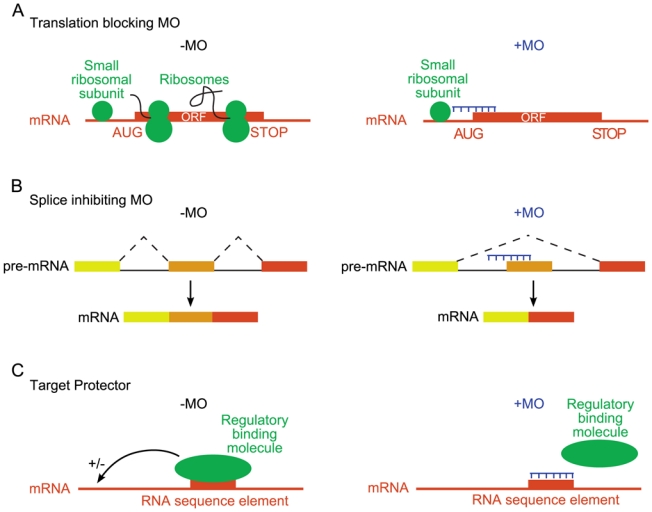
A, Translation blocking. In the absence of a morpholino (left), the small ribosomal subunit scans the 5′ untranslated region. The large ribosomal subunit is recruited at the translation initiation codon (AUG), to reconstitute a functional ribosome that translates the mRNA. If a morpholino hybridizes in the 5′ untranslated region, or in a region flanking the initiation codon (right), scanning by the small ribosomal subunit is impaired. Consequently, the mRNA is not translated. B, Splice inhibiting. A morpholino hybridizes on the pre-mRNA at an intron-exon junction (right). Consequently, this junction is no longer used and the corresponding exon (orange) is skipped. C, Target protector. The mRNA binds a regulatory molecule (protein or miRNA) that controls its fate (left). If a morpholino hybridizes to the mRNA sequence element normally bound by that molecule, binding is impaired (right). This design allows the developmental functions of regulatory interactions to be addressed.
4.3. Splice-inhibiting Morpholinos
Morpholinos can interfere with RNA maturation. MOs complementary to precursors of miRNAs are able to block their processing, resulting in their depletion (Flynt et al., 2007). MOs can also alter pre-mRNA splicing by preventing the binding of snRNP to exon/introns junctions or blocking the nucleophilic activity of the branch point adenosine present within the intron. According to the exon definition model that prevails in vertebrates, such targeting results in the exclusion of the exon (exon skipping) from the mature mRNA (Figure 6B). However, depending on the pre-mRNA features, the action of such morpholinos can lead to the retention of the intron or to the activation of cryptic donor or acceptor splice sites (Morcos, 2007).
Splice-inhibiting MOs may be useful to understand the functions of specific mRNA isoforms, generated through alternative splicing, by forcing splicing toward a given isoform. Furthermore, modifying the splicing pattern may result in a frameshift, leading to a premature stop codon that in turn targets the corresponding mRNA to rapid non-sense mRNA mediated decay [NMD, review (Rebbapragada and Lykke-Andersen, 2009)]. Hence, if correctly designed, splice-inhibiting MOs can be used as an alternative to translation blocking MOs to reduce the expression of a given gene.
When the goal is to specifically reduce gene expression in Xenopus or zebrafish, what are the pros and cons of translation-blocking and splice-inhibiting MOs? On the one hand, changes in splicing pattern can be readily measured by analysing mRNAs, either by conventional RT-PCR procedures, or more accurately by directly quantifying the relative ratios of the different isoforms by pyrosequencing (Mereau et al., 2009). In contrast, monitoring the effects of translation-blocking MOs requires antibodies to measure the level of the targeted protein. This is an interesting feature of splice inhibiting MOs as it overcomes the unavailability of most antibodies required for the control of translation blocking MOs. On the other hand, splice inhibiting MOs can only be designed in species whose genome has been sequenced. This is the case for zebrafish and Xenopus tropicalis, but not Xenopus laevis. Finally, transcription is silent in early embryos at the time of morpholinos injection, and embryos develop using maternally inherited mRNAs (Davidson, 1986). Translation blocking MOs inhibit the expression of these maternal mRNAs whereas splice inhibiting MOs only affect newly transcribed zygotic pre-mRNAs. Hence, whether or not maternal mRNAs have to be blocked in addition to zygotic mRNAs is an argument to direct the choice of using either translation-blocking or splice-inhibiting MOs.
