

Abstract
OBJECTIVES
Dexmedetomidine is an α2-adrenergic receptor agonist with sedative and analgesic effects in mechanically ventilated adults and children. Safety and efficacy data are limited in children. The purpose of this study is to retrospectively identify the incidence and types of adverse events noted in children receiving continuous infusions of dexmedetomidine and evaluate potential risk factors for adverse events.
METHODS
Between July 1, 2006, and July 31, 2007, data were collected on all children (< 18 years) who received continuous infusions of dexmedetomidine. Data collection included demographics, dexmedetomidine regimen, and type/number of adverse events. The primary endpoint was the total number of adverse events noted, including: transient hypertension, hypotension, neurological manifestations, apnea, and bradycardia. Secondary endpoints included categorization of each type of adverse event and an assessment of risk factors. A logistic regression model was used to assess the relationship of adverse events with independent variables including length of ICU stay, cumulative dose, peak infusion rate, duration of therapy, PRISM III score, and bolus dose.
RESULTS
Thirty-six patients received dexmedetomidine representing 41 infusions. The median age was 16 months (range, 0.1–204 months) and median PRISM III score was 2 (range, 0–18). Eighteen (43.9%) patients received a bolus dose of dexmedetomidine. The median cumulative dose (mcg/kg) and peak dose (mcg/kg/hr) were 8.5 (range, 2.2–193.7) and 0.5 (range, 0.2–0.7), respectively. Dexmedetomidine was continued for a median of 20 (range, 3–263) hours. Six (14.6%) patients were slowly tapered off the continuous infusions. Twenty-one adverse events were noted in 17 patients, including 4 neurologic manifestations. Fourteen patients required interventions for adverse events. ICU length of stay was the only independent risk factor (p=0.036) for development of adverse events.
CONCLUSIONS
Several potential adverse events were noted with dexmedetomidine continuous infusions including possible neurological manifestations. Further studies are needed looking at adverse events associated with dexmedetomidine use in the pediatric population.
Keywords: adverse events, children, dexmedetomidine, intensive care unit, sedation
INTRODUCTION
Dexmedetomidine (Precedex; Hospira, Lake Forest, IL) was approved in 2004 for sedation of mechanically ventilated adults during treatment in the intensive care unit (ICU).1 It is a selective alpha2-adrenergic agonist that provides sedation with minimal effects on respiratory function. The recommended dosage range is 1 mcg/kg loading dose followed by a continuous infusion of 0.2–0.7 mcg/kg/hr for no longer than 24 hours. In adult studies, dexmedetomidine enabled the doses of narcotics and benzodiazepines to be reduced.1 The positive results in adults led researchers to evaluate efficacy in the pediatric population. Tobias and Berkenbosch conducted a prospective, randomized trial in a pediatric ICU comparing dexmedetomidine to midazolam for sedation during mechanical ventilation.2 Dexmedetomidine provided more effective sedation as demonstrated by the administration of fewer morphine doses. Infants and children randomized to the midazolam group received 36 morphine boluses versus 29 and 20 morphine boluses administered, respectively, to the 0.25 and 0.5 mcg/kg/hr dexmedetomidine groups (p=0.02 for midazolam versus 0.5 mcg/kg/h of dexmedetomidine).2
Limited data that describe the efficacy and safety of dexmedetomidine in children are available.2–5 In a phase III study of dexmedetomidine in adults (n=401), the most common adverse events were hypotension (30%), hypertension (12%), nausea (11%), and bradycardia (9%).6,7 Severe bradycardia and cardiac arrhythmias with rare reports of asystole or cardiac arrest have been reported in patients requiring mechanical ventilation on dexmedetomidine.1,8,9 Apnea and neurological abnormalities (e.g., somnolence, facial drooping) have been noted in some patients.1 Despite the limited safety data of dexmedetomidine use in children, prescribers have used this agent in critically ill children as an adjunct sedative agent and frequently administer it for longer than 24 hours. This study was initiated to identify the epidemiology and risk factors of adverse events in critically ill children receiving dexmedetomidine in the pediatric ICU.
