

Abstract
Amyloid fibrils are found in association with at least two dozen fatal diseases. The tendency of numerous proteins to convert into amyloid-like fibrils poses fundamental questions for structural biology, and for protein science in general. Among these are: What is the structure of the cross-β spine, common to amyloid-like fibrils? Is there a sequence signature for proteins that form amyloid-like fibrils? What is the nature of the structural conversion from native to amyloid states, and do fibril-forming proteins have two distinct, stable states, the native state and the amyloid state? What is the basis of protein complementarity, in which a protein chain can bind to itself? We offer tentative answers here, based on our own recent structural studies, recognizing that there is much complementary research in the field, some of which is summarized in other papers in this issue, as well as elsewhere1–11.
Keywords: Cross beta spine, amyloid, X-ray crystallography
Amyloid and amyloid-like fibrils
Protein aggregation diseases are pathologies accompanied by the deposition of aggregated proteins. The most prevalent aggregation diseases are the amyloid diseases12, associated with elongated, unbranched protein fibrils. To be defined by pathologists as an amyloid disease, the fibrils must be deposited extracellularly, and must bind the dye Congo Red, giving an “apple-green” birefringence12. As of 2005, Alzheimer’s disease and some 24 others have been found to satisfy this stringent definition12.
Biochemists and biophysicists consider a wider range of protein fibrils to be amyloid-like. The biophysical study of amyloid fibrils has revealed that they display common properties, in addition to their morphology and tendency to bind Congo Red. These other properties include the so-called cross-β X-ray diffraction pattern. This pattern consists of an X-ray reflection at ~4.8 Å resolution along the fibril direction, and another X-ray reflection at ~10–12 Å resolution perpendicular to the fibril direction13–15. This pattern reveals that the fibrils contain β-sheets parallel to the fibril axis, with their extended protein strands perpendicular to the axis. Another property of amyloid fibrils is that they form from their constituent protein molecules with cooperative, nucleation-dependent kinetics16. Because the fibrils associated with other diseases such as Parkinson’s and Huntington’s (intracellular fibrils) - display many of the same physical characteristics as the fibrils of the officially-designated amyloid diseases17, biophysical chemists often refer to these fibrils also as amyloid-like fibrils.
It has been known for decades that normal globular proteins can be transformed into amyloid-like fibrils. These are fibrils that resemble those found in amyloid diseases, but not themselves associated with pathologies. In 1935, the pioneering biophysicist William Astbury stretched poached egg white and found that it exhibited the cross-β diffraction pattern13. More recently, numerous globular proteins have been converted to amyloid-like fibrils by removing them from their native temperatures and pH values18–21.
Fundamental Questions
The finding that numerous proteins can convert from their native structures to amyloid-like fibrils having common properties raises several fundamental questions for structural biology and for protein science in general:
What is the structure of the cross-β spine – the molelcular feature that gives rise to the cross-β diffraction pattern – which is common to all amyloid and amyloid-like fibrils?
Do amyloid-forming proteins have two distinctly different stable structures, their native state and the amyloid state? Can the amyloid state retain aspects of the native structure?
What is the nature of the conversion of a protein from its native structure to the amyloid state?
Is there a sequence signal for the formation of amyloid-like fibrils, or is the structure a generic backbone structure, where sidechains do not contribute to the specificity of the structure22?
What is the origin of protein self-complementarity, in which a protein binds strongly to itself, as in amyloid-like fibrils?
We provide tentative answers to these fundamental questions below, based on recent structural studies.
Microcrystal structure of the cross-β spine of amyloid-like fibrils
We chose the yeast prion Sup35p for X-ray diffraction studies of the cross-β spine because past work had shown that its fibril formation is the basis of protein-based inheritance and prion-like infectivity23–27 Based on earlier genetic studies that localized the amyloid-forming ability of Sup35p to the N-terminal segment28,29, we were able to identify a seven-residue peptide of sequence GNNQQNY that forms amyloid-like fibrils with all of the common properties30. These properties include: an elongated, unbranched shape; the binding of the dye Congo Red and birefringence of a green-yellow hue; exhibition of the cross-β diffraction pattern, and lag-dependent aggregation of monomers into fibrils. Later we found that fibrils are formed by its subfragments NNQQNY and NNQQ.
