

Abstract
Although various schemes for interstrand crosslink (ICL) repair incorporate recombination, replication, and double-strand break intermediate steps, action of the NER system or some variation of it is a common feature of most models. In the bacterium Escherichia coli, the NER enzyme UvrABC can incise on either side of an ICL to unhook the crosslink, and repair can proceed via a subsequent recombination step. The relevance of NER to ICL repair in mammalian cells has been challenged. Of all NER mutants, it is clear that ERCC1 and XPF-defective cells show the most pronounced sensitivities to ICL-inducing agents, and defects in ICL repair. However, there is good evidence that cells defective in NER proteins including XPA and XPG are also more sensitive than normal to ICL-inducing agents. These results are summarized here, together with evidence for defective crosslink removal in NER-defective cells. Studies of incision at sites of ICL by cell extracts and purified proteins have been done, but these studies are not all consistent with one another and further research is required.
Keywords: DNA crosslinks, UvrABC, XP proteins, psoralen ICL, NER
Interstrand crosslinks and their significance
DNA interstrand crosslinks (ICL) are among the most toxic forms of DNA damage because they disrupt genetic information on both strands. They present potent blocks to DNA replication and transcription, and lead to toxic and mutagenic effects that are outlined throughout this special issue. Some environmental and endogenous agents can form ICLs and so there has been significant selection during evolution for the development of mechanisms that allow cells to repair and tolerate these lesions. Understanding the repair of ICLs is also very relevant because agents that form ICLs are widely used in cancer chemotherapy and phototherapy. Clinically useful interstrand crosslinking agents include nitrogen mustards and derivatives (melphalan, chlorambucil), psoralens, mitomycin C, platinum-based compounds such as cisplatin, and nitrosoureas such as BCNU (Dronkert and Kanaar 2001; Noll et al. 2006).
Naturally occurring environmental and endogenous agents also lead to ICLs. Such agents include the furocoumarins present in many plants (Scott et al. 1976), nitrous acid in food (Edfeldt et al. 2004); endogenous agents include products of lipid peroxidation, and nitric oxide (Caulfield et al. 2003; Schärer 2005). Further research is warranted towards development of methods to detect the formation and repair of crosslinks caused by these agents in cells.
There may be several mechanisms of crosslink repair in eukaryotes, operating differently in different phases of the cell cycle or perhaps in different tissues. As described in other articles in this issue, proteins from multiple pathways are implicated, including homologous recombination, the Fanconi anemia network, mismatch repair, and translesion DNA synthesis. Most models for ICL repair implicate nucleotide excision repair (NER) in one or more steps (Fig. 1).
Figure 1.
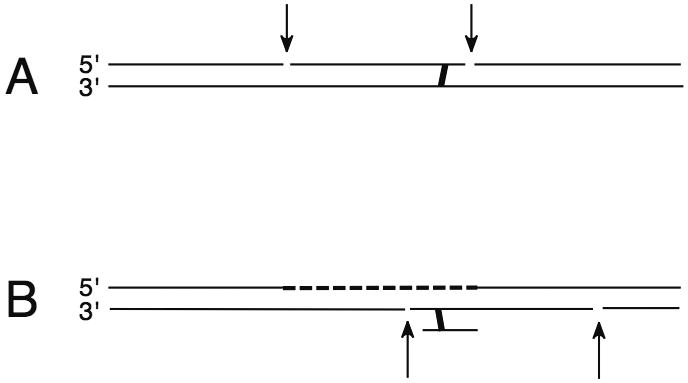
Two of the possible roles for NER in processing of ICL. A, dual incision on either side of a crosslink; B, dual incision to remove a crosslink unhooked on one strand, for example after translesion DNA synthesis has re-synthesized DNA on an initially processed strand.
Interstrand Crosslink Repair in E. coli
UvrABC endonuclease can incise DNA at sites of interstrand crosslinks
The E. coli NER UvrABC endonuclease can act on ICL formed in DNA by photoactivated psoralen. Thymine-psoralen-thymine ICL have one strand linked via the furan ring of the psoralen and the other strand attached to the pyrone ring (Hearst et al. 1984). Incision of this asymmetrical lesion has been observed at the ninth phosphodiester bond 5′ to the furan-adducted strand and at the third bond 3′ to the adduct on the same strand, with the pyrone strand remaining intact (Van Houten et al. 1986a; Van Houten et al. 1986b). This biased incision reaction apparently reflects a conformational constraint, because no preference for furan ring adducts is exhibited for incision of psoralen monoadducts (Van Houten et al. 1986b). The incision of crosslinked DNA by the UvrABC endonuclease is also influenced by the superhelical density of substrate DNA. The rate of incision of supercoiled DNA containing a psoralen furan side monoadduct is relatively insensitive to superhelix density, but incision of DNA crosslinked by psoralen is most efficient with underwound DNA (Munn and Rupp 1991).