MATERIALS AND METHODS
Study Design
This retrospective study was conducted at a 230-bed tertiary care children's hospital located within an academic medical center. Following institutional review board approval, patients less than 18 years of age who received dexmedetomidine between June 1, 2006 and July 31, 2007 in the ICU for greater than two hours were included in analysis. Patients were identified through the Meditech (Medical Information Technology, Inc., Westwood, MA) database. Given the short half-life of dexmedetomidine (approximately 2 hours), data for patients receiving more than one course of dexmedetomidine per hospital admission were evaluated as separate events provided the time between each infusion period was more than 12 hours. Data for patients with incomplete medical records and those who received dexmedetomidine outside the ICU (i.e., operating room use) or for procedural sedation were not used.
Endpoints and Data Collection
Data were collected from progress notes, physician orders, nursing notes, and/or pharmacy records. Information obtained included age at time of admission, sex, race, ICU length of stay, primary diagnosis at the time of admission to the pediatric ICU (e.g., post-operative/trauma, septic shock, pneumonia, pulmonary disease), and PRISM III (Pediatric Risk of Mortality)10 score. PRISM III was used to quantify illness severity. The dexmedetomidine dosage regimen (mcg/kg/hr), cumulative dose (mcg/kg), bolus dose administered (yes or no), duration of therapy (hours), and use of taper at the completion of therapy were also recorded.
Information was also collected on the use of concomitant sedative and analgesic agents. Continuous infusions of sedative and analgesic agents, including narcotics (fentanyl, morphine), ketamine, and benzodiazepines (lorazepam, midazolam), were assessed. Additionally, the number of intermittent bolus doses of sedative and analgesic agents was also noted. These medications were collected to identify if adverse events correlated with concomitant dexmedetomidine administration.
The potential for adverse cardiovascular effects was evaluated by monitoring heart rate, systolic and diastolic blood pressure at one hour before infusion, in the middle of the infusion, one hour post-infusion, and 12 hours after discontinuation of dexmedetomidine. The pre-infusion and post-infusion measurements were collected to identify any possible changes in heart rate or rebound hypertension. Given dexmedetomidine terminal half-life of approximately 1.8 hours in children, the 12-hour post-infusion measurement was estimated to represent the time of total or almost-total elimination of the drug.11 Hemodynamic changes throughout the entirety of all infusions were also documented, including any additional treatment measures required (e.g., dexmedetomidine discontinuation, infusion rate reduction, or volume replacement).
The primary outcome was to identify the number of adverse events in patients receiving dexmedetomidine. A standard data collection form approved by the Institutional Review Board was used by the investigators. Any potential adverse effect was identified by one investigator (BLH) and confirmed by a second investigator (PNJ). An adverse event was defined as documented hypotension, rebound hypertension, bradycardia, apnea, or neurological abnormalities. For the purpose of this study, bradycardia was defined as a heart rate < 100 beats/minute in a newborn and < 60 beats/minute in an infant or child.12 Hypotension was defined as a systolic blood pressure (SBP) for term neonates (age 0 to 28 days) < 60 mmHg, a SBP for infants (age 1 to 12 months) < 70 mmHg, a SBP for children (age 1 to 10 years) < 70 + 2 × age in years mmHg, and for older children (age 10 to 17 years) a SBP < 90 mmHg.13 Transient hypertension was defined as an unexplained change in SBP that was more than 30 mmHg above baseline and persisted for 10 minutes.14
In addition to these adverse events, apnea was defined as a respiratory pause lasting more than 20 seconds or a shorter pause associated with cyanosis, pallor, hypotonia, or bradycardia < 100 beats per minute. 12 Data collection also included an assessment of neurological abnormalities that may have occurred during dexmedetomidine continuous infusion, as a result of extended use, or associated with discontinuation of therapy. This was defined as a change in verbal and motor function or an increased incidence of agitation and nervousness from baseline.
Several secondary outcomes were also assessed. First, each individual adverse event was categorized according to the number and type of event. Secondary analysis included an assessment of potential risk factors for adverse events including length of ICU stay, PRISM III score, duration of infusion, peak dexmedetomidine dose, and cumulative dexmedetomidine dose.