All three of these short peptides formed microcrystals as well as fibrils, enabling the determination of crystal structures. The dimensions of these microcrystals never exceeded ~50 × 4 × 4 μm, despite much effort to enlarge them. Fortunately, advances in microcrystallography31 now make it possible to collect diffraction data from sturdy crystals of this size, and we were able to determine structures for GNNQQNY and Zn-NNQQNY to 1.8 and 1.3 Å resolution, respectively32. These high resolution structures offered, for the first time, objective, refined atomic models for the cross-β spine of amyloid fibrils. While here we emphasize the results from this study, important contributions to understanding amyloid structure have come from other methods. These include solid state NMR studies of various fibrils by the groups of R.G. Griffin33, D.S. Lynn34, R. Tycko35,36 and B. Meier9,37; EPR studies of fibrils by the groups of W. Hubbell, T.O. Yeates38 and R. Langen39; hydrogen/deuterioum exchange studies by of the groups of Y. Goto40 and R. Riek9,10; mutagenesis studies by the group of R. Wetzel11,41; electron microscopy studies by the groups of H.R. Saibil42,43 and U. Aebi44; spectroscopic studies by the groups of S. Radford45 and S. Lindquist5; X-ray fiber diffraction studies of the groups of L. Serpell2, E. Atkins1, and K. Namba4; and peptide design work of the group of L. Serrano46. Elsewhere we have reviewed the various models for amyloid that have come from this body of work by many investigators47.
The structure for the cross-β spine formed from GNNQQNY is shown in Fig. 1. The structure is essentially the same as that of Zn-NNQQNY (not shown here) in which the Zn ion occupies the position of the N-terminal glycyl residue in the longer peptide. Each GNNQQNY peptide is extended and forms one strand of a parallel, in-register β-sheet, which extends upwards and downwards for the entire length of the elongated crystal. Each β-sheet is paired with a second sheet around a completely dry interface that we call the steric zipper of the cross β-spine. These two sheets are related by the 21 axis shown in the figure: that is, one sheet can be superimposed on the other by a rotation about the axis by 180° and translation along the axis of one-half the inter-strand spacing of 4.8 Å. The 21 symmetry relationship brings identical faces together, so that the glutamine (Q) and asparagine (N) sidechains protruding from the two sheets are tightly intermeshed, forming the steric zipper, as shown in Fig. 1d.
Figure 1. (from Reference 32) Structure of GNNQQNY.

a, The pair-of-sheets structure, showing the backbone of each β-strand as an arrow, with side chains protruding. The dry interface is between the two sheets, and the wet interfaces are on the outside surfaces. Side chains Asn 2, Gln 4 and Asn 6 point inwards, forming the dry interface. The 21 screw axis of the crystal is shown as the vertical line. It rotates one of the strands of the near sheet 180° about the axis and moves it up ½ × 4.87 Å so that it is superimposed on one of the strands of the far sheet. b, The steric zipper viewed edge on (down the a axis). Note the vertical shift of one sheet relative to the other, allowing interdigitation of the side chains emanating from each sheet. The amide stacks of the dry interface are shaded in gray at the center, and those of the wet interface are shaded in pale red on either side, c, The GNNQQNY crystal viewed down the sheets (from the top of panel a, along the b axis). Six rows of β-sheets run horizontally. Peptide molecules are shown in black and water molecules are red plus signs. The atoms in the lower left unit cell are shown as spheres representing van der Waals radii, d, The steric zipper. This is a close-up view of a pair of GNNQQNY molecules from the same view as panel c, showing the shape complementarity of the Asn and Gln side chains protruding into the dry interface. 2F0−Fc electron density is shown, and the position of the central screw axis is indicated, e, Views of the β-sheets from the side (down the c axis), showing three β-strands with the inter-strand hydrogen bonds. Side-chain carbon atoms are yellow. Backbone hydrogen bonds are shown by purple or gray dots and side-chain hydrogen bonds by yellow dots. Hydrogen bond lengths are noted in Å. The views of the interfaces are close to the views of panel a. The left-hand set is viewed from the center of the dry interface; the right-hand set is viewed from the wet interface. Note the amide stacks in both interfaces. Carbon atoms are purple or gray, oxygen is red, and nitrogen is blue, unless noted otherwise.