Strand selection of ICL incision may be influenced by sequence context. For a psoralen ICL, enrichment of the G+C content of the region immediately 5′ to the modified thymine residue on the furan strand results in preferential incision of that strand, but incision occurs on either the furan or pyrone strands when the 3′ side of this lesion is enriched for G+C (Jones and Yeung 1990; Ramaswamy and Yeung 1994). Cleavage of both strands of an ICL to produce a double-strand break has also been observed. Peng et al (2010) examined incision of an A-T crosslink formed by reaction of a 5-(2′-deoxyuridinyl)methyl radical with the opposing 2′-deoxyadenosine in DNA. This lesion is one of the products formed by reactive oxygen species following ionizing radiation-mediated radiolysis of water. Using recombinant UvrABC derived from thermophilic bacteria, it was found that the strand containing the crosslinked A was preferentially incised. Both strands were cleaved in 25% or more of the events, apparently from two rounds of incision before dissociation of the enzyme.
More complex adducts can also be cleaved by bacterial UvrABC. A thymine-psoralen-thymine ICL directed to DNA by a triplex-forming oligonucleotide (TFO) was a substrate for UvrABC (Christensen et al. 2008). The triplex was disrupted by UvrAB during the cleavage reaction, but the TFO remained covalently linked to the excised fragment. Sczepanski et al (2009) examined cleavage of an ICL produced by reaction of a C4′-oxidized abasic site. Such oxidized abasic sites are formed by several DNA damaging agents including bleomycin, and can react in duplex DNA to form an ICL and a nick in the opposite strand. On such a compound lesion, UvrABC preferentially incised the ICL on the strand containing the nick, and in approximately 15% of the molecules, the strand opposite the nick was incised, resulting in a double-strand break.
NER and recombination steps allow complete repair of an ICL in E. coli
Complete ICL repair may be achieved by combining NER and recombination steps (Cole 1973). NER-mediated incision of an ICL generates an “unhooked crosslink” that is covalently attached to DNA through the crosslink. Isomerization of the oligonucleotide would generate a gapped structure that could be repaired by E. coli RecA-catalyzed recombinational events (Sinden and Cole 1978a; Sinden and Cole 1978b).
Biochemical experiments support this model (Sladek et al. 1989a). After incisions on one strand are made by the UvrABC endonuclease on each side of a psoralen ICL, a gap flanking the incised region can be generated in vitro by the exonucleolytic action of the 5′ to 3′ exonuclease of E. coli Pol I. Such a gap is required for the initiation of polymerization by the RecA protein. The RecA-mediated product includes a short triple-stranded region due to the presence of the crosslink (Sladek et al. 1989b). Another way in which a gap may be formed to facilitate RecA-dependent homologous recombination might be through the action of UvrABC itself. In some instances, UvrABC can make additional 5′ cuts in DNA flanking the initial 5′ incision (Gordienko and Rupp 1998).
Following recombination, a second NER-mediated incision on the other strand may release the crosslinked oligonucleotide and leave a second gap to be repaired by conventional repair synthesis and DNA ligation. The UvrABC endonuclease is able to incise a three-stranded structure in which one of the strands is an oligonucleotide covalently attached to double-stranded DNA (Cheng et al. 1988). In the presence of RecA protein and a third strand positioned at the crosslinked site (provided by an oligonucleotide homologous to the furan-side strand), the UvrABC endonuclease catalyzes incision of the pyrone side strand (Cheng et al. 1991).
ICL repair and mammalian NER proteins
The mammalian NER system
The defining event for NER (Figure 2) is the excision of damage in a short oligonucleotide, by cleavage of the damaged strand on either side of a lesion in DNA. Chapters 8, 9 and 10 of Friedberg et al. (2006) remain a comprehensive summary of the process, and some more recent references and updates are given below. Adducts in DNA initially cause some local distortion or unusual deformability of the helix. This distortion appears to be the first structural feature recognized. In mammalian cells, initial distortion recognition generally requires binding of the XPC-RAD23B complex. This complex binds damaged DNA in two stages (Clement et al. 2009). The first is very rapid binding to non-hydrogen bonded regions in duplex DNA via two β-hairpin domains in XPC (Trego and Turchi 2006; Camenisch et al. 2009), followed by anchoring more tightly to the nondamaged strand of a region containing an adduct (Min and Pavletich 2007) with the aid of a third β-hairpin domain (Camenisch et al. 2009). Binding of the XPC complex to a region containing a few DNA bases of single-stranded character facilitates the conveyance of TFIIH to the distorted site (Li et al. 1998; Yokoi et al. 2000; Araújo et al. 2001). XPC-RAD23B interacts strongly with TFIIH and TFIIH may be able to carry out a 5′ to 3′ search for a short-distance before anchoring to a site of DNA damage in cooperation with the other protein components of the preincision complex (Sugasawa et al. 2009).
Figure 2.