Statistical Analysis
Data were summarized using the mean, median, and range for continuous data and percentages for categorical information when appropriate. A stepwise forward multivariate logistic regression was used to determine factors (independent variables) that were associated with the occurrence of an adverse event. Regression model used adverse event/no adverse event as the dependent variable. Independent variables included length of PICU stay, PRISM III score, peak dose, duration of infusion, and total cumulative dose. This approach helped to ensure that each variable provided its own unique contribution to the overall model. Data were analyzed using SPSS for Windows (v14.0), with the a priori level of significance set at p ≤ 0.05.
RESULTS
Patient Demographics
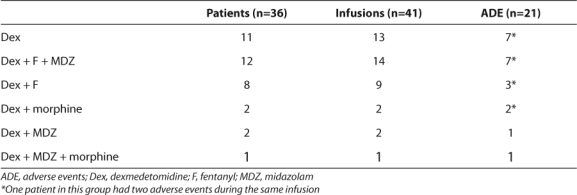
Four hundred fifty- five patients were screened for enrollment. Because the duration of dexmedetomidine infusion was less than 2 hours or the drug was used for procedural sedation, 400 patients were excluded. Although 60 infusions continued for longer than 2 hours in 55 patients, 19 of these patients were excluded due to incomplete medical records. Forty-one infusions, representing 36 patients, were included for analysis (Table 1). Five of these patients received two separate dexmedetomidine infusions during the same admission with at least 12 hours between infusions.
Table 1.
Summary of Medications Used for Sedation and Analgesia and Associated Number of Adverse Events
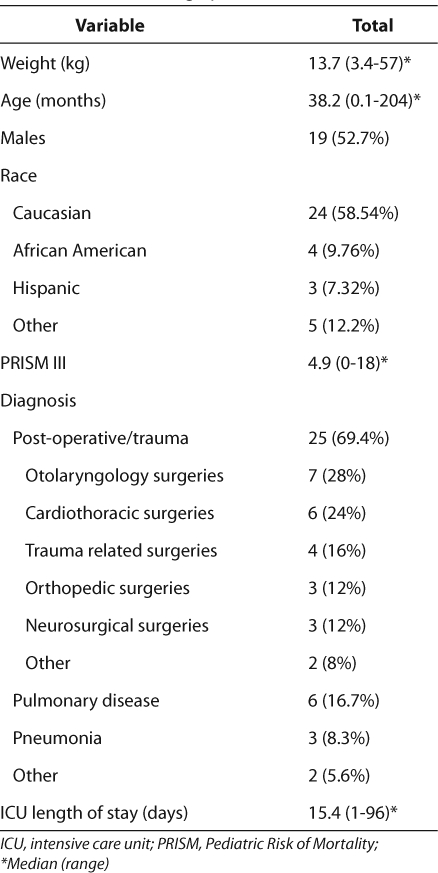
Baseline patient demographic data are shown in Table 2. The majority of patients were Caucasian males, and the average age was 38.2 ± 47.6 months. Although patients were admitted to the PICU for various reasons, the main diagnosis was post-operative/trauma. The mean calculated PRISM III score was 4.9 ± 4.7 and the mean length of stay in the ICU was15.4 ± 20.8 days.
Table 2.
Baseline Demographics (n=36)
A number of patients were receiving concomitant sedative and analgesic agents at the start of dexmedetomidine therapy. Both midazolam and fentanyl were given concurrently with dexmedetomidine in 14 of 41 infusions. Either fentanyl (n=9) or midazolam (n=2) were given at the initiation of dexmedetomidine infusion. The majority of patients were started on dexmedetomidine to facilitate extubation.
Dexmedetomidine Dosage Regimen
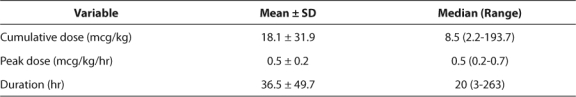
Table 3 contains information describing the dexmedetomidine dosage regimens. Eighteen (43.9%) infusions contained bolus doses to initiate dexmedetomidine therapy. The median cumulative dose (mcg/kg) and peak dosage (mcg/kg/hr) were 8.5 and 0.5, respectively. There was great variability in duration of dexmedetomidine (Table 2), with one patient receiving therapy for approximately 11 days. In order to prevent possible withdrawal symptoms, 6 (14.6%) infusions were tapered slowly at the discretion of the PICU physician.