4.4. Target-protectors
A third design of MOs is as target-protector. These MOs hybridize to a region of an mRNA that is normally recognized by a regulatory molecule, such as a miRNA or an RNA-binding protein (Figure 6C). Since MOs do not activate RNase H, targeted mRNAs are not degraded. Target protector MOs relieve the regulation that is normally exerted by the miRNA or RNA-binding protein on the mRNA. The functions of specific regulatory interactions may therefore be investigated with that strategy (Choi et al., 2007; Cibois et al., 2010). Target-protector MOs are also an alternative to MOs targeting intron/exons borders to modify splice patterns. MOs directed against a splicing regulator element involved in the activation or repression of a specific exon modify splicing by preventing the interaction with a regulatory RNA-binding proteins (Anquetil et al., 2009; Bruno et al., 2004).
4.5. Limitations of antisense approaches
The drawbacks of MO-mediated antisense approaches are very similar to those of RNAi approach: the occurrence of non-specific effects, a lack of transmission of the modifications of gene activity to the next generation, and the incomplete and sometimes not reproducible gene silencing. In addition, MOs being injected in early embryos, the time frame in which these experiments can be run is limited (if the protein encoded by the targeted mRNA is already expressed at that time, the phenotypic consequences of MO injection can only be observed after clearance of that protein, and thus depends on the stability of the protein).
Non-specific, off-target effects of MOs have been described. In zebrafish, a relatively large number of MOs cause cell death. This effect is non-specific, in as much as mutants of the corresponding genes do not show this phenotype. It is due to ectopic upregulation of the p53-dependent apoptosis pathway (Robu et al., 2007). That such non-specific effects can occur with MOs implies that several controls are required. The proposed controls (Eisen and Smith, 2008) are similar to those used for RNAi in cultured cells (See Paragraph 3.3): i) check the effect of the MO upon the expression of the targeted gene, at the mRNA (splice inhibiting MO) or protein (translation blocking MO) level; ii) use control MOs such as MOs presenting several mismatches, depending on the number of mismatches the MO should have a weaker activity or no activity; iii) use several MOs against the same target that should yield a similar phenotype when injected individually, and should have a synergistic effect when co-injected; and iv) restore the wild-type phenotype by co-injecting a molecule with the MO (“rescue” experiment). When the MO is aimed at knocking-down an mRNA, the rescuing co-injected molecule is an mRNA encoding the same protein, but whose sequence is modified so as to avoid targeting by the MO. In a target-protection experiment where the protector MO inhibited the binding of a protein that directs rapid degradation, the corresponding encoded protein was overexpressed. In that case, the rescuing molecule was a translation-blocking MO aimed at limiting that overexpression (Cibois et al., 2010).
Concluding remarks
In Eukaryotes, four main strategies were elaborated to specifically modify the expression of a gene, random mutagenesis followed by gene screening, gene engineering by homologous recombination, RNAi and antisense approaches. The decision to adopt any of these four strategies is largely imposed by the species and the biological question being studied both of which are dependent on the particular biological properties of each model organism (Table 1). Care must be taken when interpreting phenotypes, as most approaches are likely to have off-target effects. Whenever possible, it would be recommended to confirm the results obtained with one approach by another method. For species most amenable to forward genetics (zebrafish, Drosophila, C. elegans), there exist collections of mutants, and comparisons can be made between the phenotypes of these mutants and the phenotypes arising from reverse genetics.