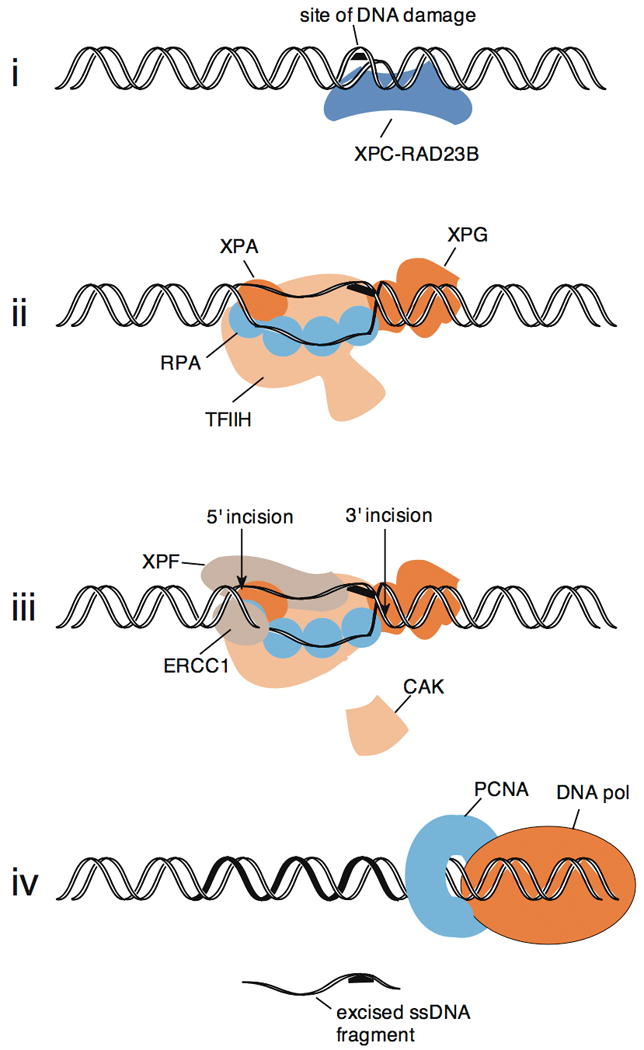
Diagram of events leading to dual incision in the human NER system for an adduct on one strand, such as a UVC-induced (6-4) photoproduct. The proteins are shown roughly to scale, relative to DNA. In step (i), XPC-RAD23B binds preferentially to a site of DNA where base pairing is disrupted by lesion. At least 17 bp of DNA are contacted, including the nondamaged DNA strand opposite the lesion (Min and Pavletich 2007). XPC-RAD23B binds TFIIH, which enters the DNA with other components in step (ii), where a pre-incision complex is formed including TFIIH of dimensions ∼ 16 nm × 12.5 nm × 7.5 nm, (Schultz et al. 2000), XPA, RPA (which includes 4 active DNA-binding OB fold domains amongst its 3 subunits), and XPG. The DNA strands are separated in this step in an ATP-dependent reaction (Evans et al. 1997a). In step (iii), the XPF-ERCC1 nuclease complex produces a 5′ incision and the XPG nuclease the 3′ incision. The two incisions take place nearly simultaneously after formation of a productive pre-incision complex. The CDK7-activating (CAK) subunits of TFIIH are not present in the fully formed pre-incision complex and detach from core TFIIH (Coin et al. 2008). In step (iv), after dual incision, a DNA polymerase holoenzyme utilizing the sliding clamp PCNA fills in the gap created by incision, and a single-stranded DNA piece containing the lesion is released.
The next key event is the formation of an unwound DNA structure around a lesion in a preincision assembly of protein factors generating an “open complex.” This opening confers single-stranded character to approximately 25 to 30 DNA bases in the vicinity of the lesion (Evans et al. 1997a). Proteins necessary and sufficient to form a productive, open preincision complex are TFIIH, XPA, RPA, and XPG (Evans et al. 1997b). On a TFO-directed psoralen crosslink, XPC-RAD23B may cooperate in binding to damage with these other NER proteins (Thoma et al. 2005). The catalytic activity for opening the DNA strands is provided by TFIIH, which among its 10 subunits includes the two ATP-dependent helicases XPB and XPD. The catalytic activity of the XPD helicase is most important for NER. Because of its size, extensive helicase activity on the DNA is not needed to create the opened region in the preincision complex, and only a modest conformational change is likely necessary to promote unwinding (Fig. 2). XPA binds mostly tightly to junctions between single-stranded and duplex DNA, and to helically-kinked DNA (Camenisch et al. 2006). XPA also acts as a protein scaffold for binding to TFIIH, RPA, and ERCC1-XPF (Friedberg et al. 2006). The entry of XPA into the preincision complex further promotes the release of three subunits of TFIIH (Coin et al. 2008), the “CAK complex”, which are unnecessary in the catalysis of NER (Araújo et al. 2000). Rejoining of core TFIIH with the CAK complex occurs following NER, ensuring that active TFIIH is available for its transcription initiation function (Coin et al. 2008).