Table 3.
Dexmedetomidine Dosage Regimen (n=41)
Adverse Events
Twenty-one adverse events occurred during 41 infusions (51%) (Table 1). Collectively, 13 of the 36 patients experienced 1 adverse event and 4 patients experienced more than 1 event. Adverse events were noted in 7 of 14 separate infusions (42.8%) in those who received dexmedetomidine, fentanyl, and midazolam concomitantly. Four adverse were reported during 11 different infusions in patients receiving fentanyl or midazolam and dexmedetomidine. The remaining five adverse events occurred in patients receiving dexmedetomidine alone.
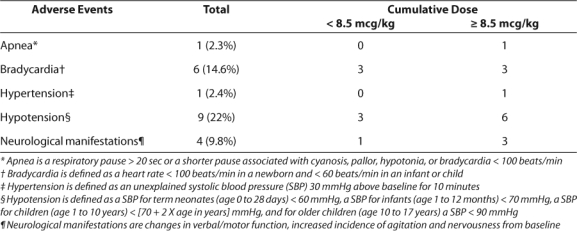
Hypotension and bradycardia were observed most often, occurring in 9 and 6 infusions, respectively (Table 4). Two patients experienced both hypotension and bradycardia and another showed neurological abnormalities and hypotension. Another patient suffered from both apnea and neurological abnormalities. One patient experienced hypertension and apnea; however, the events occurred during separate infusions.
Table 4.
Dexmedetomidine Adverse Events
There was a slight difference in mean arterial pressures (MAP) before and after the start of dexmedetomidine (66.6 ± 13.1 and 71.6 ± 13.7 mmHg). The MAP at the mid-point of the infusion was decreased (64.2 ± 12.2 mmHg) as expected. The MAP at the end of the infusion and the value obtained 12 hours after discontinuation were not markedly different (71.6 ± 13.7 and 70.2 ± 13.8 mmHg, respectively). There were no meaningful differences between mean ± SD heart rates one hour before and after the start of dexmedetomidine (126.5 ± 28.1 and 126.4 ± 24.2 beats per minute, respectively). Although heart rate decreased during the infusion (114.3 ± 23.6), it returned to pretreatment baseline at the one and 12 hour post-infusion times.
Of concern, neurological abnormalities were noticed with 4 separate infusions (9.8%) in four patients whose age ranged from 8 months to 11 years of age. The patient receiving dexmedetomidine for 11 days suffered transient neurological abnormalities (i.e., decreased verbal communication, facial drooping, and unilateral pupil dilation) upon discontinuation of the medication. This patient did not receive a bolus dose but was continued on a dose of 0.7 mcg/kg/hr of dexmedetomidine throughout the duration of therapy. He was not tapered off dexmedetomidine. The other three children who suffered from transient neurological abnormalities (i.e., increased agitation, abnormal chewing motions, non-reactive pupils, slow rhythmic jerking motions, and abnormal head turning) received dexmedetomidine from 6 to 49 hours. Two of these patients were tapered off dexmedetomidine.
To determine if there was an association between the total amount of dexmedetomidine infused and adverse events, the patients were stratified into those receiving < 8.5 mcg/kg and those given ≥ 8.5 mcg/kg (Table 3). There appears to be a larger number of adverse events in patients who received a larger cumulative dose of dexmedetomidine or a longer duration or therapy.
Fourteen of 17 patients (82.4%) or 14 of 21 infusions (66.7%) required interventions due to adverse effects. Dexmedetomidine was discontinued in 9 patients and the dose was decreased in 2 patients. Three patients received fluid boluses with crystalloids (0.9% Normal Saline, USP).