This tight, dry interface between the two sheets of a pair-of-sheets motif is different in character from the crystal contacts between one of these pair-of-sheet motifs and its surrounding pairs (Fig. 1c). The latter are wet interfaces, resembling the intermolecular contacts in protein crystals: they contain water molecules and there are few contacts made between protein atoms in different pairs. Thus we regard a pair of β-sheets mating in a dry steric zipper as the fundamental structural motif of the cross-β spine.
The view of the pair-of-sheets unit shown in Fig. 1c is down the sheets. Notice the tight interdigitation of the Gln and Asn sidechains from positions 2, 4, and 6 of each strand with the same sidechains from the mating sheet. A measure of the structural complementation of one protein surface with another is given by the SC parameter of Lawrence and Colman48, a quantity that can vary from 0 up to 1 for perfect complementation of two surfaces. The two surfaces of the mating sheets in GNNQQNY have a value of 0.86, considerably higher than the tight surfaces between proteases and their protein inhibitors, which receive an average value of SC of 0.73 ± 0.03 (ref. 48), or of the average value of SC of 0.66 ± 0.02 for the binding surfaces of antibodies to their antigens. In short, the surfaces of the two mating sheets are unusually tight-fitting as shown by the black van der Waals representation in the lower left unit cell of Fig. 1c.
The GNNQQNY peptides in one sheet form no hydrogen bonds to the peptides in the mating sheet, but each forms 11 hydrogen bonds to the identical molecules above and below it in the same sheet. Five of these are backbone N-H···O hydrogen bonds, and five are hydrogen bonds between side chains. Notice that the sidechain amide groups of each molecule are oriented in the same direction as those above and below, creating columns of hydrogen bonds running up and down the sheets. These are reminiscent of the “polar zipper” hydrogen bonds proposed for polyglutamine fibrils by Perutz et al49,50 and Sikorski and Atkins1.
A view of the pair-of-sheets in the direction down the strands is given in Fig. 1b. This shows the interdigitation of the sidechains of the Gln residues, meshing much like the teeth of the zipper. Because of their tightly complementary fit, we term this the steric zipper. Notice that the steric zipper is completely dry. The water molecules in the crystal are found on the outer surface of the pair-of-sheets motif.
Energetics of the cross-β spine
Knowing the structure of the cross-β spine allows us to understand some of the energetics and kinetics of formation. The structure suggests three levels of organization within the fibrils. The first level is the alignment of individual GNNQQNY molecules to form a β-sheet. The second level is the mating of two sheets, forming the pair-of-sheets structure, with its dry interface. The third level winds the pair-of-sheets structures around each other to form a fibril. The non-covalent forces involved in this third level are probably weaker than those driving the formation of the first two levels.