This opened intermediate is the substrate for cleavage by the structure-specific NER nucleases. At the border between duplex DNA and the opened region, XPG nuclease cuts 3′ to the lesion on the damaged DNA strand, and ERCC1-XPF nuclease cleaves on the 5′ side (Friedberg et al. 2006; Staresincic et al. 2009). This dual incision event in NER releases a fragment of approximately 24–32 nt containing the damage. DNA synthesis then forms a DNA repair patch of this size. The patch is sealed by a DNA ligase, either LIG3 or LIG1 (Moser et al. 2007).
As summarized later, NER mutants are in general hyper-sensitive to agents that form ICLs. The most pronounced sensitivity is displayed by cells defective in ERCC1 and XPF. This is consistent with an additional role for the structure-specific nuclease formed by ERCC1 and XPF proteins in ICL repair. Examples of relevant activities are the ability to cleave near a crosslink at the junction of a forked structure (Kuraoka et al. 2000). A similar reaction has been confirmed with a crosslink in duplex DNA near two duplex arms simulating a Y-shaped stalled replication fork (Fisher et al. 2008). ERCC1-XPF may participate in a recombination pathway where it clips off strands with 3′ tails, by analogy with the homologous Rad1-Rad10 enzyme in yeast (Bardwell et al. 1994; Paques and Haber 1999). These roles are discussed in detail by Nairn et al in this issue.
Evidence for involvement of mammalian NER enzymes in ICL repair
There is a possible conceptual difficulty with visualizing how dual incision of an ICL might work. During an NER reaction on a unistranded adduct, some separation of the DNA strands occurs on both sides of a lesion. An ICL prevents full strand separation but this is not a problem in practice for the E. coli UvrABC system, as described above. UvrABC also works by a mechanism involving an ATP-dependent strand separation step [reviewed in (Batty and Wood 2000; Croteau et al. 2006)], and it is able to cleave on both sides of an ICL. With regard to the mammalian system, it is possible that unwinding could take place on both sides of an ICL. It is also possible that unwinding to create an open structure might occur only on one side, but that dual incisions flanking the open structure could still flank the ICL. ERCC1-XPF is able to cleave several bases 5′ of an unpaired junction, and depending on the placement of an ICL in a duplex with respect to two single-stranded arms, is able to cut on either side of an ICL (Kuraoka et al. 2000). Similarly, XPG may cut a few bases 3′ to a junction between duplex and single-stranded DNA (Evans et al. 1997a). It is also possible that additional nucleases might be used in ICL unhooking in some situations.
Sensitivity of mammalian NER mutants to ICL-inducing agents
It is important to consider carefully whether NER is relevant to ICL repair in cells. It is widely agreed that the marked hypersensitivity of ERCC1-XPF mutants to ICL-inducing agents indicates that this enzyme has an additional function in ICL repair that is separate from its role in NER. Nevertheless, almost all models of ICL incorporate NER into the process at one or more steps, whether for G0/G1 phase or S phase. There is ample evidence that some repair of ICL takes place outside of S-phase and does not require replication of the substrate, as shown in yeast (Sarkar et al. 2006) and mammalian cells (Ben-Yehoyada et al. 2009; Shen et al. 2009). There is, however, some recent tendency to dismiss the importance of the NER process in the processing of ICL in mammalian cells. For example, an opinion article discussing ERCC1-XPF function in ICL repair (Bergstralh and Sekelsky 2008) contains the following statement:
“Mammalian cell lines with mutations in ERCC1 or XPF are hypersensitive to crosslinking agents, but those mutant for XPB, XPD or XPG are not (Andersson et al. 1996; De Silva et al. 2000; Niedernhofer et al. 2004; Clingen et al. 2005). Likewise, fibroblast cells derived from XP-F patients show enhanced sensitivity to MMC and HN2, whereas those from XP-A patients do not, even though both are deficient in NER (Niedernhofer et al. 2006; Clingen et al. 2007). Moreover, ICL DNA is efficiently replicated in vitro in the absence of XPA (Cipak et al. 2006).”
Is it correct that there is a lack of sensitivity to ICL inducing agents in cells with NER deficiencies other than ERCC1-XPF? Examination of the evidence in each of the above citations shows otherwise. Andersson and co-workers (Andersson et al. 1996) investigated the sensitivity of NER-defective CHO cell lines to cisplatin and to cyclophosphamide derivatives. For cisplatin, the relative drug ratio at 0.1 survival compared to wild type was 50 for ERCC1 and XP-F mutants; 10 for XP-G, and 7 for XP-B and XP-D. Thus all of the NER-defective mutants are hypersensitive to cisplatin, with ERCC1 and XPF of course the most sensitive. With cyclophosphamide derivatives, the relative sensitivity of ERCC1 and XPF mutants was more pronounced, but XPB (ERCC3) and XPG (ERCC5) mutants were more sensitive than NER-proficient cells. De Silva et al. (De Silva et al. 2000) used a cell counting assay to measure sensitivity of CHO cells to nitrogen mustard (HN2). ERCC1 and XPF-defective cells were the most sensitive, but XPB, XPD and XPG-defective cells exhibited measurable sensitivity compared to normal. In a further study, De Silva et al. (De Silva et al. 2002) found that all CHO cell NER mutants tested were somewhat more sensitive than normal to cisplatin (with ERCC1 and XPF the most sensitive). Niedernhofer et al. (2004) contains no information comparing ICL agent sensitivity of ERCC1 or XPF mutant cells to other NER mutant cells, but Clingen et al. (2005) showed that XPB, XPD, and XPG defective cells are more sensitive than normal cells to the ICL-inducing agent melphalan (with ERCC1 and XPF cells even more sensitive).