Logistic Regression Analysis
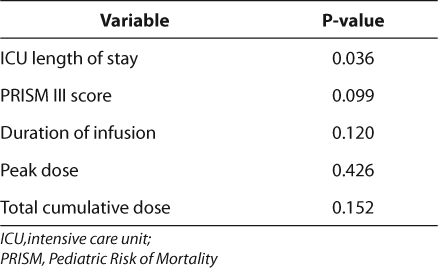
A logistic regression analysis was completed with adverse events as the dependent variable comparing the ICU length of stay, PRISM III score, infusion, peak dose, duration of dexmedetomidine, and total cumulative dose as independent variables (Table 5). Length of ICU stay was the only statistically significant independent risk factor (OR=4.193, p<0.05) for the development of adverse events.
Table 5.
Regression Analysis (n=41)
DISCUSSION
Dexmedetomidine is currently being used for sedation and analgesia in children in the ICU. This agent provides both sedative and analgesic effects without the clinically significant respiratory depression seen with other agents (e.g., fentanyl or morphine). Numerous case series show the value of dexmedetomidine in the pediatric population.15–18 Buck and Willson prospectively observed 17 infants and children who received dexmedetomidine in the PICU. 3 They concluded that dexmedetomidine allowed for reduction or elimination of midazolam, and it was particularly useful in specific subsets of the population in whom traditional sedatives fail to produce the desired response (i.e., chronic neurologic impairment). 3 Dexmedetomidine has also been useful for reducing the amount of narcotics.2 In addition, dexmedetomidine is utilized in some patients for facilitation of extubation, and recent reports have described the potential utility of dexmedetomidine for this indication. Carroll and colleagues described dexmedetomidine use in 41% (n=74) of pediatric cases as a bridge to extubation while other sedatives were weaned or discontinued and in 53% of cases to supplement other sedatives judged to be inadequate.4
The primary indication for the initiation of dexmedetomidine in this study was difficult to determine due to its retrospective nature. We noted a trend in reduction of fentanyl and other sedatives following dexmedetomidine initiation. The dosage regimens seen in our patients are listed in Table 2. The dosing observed in this study is within the Food and Drug Administration (FDA) approved range for adults. For adult patients, dexmedetomidine can be initiated with a loading infusion of 1 mcg/kg, followed by a maintenance infusion of 0.2 to 0.7 mcg/kg/hr.1 The dosage range of 0.2 to 0.7 mcg/kg/hr is consistent in our study and is comparable to other published dexmedetomidine literature in pediatrics.2–5,19
Duration of dexmedetomidine therapy varies significantly between studies. The manufacturer urges against dexmedetomidine use greater than 24 hours.1 Our average infusion time was 36.5 hours with a range of 3 to 263 hours. To our knowledge, there are two other case series that describe the use of dexmedetomidine for longer than 24 hours. Carroll et al. described a patient receiving dexmedetomidine for 451 hours (3–451 hours).4 Walker et al. reported pediatric burn patients on dexmedetomidine for an average duration of 11 days (2–50 days).5 The safety and efficacy of dexmedetomidine for more than 24 hours has not been evaluated. It is possible that these patients could have a higher incidence of some types of adverse events. We noted, in our institution, a higher incidence of adverse events in children receiving dexmedetomidine for a prolonged duration or at a higher cumulative dose.
This is a novel study looking directly at adverse events of dexmedetomidine in children. Hemodynamic effects of dexmedetomidine (i.e., hypotension, bradycardia, and transient hypertension) are well known and documented. Carroll and colleagues reported a case series of 60 patients receiving 74 dexmedetomidine infusions.4 They noted hypotension (9%), hypertension (8%), and bradycardia (3%) as the most common adverse events seen with dexmedetomidine. The incidence of hypotension (22%) and bradycardia (14.6%) found in our study were higher than that found in previous studies. Due to our study design and small sample size it is difficult to determine if the adverse events were related to individual patient characteristics, severity of illness, and/or duration of dexmedetomidine therapy.