β-sheets form rapidly51,52 and reversibly, so we assume that the individual sheets form more rapidly than the pairing of sheets. The pairing is likely to be slower because the amide side chains must acquire their proper rotamers to allow interdigitation with the opposite sheet. We believe the entropy reduction accompanying this step creates the barrier to fibril formation, which is seen in the lag-dependent cooperative formation. Once a nucleus of the cross-β spine has formed, other molecules can be added more rapidly, provided the concentration of monomers is high enough. We have argued from the structure that the nucleus for the pair-of-sheets structure is about four molecules, so that the transition–state complex on the path to the nucleus is approximately three molecules32. From crude energy calculations32, we estimate that the free energy of forming this complex is ~8 kcal mol-of-monomer−1. Thus if there are three molecules in the transition-state complex, the barrier is ~24 kcal mol−1. A transition-state barrier of the order of 24 kcal mol−1 is substantial enough to make nucleation a rare event, and hence to give an appreciable lag time before fibers start to grow
Our crude analysis of the standard free energy change of the order of +8 kcal mol−1 for the addition of a monomer to the growing of the nucleus32 suggests that fibrils are not “intrinsically stable” in the sense of having a sizable negative free energy change for addition of monomers to the fibril. According to this analysis, the fibrillar state becomes the stable state only at high monomer concentration. If this is so, fibril formation from monomers is different from the formation of an oligomeric enzyme from its monomers: in the latter case there is a more negative free energy change, so that oligomerization from monomers takes place at lower monomer concentrations. There is, however, a kinetic barrier to cross either in the formation or the dissolution of cross-β spine. This means that once formed, amyloid structure could take a long time to dissolve, even when the monomer concentration is reduced, because the molecules would be kinetically trapped in the fibril state.
Designed amyloid-like fibrils of ribonuclease A have a three-dimensional, domain-swapped, native-like structure
To gain insight into the structural changes that take place when entire proteins enter amyloid-like fibrils, we designed an amyloid-like fibril based on ribonuclease A (RNase A, shown in Fig. 2a). We selected RNase A because it is well characterized, it is tightly cross-linked by four disulfide bonds, and it forms a domain-swapped dimer when concentrated in acetic acid 53–55. Domain swapping is a mechanism for forming oligomers by the exchange of protein domains. The swapped domain is in some cases a helix or β-strand, and other cases an entire tertiary domain, and is linked to the rest of the protein by a segment of chain called the hinge loop. From the structure of the RNase A dimer, shown in Fig. 2b, we reasoned53 that expanding the hinge loop connecting the core domain with the swapped domain by inserting an amyloidogenic segment might permit the formation of a domain-swapped amyloid-like fibril. A schematic diagram of such a structure is shown in Fig. 2c. It contains a cross-β spine in the center, in this case formed from an expansion of 10 Gln residues. At the periphery of the spine are RNase A molecules, each formed from a core domain and a complementing C-terminal β-strand from another RNase A molecule. If such a structure forms, we would expect that it would be fibrous and. would contain native-like domains capable of enzymatic activity.
Figure 2. (from Reference 56) Ribonuclease A monomer and domain-swapped C-terminal dimer and the 3D Domain-Swapped Zipper-Spine model.
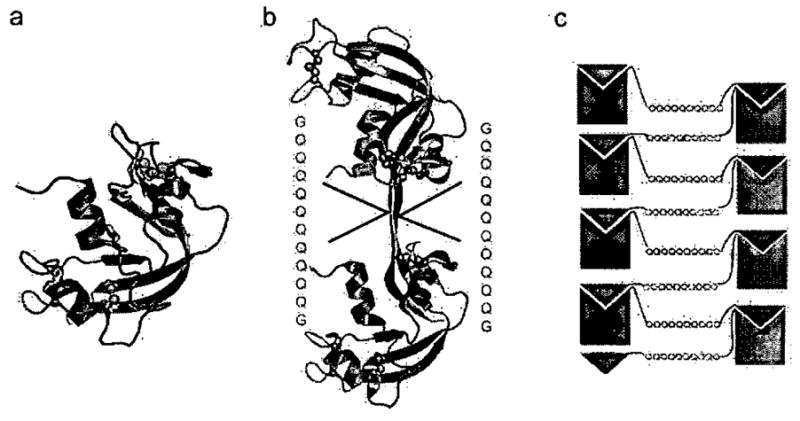
a, The Ribonuclease A monomer is stabilized by four disulfide bonds between Cys26-Cys84, Cys40-Cys95, Cys58-Cysl 110, and Cys65-Cys72, hindering conformational changes. His 12 in the core of the protein and His 119 on the β-strand that is swapped (shown by sticks), are active site residues mutated to test for activity by complementation, b, The C-terminal domain-swapped dimer is formed by exchanging the C-terminal β-strands between two monomers. The hinge-loop (residues 112–115) has been expanded by inserting the sequence −GQ10G-. c, Schematic model for amyloid-like fibril formation in RNase A with Q10 expansion, leading to a runaway domain swap. In blue are the Q10-H12A mutants and in green the Q10-H119A mutants. Domain swapping between two mutants complements active sites.