The price of reverse genetics approaches are comprised within a large range. Obtaining one custom organism with an altered expression of a targeted gene will cost from some tens of Euros in yeast (homologous recombination with PCR products) to some hundreds of Euros in Xenopus and zebrafish (Morpholinos) and some tens of thousands of Euros in mice (gene targeting by homologous recombination and animal facilities). The publication, within the last decade, of several collections of targeted mutants, strongly reduces the price of each individual mutants.
Acknowledgments
We thank H Beverley Osborne for critical reading of the manuscript. Work in the authors’ lab is supported by grants from the Association pour la Recherche Contre le Cancer (ARC4003, to LP), Agence Nationale de la Recherche (ANR-07-JCJC-0097-01, to LP) and Agence Française contre les Myopathies (AFM G12999A to SH). VL is a staff member of the INSERM (Institut National de la Santé Et de la Recherche Médicale).
Abbreviations
- cKO
conditional knock-out
- DSB
double-strand break
- dsRNA
double-stranded RNA
- ES
embryonic stem
- FRT
FLP recombination target
- HR
homologous recombination
- KI
Knock-in
- KO
Knock-out
- MO
Morpholino
- MosTIC
Mos1 excision-induced transgene instructed gene conversion
- RISC
RNA-induced silencing complex
- siRNA
small interfering RNA
- shRNA
short hairpin RNA
- SSR
Site-specific recombinase
- TILLING
Targeting induced local lesions in genomes
- UAS
upstream activating sequence
References
- Alcazar RM, Lin R, Fire AZ. Transmission dynamics of heritable silencing induced by double-stranded RNA in Caenorhabditis elegans. Genetics. 2008;180:1275–88. doi: 10.1534/genetics.108.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, Gadrinab C, Heller C, Jeske A, Koesema E, Meyers CC, Parker H, Prednis L, Ansari Y, Choy N, Deen H, Geralt M, Hazari N, Hom E, Karnes M, Mulholland C, Ndubaku R, Schmidt I, Guzman P, Aguilar-Henonin L, Schmid M, Weigel D, Carter DE, Marchand T, Risseeuw E, Brogden D, Zeko A, Crosby WL, Berry CC, Ecker JR. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301:653–7. doi: 10.1126/science.1086391. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–7. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anquetil V, Le Sommer C, Mereau A, Hamon S, Lerivray H, Hardy S. Polypyrimidine tract binding protein prevents activity of an intronic regulatory element that promotes usage of a composite 3′-terminal exon. J Biol Chem. 2009;284:32370–83. doi: 10.1074/jbc.M109.029314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PL, Fleming JT, Gobel V. Targeted gene alteration in Caenorhabditis elegans by gene conversion. Nat Genet. 2004;36:1231–7. doi: 10.1038/ng1459. [DOI] [PubMed] [Google Scholar]
- Bazopoulou D, Tavernarakis N. The NemaGENETAG initiative: large scale transposon insertion gene-tagging in Caenorhabditis elegans. Genetica. 2009;137:39–46. doi: 10.1007/s10709-009-9361-3. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, Hoskins RA, Spradling AC. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–81. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Bargmann CI, Plasterk RH. Homologous gene targeting in Caenorhabditis elegans by biolistic transformation. Nucleic Acids Res. 2004;32:e40. doi: 10.1093/nar/gnh033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birling MC, Gofflot F, Warot X. Site-specific recombinases for manipulation of the mouse genome. Methods Mol Biol. 2009;561:245–63. doi: 10.1007/978-1-60327-019-9_16. [DOI] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet. 1984;197:345–6. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Bruno IG, Jin W, Cote GJ. Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet. 2004;13:2409–20. doi: 10.1093/hmg/ddh272. [DOI] [PubMed] [Google Scholar]