The developments of neurological abnormalities and symptoms following abrupt withdrawal of dexmedetomidine are rare, but have been reported with dexmedetomidine administration.3,20 Identification of these symptoms is difficult to assess, especially in children. The manufacturer warns about the potential for withdrawal symptoms if dexmedetomidine is administered chronically and abruptly stopped.1 Withdrawal symptoms (e.g., nervousness, agitation, headaches, and rapid rise in blood pressure) have been reported extensively with clonidine, another alpha-2-adrenergic agent.1 In a prospective study, Buck and colleagues monitored for alterations in mental status.3 The trial had 10 patients (59%) with chronic neurologic impairments (e.g., Down syndrome). This study evaluated the use of prolonged dexmedetomidine infusions with an average duration of 32 ± 21 hours, and authors did not find any neurologic adverse events attributable to dexmedetomidine. The authors concluded that it was unclear if the success of therapy and limited adverse events was from patient selection or a conservative dosing strategy. Unlike the previous trial, Venn et al. described an adult who developed dystonic movements while receiving 1.5 mcg/kg/hr of dexmedetomidine for 24 hours.20 Future studies and close patient assessment are needed to evaluate this potential adverse event.
There are several limitations to our study. First, the data were collected retrospectively, and some patients were excluded due to incomplete medical records. The retrospective design is not the most conducive method for identification of adverse events. Some adverse events could be overlooked or misinterpreted strictly secondary to this study design. Second, our patients were all in the pediatric ICU and were severely ill. The use of a control group comprised of PICU patients that did not receive dexmedetomidine would enable one to evaluate whether the number of adverse events observed in the study was due to the patients' co-morbid conditions, concomitant medications, and/or the prolonged pediatric ICU length of stay. An additional limitation of the study is the small sample size. Due to these limitations, it is difficult to ascertain whether the adverse events seen were related to dexmedetomidine therapy or critical illness.
CONCLUSION
In this retrospective analysis, dexmedetomidine was associated with a number of adverse events in our pediatric population. While we noted changes in MAP and heart rate, these adverse events were for the most part not clinically significant and responded to a decrease in dose or a normal saline bolus. Although rare, a variety of transient and reversible neurological events have been reported. Clinicians should be knowledgeable about the potential of dexmedetomidine to cause neurological abnormalities upon abrupt discontinuation of therapy and should monitor patients for an increase in agitation, speech abnormalities, neuralgia, hallucinations/delirium, etc. Because symptoms can occur following abrupt withdrawal, it is also prudent to taper dexmedetomidine in any patient who has received therapy > 24–48 hours. A reasonable approach is to wean dexmedetomidine by 0.1 mcg/kg/hr every 12–24 hours. Future studies should evaluate the safety profile of dexmedetomidine, specifically assessing the potential for possible neurological abnormalities.
Acknowledgments
The authors would like to thank Beth Resman-Targoff, PharmD for her meticulous review of this article. Platform presentations at the Pediatric Pharmacy Advocacy Group Spring Meeting, Little Rock, AR, April 2008, and ALCALDE Residency Conference, Dallas, TX, April 2008; Poster Presentation at the Society of Critical Care Medicine's 38th Critical Care Congress, Nashville, Tennessee, February 2009.
ABBREVIATIONS
- ICU
intensive care unit
- PRISM
Pediatric Risk of Mortality score
- MAP
mean arterial pressures
- SBP
systolic blood pressure
Footnotes
see Editorial on page 4
DISCLOSURE The authors declare no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria.