These expectations were fulfilled by the construction of such an expanded RNase A molecule56. RNase A molecules into which we inserted a Q10 expansion in the C-terminal hinge loop in fact formed amyloid-like fibrils that bind Congo Red and display the cross-β diffraction pattern. RNase A with a GQ7G expansion or with an expansion of the Sup35p sequence GNNQQNY also forms amyloid-like fibrils. In contrast, wild type RNase A did not form fibrils, nor did a polyglycine (G9) expansion. In short, amyloidogenic sequences inserted into the C-terminal hinge loop of RNase A produce amyloid-like fibrils.
Are functional RNase A molecules formed by domain-swapping in these fibrils? To answer this question, we measured the enzymatic activities of mutant RNase A fibrils, both alone and mixed. For more than 40 years it has been known that the active site of RNase A contains two histidyl residues: His 12, in the core domain, and His 119, in the C-terminal beta strand54. This strand is swapped into the core of a second molecule in the domain-swapped dimer shown in Fig. 2c. We used this old finding to establish that domains are swapped in the fibrils. The mutant RNase A molecules H12A and HI 19A were separately prepared for RNase with Q10 expansion. Both of these molecules form fibrils, but lack enzymatic activity, because neither has a complete active site. When the two molecules are mixed, and fibrils are formed, about 1/8 of the wild type activity returns. We presume this activity of the mixed fibril comes from complemented active sites, which have both H12 and HI 119. If the reconstitution of the active sites were perfect, then we would expect ¼ of full activity. Presumably there are losses because of imperfect refolding, and imperfect stoichiometry of mixing. Nevertheless, the observed activity is enough to conclude that there are active RNase A molecules in the fibrils and that these are active because of domain swapping.
Because enzymatic activity is perhaps the best measure of native structure, we are forced to conclude that the RNase A fibrils contain molecules in their native state, and that the fibril contains both a cross-β spine and domain-swapped molecules. Based on this conclusion, we built a hypothetical atomic model for the RNase A amyloid-like fibril shown in Fig. 3, which contains domain-swapped functional units. In this model, the spine of the structure is a twisted pair of antiparallel β-sheets. Each β-strand is the Q10 insertion in the hinge-loop of RNase A. These polyglutamine β-strands are stacked 4.8 Å apart along the fibril axis (Fig. 3c). A slight twist is introduced between successive segments of the spine to be consistent with the measured twist of the fibrils. Each Q10, segment forms hydrogen bonds to identical segments above and below within each sheet, but not between sheets. The two sheets are held together by a steric zipper of the Q10 sidechains. This model depicts the native fold of RNase A as being essentially retained with only a small segment of the protein (the Q10 hinge loop) forming the cross β-spine. Evidence of native-like character in the fibril forms of Ure2p57–59 and β2-microglobulin60 has been uncovered in other laboratories.
Figure 3. (from Reference 56) Domain-swapped Zipper-Spine Model for the RNase A Protofibril.