- Byrne AB, Weirauch MT, Wong V, Koeva M, Dixon SJ, Stuart JM, Roy PJ. A global analysis of genetic interactions in Caenorhabditis elegans. J Biol. 2007;6:8. doi: 10.1186/jbiol58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507–12. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzin C, Ring CJ. Recent advances in the manipulation of murine gene expression and its utility for the study of human neurological disease. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbadis.2009.11.005. [DOI] [PubMed] [Google Scholar]
- *.Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–4. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- Cibois M, Gautier-Courteille C, Vallee A, Paillard L. A strategy to analyze the phenotypic consequences of inhibiting the association of an RNA-binding protein with a specific RNA. Rna. 2010;16:10–5. doi: 10.1261/rna.1742610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert T, Till BJ, Tompa R, Reynolds S, Steine MN, Yeung AT, McCallum CM, Comai L, Henikoff S. High-throughput screening for induced point mutations. Plant Physiol. 2001;126:480–4. doi: 10.1104/pp.126.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Finnell RH, Rossant J, Wurst W. A new partner for the international knockout mouse consortium. Cell. 2007a;129:235. doi: 10.1016/j.cell.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Collins FS, Rossant J, Wurst W. A mouse for all reasons. Cell. 2007b;128:9–13. doi: 10.1016/j.cell.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Cooper JL, Till BJ, Henikoff S. Fly-TILL: reverse genetics using a living point mutation resource. Fly (Austin) 2008;2:300–2. doi: 10.4161/fly.7366. [DOI] [PubMed] [Google Scholar]
- Cretekos CJ, Wang Y, Green ED, Martin JF, Rasweiler JJt, Behringer RR. Regulatory divergence modifies limb length between mammals. Genes Dev. 2008;22:141–51. doi: 10.1101/gad.1620408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daee DL, Mertz TM, Shcherbakova PV. A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci U S A. 2009;107:157–62. doi: 10.1073/pnas.0907526106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Nybakken K, Booker M, Mathey-Prevot B, Gonsalves F, Changkakoty B, Perrimon N. A case study of the reproducibility of transcriptional reporter cell-based RNAi screens in Drosophila. Genome Biol. 2007;8:R203. doi: 10.1186/gb-2007-8-9-r203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH. The nature and function of maternal transcripts. Academic Press; New York: 1986. [Google Scholar]
- *.Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–6. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- *.Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. Systematic mapping of genetic interaction networks. Annu Rev Genet. 2009;43:601–25. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- Duverger Y, Belougne J, Scaglione S, Brandli D, Beclin C, Ewbank JJ. A semi-automated high-throughput approach to the generation of transposon insertion mutants in the nematode Caenorhabditis elegans. Nucleic Acids Res. 2007;35:e11. doi: 10.1093/nar/gkl1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckfeldt CE, Mendenhall EM, Flynn CM, Wang TF, Pickart MA, Grindle SM, Ekker SC, Verfaillie CM. Functional analysis of human hematopoietic stem cell gene expression using zebrafish. PLoS Biol. 2005;3:e254. doi: 10.1371/journal.pbio.0030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Eisen JS, Smith JC. Controlling morpholino experiments: don’t stop making antisense. Development. 2008;135:1735–43. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- Elbashir S, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature (London) 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Enard W, Gehre S, Hammerschmidt K, Holter SM, Blass T, Somel M, Bruckner MK, Schreiweis C, Winter C, Sohr R, Becker L, Wiebe V, Nickel B, Giger T, Muller U, Groszer M, Adler T, Aguilar A, Bolle I, Calzada-Wack J, Dalke C, Ehrhardt N, Favor J, Fuchs H, Gailus-Durner V, Hans W, Holzlwimmer G, Javaheri A, Kalaydjiev S, Kallnik M, Kling E, Kunder S, Mossbrugger I, Naton B, Racz I, Rathkolb B, Rozman J, Schrewe A, Busch DH, Graw J, Ivandic B, Klingenspor M, Klopstock T, Ollert M, Quintanilla-Martinez L, Schulz H, Wolf E, Wurst W, Zimmer A, Fisher SE, Morgenstern R, Arendt T, de Angelis MH, Fischer J, Schwarz J, Paabo S. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell. 2009;137:961–71. doi: 10.1016/j.cell.2009.03.041. [DOI] [PubMed] [Google Scholar]