REFERENCES
- 1.Precedex [package insert] Lake Forest, IL: Hospira; 2004. [Google Scholar]
- 2.Tobias JD, Berkenbosch JW. Sedation during mechanical ventilation in infants and children: dexmedetomidine versus midazolam. South Med J. 2004;97:451–455. doi: 10.1097/00007611-200405000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Buck ML, Willson DF. Use of dexmedetomidine in the pediatric intensive care unit. Pharmacotherapy. 2008;28:51–57. doi: 10.1592/phco.28.1.51. [DOI] [PubMed] [Google Scholar]
- 4.Carroll CL, Krieger D, Campbell M, et al. Use of dexmedetomidine for sedation of children hospitalized in the intensive care unit. J Hosp Med. 2008;3:142–147. doi: 10.1002/jhm.282. [DOI] [PubMed] [Google Scholar]
- 5.Walker J, MacCallum M, Fischer C, et al. Sedation using dexmedetomidine in pediatric burn patients. J Burn Care Res. 2006;27:206–210. doi: 10.1097/01.BCR.0000200910.76019.CF. [DOI] [PubMed] [Google Scholar]
- 6.Gerlach AT, Dasta JF. Dexmedetomidine: an updated review. Ann Pharmacother. 2007;41:245–254. doi: 10.1345/aph.1H314. [DOI] [PubMed] [Google Scholar]
- 7.Bhana N, Goa KL, McClellan KJ. Dexmedetomidine. Drugs. 2000;59:263–268. doi: 10.2165/00003495-200059020-00012. [DOI] [PubMed] [Google Scholar]
- 8.Ingersoll-Weng E, Manecke GR, Thistlewaite PA. Dexmedetomidine and cardiac arrest. Anesthesiology. 2004;100:738–739. doi: 10.1097/00000542-200403000-00040. [DOI] [PubMed] [Google Scholar]
- 9.Peden CJ, Cloote AH, Stratford N, Prys-Roberts C. The effect of intravenous dexmedetomidine premedication on the dose requirement of propofol to induce loss of consciousness in patients receiving alfentanil. Anaesthesia. 2001;56:408–413. doi: 10.1046/j.1365-2044.2001.01553.x. [DOI] [PubMed] [Google Scholar]
- 10.Pollack MM, Patel KM, Ruttimann UE. PRISM III: an updated Pediatric Risk of Mortality score. Crit Care Med. 1996;24:743–752. doi: 10.1097/00003246-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Petroz GC, Sikich N, James M, et al. A phase 1, two-centered study of the pharmacokinetics and pharmacodynamics of dexmedetomidine in children. Anesthesiology. 2006;105:1098–1110. doi: 10.1097/00000542-200612000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Gunn VL, Nechyba C, editors. The Harriet Lane Handbook. 16th ed. Philadelphia: Mosby; 2002. [Google Scholar]
- 13.Zaritsky AL, Nadkarni VM, Hickey RW, et al., editors. PALS provider manual. Texas: American Heart Association; 2002. [Google Scholar]
- 14.Siddappa R, Fletcher JE, Heard AMB, et al. Methadone dosage for prevention of opioid withdrawal in children. Pediatr Anesth. 2003;13:805–810. doi: 10.1046/j.1460-9592.2003.01153.x. [DOI] [PubMed] [Google Scholar]
- 15.Chrysostomou C, Zeballos T. Use of dexmedetomidine in a pediatric heart transplant patient. Pediatr Cardiol. 2005;26:651–654. doi: 10.1007/s00246-005-0683-3. [DOI] [PubMed] [Google Scholar]
- 16.Chrysostomou C, Di Filippo S, Manrique A, et al. Use of dexmedetomidine in children after cardiac and thoracic surgery. Pediatr Crit Care Med. 2006;7:126–131. doi: 10.1097/01.PCC.0000200967.76996.07. [DOI] [PubMed] [Google Scholar]
- 17.Hammer GB, Philip BM, Schroeder AR, Rosen FS, Koltai PJ. Prolonged infusion of dexmedetomidine for sedation following tracheal resection. Pediatr Anesth. 2005;15:616–620. doi: 10.1111/j.1460-9592.2005.01656.x. [DOI] [PubMed] [Google Scholar]
- 18.Kalyanaraman M, Costello JL, Starr JP. Use of dexmedetomidine in patients with Trisomy 21 after cardiac surgery. Pediatr Cardiol. 2007;28:396–399. doi: 10.1007/s00246-006-0072-6. [DOI] [PubMed] [Google Scholar]
- 19.Diaz SM, Rodarte A, Foley J, Capparelli EV. Pharmacokinetics of dexmedetomidine in postsurgical pediatric intensive care unit patients: preliminary study. Pediatr Crit Care Med. 2007;8:419–424. doi: 10.1097/01.PCC.0000282046.66773.39. [DOI] [PubMed] [Google Scholar]
- 20.Venn RM, Newman PJ, Grounds RM. A phase II study to evaluate the efficacy of dexmedetomidine for sedation in the medical intensive care unit. Intensive Care Med. 2003;29:201–207. doi: 10.1007/s00134-002-1579-9. [DOI] [PubMed] [Google Scholar]