Niedernhofer et al. (2006) found that primary XPA mutant human primary fibroblasts are more sensitive than normal controls to MMC, as are mouse embryonic fibroblasts, though of course not as sensitive as XPF mutants. Clingen et al. (2007) showed that HN2 inhibits the growth of XPA mutant cells as much or more that the growth of XPF mutant cells in a short term assay. This study also found an unhooking defect in both XPA and XPF mutants. Cipak et al. (2006) does not contain any information on efficient replication of DNA containing an ICL. The conclusion of the paper is that removal of the unhooked lesion is XPA-dependent. In an older study, Hoy et al (1985) showed that XPB and XPG mutants of CHO cells are more sensitive than normal to MMC.
In conclusion, NER mutants of mammalian cells do exhibit hypersensitivity to ICL-inducing agents, though in general not as great as ERCC1- or XPF-defective mutants.
Defects in ICL repair in mammalian NER mutants
Hypersensitivity to ICL-inducing agents does not in itself prove that NER incises ICL in mammalian cells, because most agents that create ICL also form monoadducts that are potential NER substrates. However, there are several results which directly show ICL repair defects in NER-defective cells. Kaye et al (1980) used a gene-specific assay and found no removal of ICL formed by photoactivated 8-methoxypsoralen in XPA-deficient cells. Clingen et al (2007) used a comet tail assay and reported an unhooking defect in both XPA and XPF mutants. De Silva et al. (2002) used an antibody assay for removal of cisplatin-induced ICL, and found that XPG mutants exhibited a defect even greater than XPF or ERCC1 mutants. A psoralen ICL directed to a specific site in a luciferase or GFP reporter gene has also been used to assay non-recombination dependent ICL repair. Cell lines with mutations in XPA, XPB, XPD, XPG, ERCC1 and XPF were all defective in repair of the ICL, and XPC-deficient cells showed a partial defect (Wang et al. 2001).
Research with TFO-directed psoralen-DNA ICL has shown that NER proteins are involved in binding to ICLs and in their subsequent processing, as summarized by Vasquez in this issue. Replication-independent recruitment of the Fanconi anemia core complex to an ICL in DNA is dependent on the NER proteins XPA and XPC (Shen et al. 2009).
Biochemical experiments to observe incision of ICL repair in mammalian cell extracts
Incisions at site-directed ICL in mammalian cell extracts have been analyzed by several laboratories, using human cell extracts and/or purified NER proteins. A variety of outcomes have been observed. Using a both a single HMT-psoralen crosslink in a circular molecule, and a 140-mer linear duplex containing a single centrally located HMT-psoralen adduct, Bessho, Mu and Sancar found that mammalian cell extracts could carry out a “futile” NER-dependent reaction, introducing dual incisions both on the 5′ side of crosslink, principally 27 and 6 phosphodiester bonds 5′ to the cross linked base. This resulted in the removal of a 22 nt oligonucleotide, releasing an oligonucleotide that did not harbor the adduct within it (Bessho et al. 1997; Mu et al. 2000). The occurrence of this reaction is interesting as it confirms that ICL are recognized in some sense by the NER system in mammalian cell extracts. However, the existence of this reaction does not preclude the concomitant occurrence of unhooking reactions mediated by mammalian cell extracts. If this mechanism were to be productive, a 3′ incision occurring very close to the adduct could conceivably mediate further processing, for example facilitating DSB formation near the lesion.
Smeaton et al. (2008) observed a similar “futile” 5′ incision reaction using completely different chemically synthesized crosslinks, particularly an N4C-ethyl-N4C DNA interstrand crosslink placed in a duplex ∼150 bp long. This study reported additional cleavage positions surrounding the crosslink. Although not mapped precisely, the main positions of postulated incisions were 1-5 nt on the 5′ side and 3-5 nt on the 3′ side. The incisions were still present in NER-defective XP cell extracts, including ERCC1-XPF defective cells. To date, no cell extracts defective in producing these incisions have been found. It is not yet clear whether these events represent primary events, or whether they represent endo/exonucleolytic processing of the linear substrate, not related to repair-mediated incisions.