- Feil R. Conditional somatic mutagenesis in the mouse using site-specific recombinases. Handb Exp Pharmacol. 2007:3–28. doi: 10.1007/978-3-540-35109-2_1. [DOI] [PubMed] [Google Scholar]
- *.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans [see comments] Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet. 2007;39:259–63. doi: 10.1038/ng1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hollopeter G, Taylor J, Harris TW, Nix P, Lofgren R, Prestgard-Duke M, Bastiani M, Moerman DG, Jorgensen EM. Targeted gene deletions in C. elegans using transposon excision. Nat Methods. 2010;7:451–3. doi: 10.1038/nmeth.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–83. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier-Courteille C, Le Clainche C, Barreau C, Audic Y, Graindorge A, Maniey D, Osborne HB, Paillard L. EDEN-BP-dependent post-transcriptional regulation of gene expression in Xenopus somitic segmentation. Development. 2004;131:6107–17. doi: 10.1242/dev.01528. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–7. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Gengyo-Ando K, Mitani S. Characterization of mutations induced by ethyl methanesulfonate, UV, and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem Biophys Res Commun. 2000;269:64–9. doi: 10.1006/bbrc.2000.2260. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Gilchrist EJ, O’Neil NJ, Rose AM, Zetka MC, Haughn GW. TILLING is an effective reverse genetics technique for Caenorhabditis elegans. BMC Genomics. 2006;7:262. doi: 10.1186/1471-2164-7-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda T, Abu-Daya A, Carruthers S, Clark MD, Stemple DL, Zimmerman LB. Genetic screens for mutations affecting development of Xenopus tropicalis. PLoS Genet. 2006;2:e91. doi: 10.1371/journal.pgen.0020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P, Echeverri G, Oegema K, Coulson A, Jones SJ, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlu N, Bork P, Hyman AA. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000;408:331–6. doi: 10.1038/35042526. [DOI] [PubMed] [Google Scholar]
- Gong WJ, Golic KG. Ends-out, or replacement, gene targeting in Drosophila. Proc Natl Acad Sci U S A. 2003;100:2556–61. doi: 10.1073/pnas.0535280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger L, Martin E, Segalat L. Mos as a tool for genome-wide insertional mutagenesis in Caenorhabditis elegans: results of a pilot study. Nucleic Acids Res. 2004;32:e117. doi: 10.1093/nar/gnh111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, McGill C, Shafer B, Strathern JN. Ends-in vs. ends-out recombination in yeast. Genetics. 1993;135:973–80. doi: 10.1093/genetics/135.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilson P, Allemeersch J, Altmann T, Aubourg S, Avon A, Beynon J, Bhalerao RP, Bitton F, Caboche M, Cannoot B, Chardakov V, Cognet-Holliger C, Colot V, Crowe M, Darimont C, Durinck S, Eickhoff H, de Longevialle AF, Farmer EE, Grant M, Kuiper MT, Lehrach H, Leon C, Leyva A, Lundeberg J, Lurin C, Moreau Y, Nietfeld W, Paz-Ares J, Reymond P, Rouze P, Sandberg G, Segura MD, Serizet C, Tabrett A, Taconnat L, Thareau V, Van Hummelen P, Vercruysse S, Vuylsteke M, Weingartner M, Weisbeek PJ, Wirta V, Wittink FR, Zabeau M, Small I. Versatile gene-specific sequence tags for Arabidopsis functional genomics: transcript profiling and reverse genetics applications. Genome Res. 2004;14:2176–89. doi: 10.1101/gr.2544504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnen A, Hicks JB, Fink GR. Transformation of yeast. Proc Natl Acad Sci U S A. 1978;75:1929–33. doi: 10.1073/pnas.75.4.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–3. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–91. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–7. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Hazendonk E, Thijssen KL, Plasterk RH. Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat Genet. 1997;17:119–21. doi: 10.1038/ng0997-119. [DOI] [PubMed] [Google Scholar]