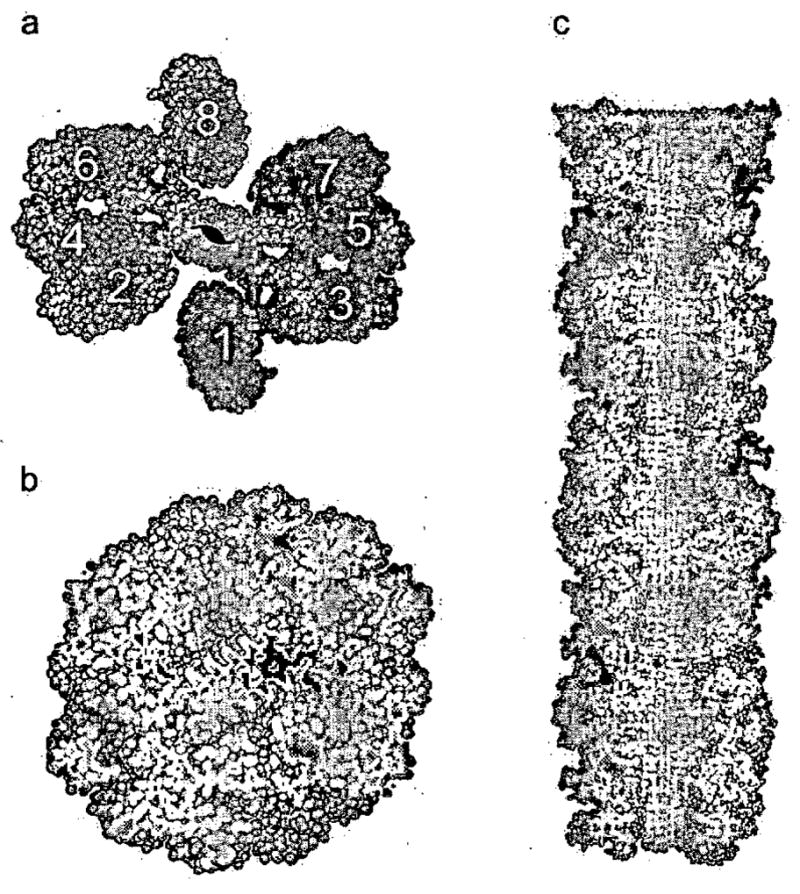
a, The model is a ‘runaway’ domain swap between the RNase A monomers with swaps occurring within one half protofibril but not between half protofibrils. Monomers 1–4 compose half the protofibrillar unit and are coloured as in Fig. 1c to emphasize domain swapping. The C-terminal β-strand of monomer 1 swaps into 2, 2 swaps into 3, and 3 swaps into 4, rising along the axis of the fibril. Q10 segments from these monomers form one antiparallel β-sheet in the spine. Monomers 5–8 form the other β-sheet, related to the monomers 1–4 by a 21 axis along the fibril. Eight RNase A monomers comprise the asymmetric unit of the fibril. A similar model can be built from domain-swapped dimers, and currently available data do not favour one of these models over the other, b, The protofibril cross-section reveals the steric zipper, the interdigitation of Gln sidechains in the spine of the fibril, modelled on the structure of GNNQQNY. c, The protofibril model in longitudinal cross-section. The zipper-spine is seen at the center of the protofibril.
Summary and Tentative answers to the fundamental questions
Based on the studies described above, we offer tentative answers to the fundamental questions raised at the start of this review:
What is the structure of the cross-β spine common to all amyloid and amyloid-like fibrils? Both GNNQQNY and NNQQNV form fibrils and microcrystals, each with their β-strands perpendicular to the long axis. The atomic-level structures of the two microcrystals offer objective pictures of the cross-β spine. In both structures, the spine consists of a pair of β-sheets, mated tightly together, with their sidechains intermeshed in what is termed a steric zipper. Each peptide forms one β-strand of a sheet. The sheets are parallel with strands in register. Also the two sheets are in register. There seems to be no fundamental reason that other amyloid structures need to be built of parallel sheets; they could instead be built from antiparallel sheets. Nor is there any basic reason that the sheets need to be in register with one another; they could slip along the strand directions. The more fundamental feature appears to be the dry steric zipper motif.