In other studies, Lambert's group reported that human cell extracts can incise weakly, both 5′ and 3′ of a trimethylpsoralen crosslink in a 132-140 bp segment of linear duplex DNA (Kumaresan et al. 1995). Nicks were observed 9 nt apart on the furan side of the crosslink. A later study indicated that group A Fanconi anemia cells were defective in making such incisions (Kumaresan and Lambert 2000), but this result has not been confirmed by complementation with FANCA.
Legerski, Li, and co-workers used a repair synthesis assay to approach the problem (Li et al. 1999). Closed-circular DNA containing a psoralen ICL stimulated repair synthesis by human cell extracts, depending on both the mismatch binding complex MutSβ (MSH2-MSH3) and ERCC1-XPF (Zhang et al. 2002). The repair synthesis started on the 5′ side of a crosslink and extended just past the adduct on the 3′ side. It remains to be determined whether this reaction represents processing of an ICL to release a crosslink (Zheng et al. 2003), but it does provide evidence that incision can take place on both sides of an adduct. The incisions were not dependent on other NER proteins tested (XP-B or XP-G) and were also not dependent on FANCA, FANCB, or FANCC proteins. There are further results implicating MutSβ in repair of ICL, as MutSβ binds to substrates containing ICL and influences their processing (Wu et al. 2005).
The above interpretations are not all consistent with one another, and the subject of dual incision of an ICL requires further detailed investigation. Further controlled investigations are necessary to reconcile these disparate results, to discover which reactions are robustly found in circular molecules, and to resolve which (if any) are in vitro artifacts of nucleolytic processing. Careful genetic and biochemical analysis is necessary to determine how proteins from the NER, MMR, and FA pathways are involved in these events in all phases of the cell cycle.
Acknowledgments
Research on ICL repair in the author's laboratory is supported by an NIH P01 grant from the National Cancer Institute, CA09717, and by grant P30ES007784 from the National Institute of Environmental Health Sciences.
References
- Andersson BS, Sadeghi T, Siciliano MJ, Legerski R, Murray D. Nucleotide excision repair genes as determinants of cellular sensitivity to cyclophosphamide analogs. Cancer Chemother Pharmacol. 1996;38:406–16. doi: 10.1007/s002800050504. [DOI] [PubMed] [Google Scholar]
- Araújo SJ, Nigg EA, Wood RD. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol. 2001;21:2281–91. doi: 10.1128/MCB.21.7.2281-2291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araújo SJ, Tirode F, Coin F, Pospiech H, Syväoja JE, Stucki M, Hübscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH and modulation by CAK. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science. 1994;265:2082–2085. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]
- Batty DP, Wood RD. Damage recognition in nucleotide excision repair of DNA. Gene. 2000;241:193–204. doi: 10.1016/s0378-1119(99)00489-8. [DOI] [PubMed] [Google Scholar]
- Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J. Checkpoint signaling from a single DNA interstrand crosslink. Mol Cell. 2009;35:704–15. doi: 10.1016/j.molcel.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh DT, Sekelsky J. Interstrand crosslink repair: can XPF-ERCC1 be let off the hook? Trends Genet. 2008;24:70–6. doi: 10.1016/j.tig.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Bessho T, Mu D, Sancar A. Initiation of DNA interstrand cross-link repair in humans: the nucleotide excision repair system makes dual incisions 5′ to the cross-linked base and removes a 22- to 28-nucleotide-long damage-free strand. Mol Cell Biol. 1997;17:6822–30. doi: 10.1128/mcb.17.12.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch U, Dip R, Schumacher SB, Schuler B, Naegeli H. Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat Struct Mol Biol. 2006;13:278–84. doi: 10.1038/nsmb1061. [DOI] [PubMed] [Google Scholar]
- Camenisch U, Trautlein D, Clement FC, Fei J, Leitenstorfer A, Ferrando-May E, Naegeli H. Two-stage dynamic DNA quality check by xeroderma pigmentosum group C protein. EMBO J. 2009;28:2387–99. doi: 10.1038/emboj.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield JL, Wishnok JS, Tannenbaum SR. Nitric oxide-induced interstrand cross-links in DNA. Chem Res Toxicol. 2003;16:571–4. doi: 10.1021/tx020117w. [DOI] [PubMed] [Google Scholar]
- Cheng S, Sancar A, Hearst JE. RecA-dependent incision of psoralen-crosslinked DNA by (A)BC excinuclease. Nucleic Acids Research. 1991;19:657–63. doi: 10.1093/nar/19.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Van Houten B, Gamper HB, Sancar A, Hearst JE. Use of psoralen-modified oligonucleotides to trap three-stranded RecA- DNA complexes and repair of these cross-linked complexes by ABC excinuclease. J Biol Chem. 1988;263:15110–15117. [PubMed] [Google Scholar]