- *.Kamath R, Fraser A, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Bot NL, Moreno S, Sohrmann M, Welchman D, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kondo S, Booker M, Perrimon N. Cross-species RNAi rescue platform in Drosophila melanogaster. Genetics. 2009;183:1165–73. doi: 10.1534/genetics.109.106567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress C, Gautier-Courteille C, Osborne HB, Babinet C, Paillard L. Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol Cell Biol. 2007;27:1146–57. doi: 10.1128/MCB.01009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–43. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Krysan PJ, Young JC, Sussman MR. T-DNA as an insertional mutagen in Arabidopsis. Plant Cell. 1999;11:2283–90. doi: 10.1105/tpc.11.12.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, Cheung KH, Miller P, Gerstein M, Roeder GS, Snyder M. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–19. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer CC, Ejsmont RK, Schonbauer C, Schnorrer F, Tomancak P. In vivo RNAi rescue in Drosophila melanogaster with genomic transgenes from Drosophila pseudoobscura. PLoS ONE. 2010;5:e8928. doi: 10.1371/journal.pone.0008928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- Liu LX, Spoerke JM, Mulligan EL, Chen J, Reardon B, Westlund B, Sun L, Abel K, Armstrong B, Hardiman G, King J, McCague L, Basson M, Clover R, Johnson CD. High-throughput isolation of Caenorhabditis elegans deletion mutants. Genome Res. 1999;9:859–67. doi: 10.1101/gr.9.9.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Paroo Z. Biochemical Principles of Small RNA Pathways. Annu Rev Biochem. 2010;79:295–319. doi: 10.1146/annurev.biochem.052208.151733. [DOI] [PubMed] [Google Scholar]
- Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 2006;443:359–63. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- Martin B, Ramiro M, Martinez-Zapater JM, Alonso-Blanco C. A high-density collection of EMS-induced mutations for TILLING in Landsberg erecta genetic background of Arabidopsis. BMC Plant Biol. 2009;9:147. doi: 10.1186/1471-2229-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.McCallum CM, Comai L, Greene EA, Henikoff S. Targeting induced local lesions IN genomes (TILLING) for plant functional genomics. Plant Physiol. 2000;123:439–42. doi: 10.1104/pp.123.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereau A, Anquetil V, Cibois M, Noiret M, Primot A, Vallee A, Paillard L. Analysis of splicing patterns by pyrosequencing. Nucleic Acids Res. 2009;37:e126. doi: 10.1093/nar/gkp626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Shinozaki K. Genomics and bioinformatics resources for crop improvement. Plant Cell Physiol. 2010;51:497–523. doi: 10.1093/pcp/pcq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcos PA. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochem Biophys Res Commun. 2007;358:521–7. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- Nagayoshi S, Hayashi E, Abe G, Osato N, Asakawa K, Urasaki A, Horikawa K, Ikeo K, Takeda H, Kawakami K. Insertional mutagenesis by the Tol2 transposon-mediated enhancer trap approach generated mutations in two developmental genes: tcf7 and synembryn-like. Development. 2008;135:159–69. doi: 10.1242/dev.009050. [DOI] [PubMed] [Google Scholar]