- Christensen LA, Wang H, Van Houten B, Vasquez KM. Efficient processing of TFO-directed psoralen DNA interstrand crosslinks by the UvrABC nuclease. Nucleic Acids Res. 2008;36:7136–45. doi: 10.1093/nar/gkn880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipak L, Watanabe N, Bessho T. The role of BRCA2 in replication-coupled DNA interstrand cross-link repair in vitro. Nat Struct Mol Biol. 2006;13:729–33. doi: 10.1038/nsmb1120. [DOI] [PubMed] [Google Scholar]
- Clement FC, Camenisch U, Fei J, Kaczmarek N, Mathieu N, Naegeli H. Dynamic two-stage mechanism of versatile DNA damage recognition by xeroderma pigmentosum group C protein. Mutat Res. 2009 doi: 10.1016/j.mrfmmm.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Clingen PH, Arlett CF, Hartley JA, Parris CN. Chemosensitivity of primary human fibroblasts with defective unhooking of DNA interstrand cross-links. Exp Cell Res. 2007;313:753–60. doi: 10.1016/j.yexcr.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Clingen PH, De Silva IU, McHugh PJ, Ghadessy FJ, Tilby MJ, Thurston DE, Hartley JA. The XPF-ERCC1 endonuclease and homologous recombination contribute to the repair of minor groove DNA interstrand crosslinks in mammalian cells produced by the pyrrolo[2,1-c][1,4]benzodiazepine dimer SJG-136. Nucleic Acids Res. 2005;33:3283–91. doi: 10.1093/nar/gki639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell. 2008;31:9–20. doi: 10.1016/j.molcel.2008.04.024. [DOI] [PubMed] [Google Scholar]
- Cole RS. Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci USA. 1973;70:1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, DellaVecchia JJ, Skorvaga M, Van Houten B. Damage recognition by the bacterial nucleotide excision repair machinery. In: Siede W, Kow YW, Doetsch PW, editors. DNA Damage Recognition. New York: Taylor & Francis; 2006. pp. 111–138. [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–90. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002;30:3848–56. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–47. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Edfeldt NB, Harwood EA, Sigurdsson ST, Hopkins PB, Reid BR. Solution structure of a nitrous acid induced DNA interstrand cross-link. Nucleic Acids Res. 2004;32:2785–94. doi: 10.1093/nar/gkh606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Fellows J, Coffer A, Wood RD. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J. 1997a;16:625–638. doi: 10.1093/emboj/16.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997b;16:6559–6573. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher LA, Bessho M, Bessho T. Processing of a psoralen DNA interstrand cross-link by XPF-ERCC1 complex in vitro. J Biol Chem. 2008;283:1275–1281. doi: 10.1074/jbc.M708072200. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2nd. Washington, DC: ASM Press; 2006. p. 1118. [Google Scholar]
- Gordienko I, Rupp WD. A specific 3′ exonuclease activity of UvrABC. EMBO J. 1998;17:626–33. doi: 10.1093/emboj/17.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearst JE, Isaacs ST, Kanne D, Rapoport H, Straub K. The reaction of the psoralens with deoxyribonucleic acid. Quarterly Reviews of Biophysics. 1984;17:1–44. doi: 10.1017/s0033583500005242. [DOI] [PubMed] [Google Scholar]
- Hoy CA, Thompson LH, Mooney CL, Salazar EP. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985;45:1737–43. [PubMed] [Google Scholar]
- Jones BK, Yeung AT. DNA base composition determines the specificity of UvrABC endonuclease incision of a psoralen cross-link. J Biol Chem. 1990;265:3489–96. [PubMed] [Google Scholar]
- Kaye J, Smith CA, Hanawalt PC. DNA repair in human cells containing photoadducts of 8-methoxypsoralen or angelicin. Cancer Res. 1980;40:696–702. [PubMed] [Google Scholar]
- Kumaresan KR, Hang B, Lambert MW. Human endonucleolytic incision of DNA 3′ and 5′ to a site-directed psoralen monoadduct and interstrand cross-link. J Biol Chem. 1995;270:30709–16. doi: 10.1074/jbc.270.51.30709. [DOI] [PubMed] [Google Scholar]
- Kumaresan KR, Lambert MW. Fanconi anemia, complementation group A, cells are defective in ability to produce incisions at sites of psoralen interstrand cross-links. Carcinogenesis. 2000;21:741–51. doi: 10.1093/carcin/21.4.741. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. Repair of an interstrand DNA crosslink initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- Li L, Peterson CA, Lu XY, Wei P, Legerski RJ. Interstrand cross-links induce DNA synthesis in damaged and undamaged plasmids in mammalian cell extracts. Mol Cell Biol. 1999;19:5619–5630. doi: 10.1128/mcb.19.8.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RY, Calsou P, Jones CJ, Salles B. Interactions of the transcription/DNA repair factor TFIIH and XP repair proteins with DNA lesions in a cell-free repair assay. J Mol Biol. 1998;281:211–218. doi: 10.1006/jmbi.1998.1949. [DOI] [PubMed] [Google Scholar]
- Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–5. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH, Fousteri MI. Sealing of Chromosomal DNA Nicks during Nucleotide Excision Repair Requires XRCC1 and DNA Ligase IIIalpha in a Cell-Cycle-Specific Manner. Mol Cell. 2007;27:311–23. doi: 10.1016/j.molcel.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A. DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol Cell Biol. 2000;20:2446–2454. doi: 10.1128/mcb.20.7.2446-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn MM, Rupp WD. Interaction of the UvrABC endonuclease with DNA containing a psoralen monoadduct or cross-link. Differential effects of superhelical density and comparison of preincision complexes. J Biol Chem. 1991;266:24748–56. [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, et al. The structure-specific endonuclease ERCC!-XPF is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–87. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Ghosh AK, Van Houten B, Greenberg MM. Nucleotide Excision Repair of a DNA Interstrand Cross-Link Produces Single- and Double-Strand Breaks. Biochemistry. 2010;49:11–19. doi: 10.1021/bi901603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy M, Yeung AT. Sequence-specific interactions of UvrABC endonuclease with psoralen interstrand cross-links. J Biol Chem. 1994;269:485–92. [PubMed] [Google Scholar]
- Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006;25:1285–94. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schärer OD. DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. ChemBiochem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- Schultz P, Fribourg S, Poterszman A, Mallouh V, Moras D, Egly JM. Molecular structure of human TFIIH. Cell. 2000;102:599–607. doi: 10.1016/s0092-8674(00)00082-9. [DOI] [PubMed] [Google Scholar]
- Scott BR, Pathak MA, Mohn GR. Molecular and genetic basis of furocoumarin reactions. Mutat Res. 1976;39:29–74. doi: 10.1016/0165-1110(76)90012-9. [DOI] [PubMed] [Google Scholar]
- Sczepanski JT, Jacobs AC, Van Houten B, Greenberg MM. Double-strand break formation during nucleotide excision repair of a DNA interstrand cross-link. Biochemistry. 2009;48:7565–7. doi: 10.1021/bi901006b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of Fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell. 2009;35:716–23. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden RR, Cole RS. Repair of cross-linked DNA and survival of Escherichia coli treated with psoralen and light: effects of mutations influencing genetic recombination and DNA metabolism. J Bacteriol. 1978a;136:538–47. doi: 10.1128/jb.136.2.538-547.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden RR, Cole RS. Topography and kinetics of genetic recombination in Escherichia coli treated with psoralen and light. Proc Natl Acad Sci USA. 1978b;75:2373–7. doi: 10.1073/pnas.75.5.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek FM, Melian A, Howard-Flanders P. Incision by UvrABC excinuclease is a step in the path to mutagenesis by psoralen crosslinks in Escherichia coli. Proc Natl Acad Sci USA. 1989a;86:3982–6. doi: 10.1073/pnas.86.11.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek FM, Munn MM, Rupp WD, Howard-Flanders P. In vitro repair of psoralen-DNA cross-links by RecA, UvrABC, and the 5′-exonuclease of DNA polymerase I. J Biol Chem. 1989b;264:6755–65. [PubMed] [Google Scholar]
- Smeaton MB, Hlavin EM, McGregor Mason T, Noronha AM, Wilds CJ, Miller PS. Distortion-dependent unhooking of interstrand cross-links in mammalian cell extracts. Biochemistry. 2008;47:9920–30. doi: 10.1021/bi800925e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–20. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Akagi Ji, Nishi R, Iwai S, Hanaoka F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Molecular Cell. 2009;36:642–653. doi: 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
- Thoma BS, Wakasugi M, Christensen J, Reddy MC, Vasquez KM. Human XPC-hHR23B interacts with XPA-RPA in the recognition of triplex-directed psoralen DNA interstrand crosslinks. Nucleic Acids Res. 2005;33:2993–3001. doi: 10.1093/nar/gki610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trego KS, Turchi JJ. Pre-steady-state binding of damaged DNA by XPC-hHR23B reveals a kinetic mechanism for damage discrimination. Biochemistry. 2006;45:1961–9. doi: 10.1021/bi05196t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houten B, Gamper H, Hearst JE, Sancar A. Construction of DNA substrates modified with psoralen at a unique site and study of the action mechanism of ABC excinuclease on these uniformly modified substrates. J Biol Chem. 1986a;261:14135–41. [PubMed] [Google Scholar]
- Van Houten B, Gamper H, Holbrook SR, Hearst JE, Sancar A. Action mechanism of ABC excision nuclease on a DNA substrate containing a psoralen crosslink at a defined position. Proc Natl Acad Sci U S A. 1986b;83:8077–81. doi: 10.1073/pnas.83.21.8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol. 2001;21:713–20. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch repair participates in error-free processing of DNA interstrand crosslinks in human cells. EMBO Rep. 2005;6:551–7. doi: 10.1038/sj.embor.7400418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem. 2000;275:9870–9875. doi: 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ. hMutSβ is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol. 2002;22:2388–97. doi: 10.1128/MCB.22.7.2388-2397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol. 2003;23:754–61. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]