- Napoli C, Lemieux C, Jorgensen R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell. 1990;2:279–289. doi: 10.1105/tpc.2.4.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Chang PJ, Conklin BR, Cox AV, Harper CA, Hicks GG, Huang CC, Johns SJ, Kawamoto M, Liu S, Meng EC, Morris JH, Rossant J, Ruiz P, Skarnes WC, Soriano P, Stanford WL, Stryke D, von Melchner H, Wurst W, Yamamura K, Young SG, Babbitt PC, Ferrin TE. The International Gene Trap Consortium Website: a portal to all publicly available gene trap cell lines in mouse. Nucleic Acids Res. 2006;34:D642–8. doi: 10.1093/nar/gkj097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW. Multiple, tandem plasmid integration in Saccharomyces cerevisiae. Mol Cell Biol. 1983;3:747–9. doi: 10.1128/mcb.3.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, Tan LR, Winter CG, Bogart KP, Deal JE, Deal-Herr ME, Grant D, Marcinko M, Miyazaki WY, Robertson S, Shaw KJ, Tabios M, Vysotskaia V, Zhao L, Andrade RS, Edgar KA, Howie E, Killpack K, Milash B, Norton A, Thao D, Whittaker K, Winner MA, Friedman L, Margolis J, Singer MA, Kopczynski C, Curtis D, Kaufman TC, Plowman GD, Duyk G, Francis-Lang HL. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet. 2004;36:288–92. doi: 10.1038/ng1312. [DOI] [PubMed] [Google Scholar]
- Petzold AM, Balciunas D, Sivasubbu S, Clark KJ, Bedell VM, Westcot SE, Myers SR, Moulder GL, Thomas MJ, Ekker SC. Nicotine response genetics in the zebrafish. Proc Natl Acad Sci U S A. 2009;106:18662–7. doi: 10.1073/pnas.0908247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart MA, Klee EW, Nielsen AL, Sivasubbu S, Mendenhall EM, Bill BR, Chen E, Eckfeldt CE, Knowlton M, Robu ME, Larson JD, Deng Y, Schimmenti LA, Ellis LB, Verfaillie CM, Hammerschmidt M, Farber SA, Ekker SC. Genome-wide reverse genetics framework to identify novel functions of the vertebrate secretome. PLoS ONE. 2006;1:e104. doi: 10.1371/journal.pone.0000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–29. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Rana AA, Collart C, Gilchrist MJ, Smith JC. Defining synphenotype groups in Xenopus tropicalis by use of antisense morpholino oligonucleotides. PLoS Genet. 2006;2:e193. doi: 10.1371/journal.pgen.0020193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–2. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Rigola D, van Oeveren J, Janssen A, Bonne A, Schneiders H, van der Poel HJ, van Orsouw NJ, Hogers RC, de Both MT, van Eijk MJ. High- throughput detection of induced mutations and natural variation using KeyPoint technology. PLoS ONE. 2009;4:e4761. doi: 10.1371/journal.pone.0004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Robert V, Bessereau JL. Targeted engineering of the Caenorhabditis elegans genome following Mos1-triggered chromosomal breaks. Embo J. 2007;26:170–83. doi: 10.1038/sj.emboj.7601463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, Ekker SC. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, Qu H, Shales M, Park HO, Hayles J, Hoe KL, Kim DU, Ideker T, Grewal SI, Weissman JS, Krogan NJ. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322:405–10. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Rong YS, Golic KG. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–8. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K, Weisshaar B. An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol Biol. 2003;53:247–59. doi: 10.1023/B:PLAN.0000009297.37235.4a. [DOI] [PubMed] [Google Scholar]
- Sarov M, Schneider S, Pozniakovski A, Roguev A, Ernst S, Zhang Y, Hyman AA, Stewart AF. A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat Methods. 2006;3:839–44. doi: 10.1038/nmeth933. [DOI] [PubMed] [Google Scholar]
- Schulz JG, David G, Hassan BA. A novel method for tissue-specific RNAi rescue in Drosophila. Nucleic Acids Res. 2009;37:e93. doi: 10.1093/nar/gkp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivasubbu S, Balciunas D, Amsterdam A, Ekker SC. Insertional mutagenesis strategies in zebrafish. Genome Biol. 2007;8(Suppl 1):S9. doi: 10.1186/gb-2007-8-s1-s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivasubbu S, Balciunas D, Davidson AE, Pickart MA, Hermanson SB, Wangensteen KJ, Wolbrink DC, Ekker SC. Gene-breaking transposon mutagenesis reveals an essential role for histone H2afza in zebrafish larval development. Mech Dev. 2006;123:513–29. doi: 10.1016/j.mod.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Sood R, English MA, Jones M, Mullikin J, Wang DM, Anderson M, Wu D, Chandrasekharappa SC, Yu J, Zhang J, Paul Liu P. Methods for reverse genetic screening in zebrafish by resequencing and TILLING. Methods. 2006;39:220–7. doi: 10.1016/j.ymeth.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM. The Berkeley Drosophila Genome Project gene disruption project: Single P-element insertions mutating 25% of vital Drosophila genes. Genetics. 1999;153:135–77. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulc J, Wiznerowicz M, Sauvain MO, Trono D, Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat Methods. 2006;3:109–16. doi: 10.1038/nmeth846. [DOI] [PubMed] [Google Scholar]
- Tabara H, Grishok A, Mello CC. RNAi in C. elegans: soaking in the genome sequence. Science. 1998;282:430–1. doi: 10.1126/science.282.5388.430. [DOI] [PubMed] [Google Scholar]
- Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, Singh CM, Buchholz R, Demsky M, Fawcett R, Francis-Lang HL, Ryner L, Cheung LM, Chong A, Erickson C, Fisher WW, Greer K, Hartouni SR, Howie E, Jakkula L, Joo D, Killpack K, Laufer A, Mazzotta J, Smith RD, Stevens LM, Stuber C, Tan LR, Ventura R, Woo A, Zakrajsek I, Zhao L, Chen F, Swimmer C, Kopczynski C, Duyk G, Winberg ML, Margolis J. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–7. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- Till BJ, Colbert T, Tompa R, Enns LC, Codomo CA, Johnson JE, Reynolds SH, Henikoff JG, Greene EA, Steine MN, Comai L, Henikoff S. High-throughput TILLING for functional genomics. Methods Mol Biol. 2003;236:205–20. doi: 10.1385/1-59259-413-1:205. [DOI] [PubMed] [Google Scholar]
- Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- *.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–13. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Vermeire L, Ibrahimi A, Voet T, Umans L, Coddens K, Francis A, Van de Putte T, van Grunsven LA, Zwijsen A, Huylebroeck D, Nelles L. Essential validation of gene trap mouse ES cell lines: a test case with the gene Ttrap. Int J Dev Biol. 2009;53:1045–51. doi: 10.1387/ijdb.092859lv. [DOI] [PubMed] [Google Scholar]
- Wienholds E, van Eeden F, Kosters M, Mudde J, Plasterk RH, Cuppen E. Efficient target-selected mutagenesis in zebrafish. Genome Res. 2003;13:2700–7. doi: 10.1101/gr.1725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler S, Schwabedissen A, Backasch D, Bokel C, Seidel C, Bonisch S, Furthauer M, Kuhrs A, Cobreros L, Brand M, Gonzalez-Gaitan M. Target-selected mutant screen by TILLING in Drosophila. Genome Res. 2005;15:718–23. doi: 10.1101/gr.3721805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Xie HB, Golic KG. Gene deletions by ends-in targeting in Drosophila melanogaster. Genetics. 2004;168:1477–89. doi: 10.1534/genetics.104.030882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau DA, Mead PE. Transposon-mediated transgenesis in the frog: New tools for biomedical and developmental studies. Front Biosci. 2009;14:225–36. doi: 10.2741/3242. [DOI] [PubMed] [Google Scholar]
- Yu J, McMahon AP. Reproducible and inducible knockdown of gene expression in mice. Genesis. 2006;44:252–61. doi: 10.1002/dvg.20213. [DOI] [PubMed] [Google Scholar]
- Zhou D, Ren JX, Ryan TM, Higgins NP, Townes TM. Rapid tagging of endogenous mouse genes by recombineering and ES cell complementation of tetraploid blastocysts. Nucleic Acids Res. 2004;32:e128. doi: 10.1093/nar/gnh128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaal RR, Broeks A, van Meurs J, Groenen JT, Plasterk RH. Target-selected gene inactivation in Caenorhabditis elegans by using a frozen transposon insertion mutant bank. Proc Natl Acad Sci U S A. 1993;90:7431–5. doi: 10.1073/pnas.90.16.7431. [DOI] [PMC free article] [PubMed] [Google Scholar]