Do amyloid-forming proteins have two distinctly different stable structures, their native state and the amyloid state? In the case of the designed amyloid-like fibrils of RNase A, the native and amyloid states resemble each other in that both contain native-like functional units. In the amyloid form, the functional units are domain-swapped RNase A units made up of complementary domains from two RNase A molecules. The argument for native-like domains rests on the observation of enzymatic activity in the fibrils. So at least in the example of the RNase A fibrils, there is no fundamental structural change between the native and fibrillar states, except that the hinge loop that links each RNase A core domain to its swapped domain participates in the cross-β spine. Evidence for retention of native-like structure has also been demonstrated by others for fibrils of Ure2p58,61 and β2-microglobulin60. To the extent that native-like structures are found in general as part of amyloid-like fibrils, the changes in structure would be mainly confined to the segments forming the steric zipper.
What is the nature of the conversion of a protein from its native structure to the amyloid like fibrillar state? In the case of the RNase A fibrils, the process of conversion would start with a braking of non-covalent bonds between the C-terminal β-strand of the enzyme with the core, and then its swapping into the core of a second molecule. This would expose the Q10 expansion loops, which would be free to stack into a cross-β spine.
Is there a sequence signal for the formation of amyloid-like fibrils, or is the structure a generic backbone structure? The tightly complementary cross-β spine, with the intermeshing sidechains suggests that spine formation would be sequence dependent, favoring sequences that can intermesh over those that cannot. Thus low complexity sequences (such as those containing Asn and Gln residues) would be favored. Highly charged sequences for residues in the inward- pointing positions would be disfavored. Additional support for the importance of protein sequence in fibril formation has come from both computational62 and experimental46,63,64 studies.
What is the origin of protein self-complementarity, in which a protein binds strongly to itself, as in amyloid-like fibrils? Our work to date has revealed three types of self-complementarity in amyloid-like fibrils. The first is the steric zipper of the cross-β spine. It seems likely that this motif will be found in other amyloid-like fibrils. The second is the domain-swap. A domain swap is not necessary for the formation of a fibril, but it may be found in some instances3,56,60,65. Domain-swapping has the feature that it is protein specific. That is, a domain swap can occur only with a molecule having an identical domain. Thus domain swapping can account for the observation that amyloid fibrils tend to be formed from a single protein. If instead amyloid structure were independent of sidechains and had a generic backbone structure, there would be no reason that amyloid fibrils could not contain more than a single type of protein. The third type of complementarity we have seen is the stacking of side chains in “polar zippers”, such as shown in Fig. 1b. Side chain stacking, with hydrogen bonding between sidechains in strands on top of one another, can arise only for parallel, in-register β-sheets with sidechains capable of stacking, such as Asn and Gln. So this type of complementarity is not a necessary feature of amyloid, and we would expect to find it only with sequences containing “stackable” residues, such as Asn and Gln.
We expect that new features of amyloid structures will be revealed in future atomic-level structures, and that these structures will give us a more detailed and general picture of features involved.
Acknowledgments
We thank D. Anderson, M. Aposotol, D. Cascio, M. Ginergy, R. Grothe, H. McFarlan, S. Sievers, M.J. Thompson, and J. Wilzius for discussions, and NSF, NIH, and HHMI for support.
Biographies
David Eisenberg (born Chicago, 15 March, 1939). A.B. in Biochemical Sciences (Harvard, 1961); D. Phil in Theoretical Chemistry (Oxford with Charles Coulson, 1964); Postdoctoral fellowships (Princeton with Walter Kauzmann, 1964–66 and Caltech with Richard Dickerson, 1964-9); UCLA faculty 1969-present; Investigator HHMI (2001-present). Research interests: protein interactions
Rebecca Nelson (born Paris, France, 21 July, 1977). B.S. in Chemistry (Purdue University, 1999); Graduate student at UCLA, 1999-present; Research interests: protein chemistry, amyloid structure
Michael R. Sawaya (born San Diego, 30 October, 1967). B.S. in Chemistry (San Diego State University, 1989); PhD in Biochemistry (University of California, San Diego with Joseph Kraut, 1994); Postdoctoral fellowship (Harvard with Thomas Ellenberger, 1997–2000); UCLA research faculty 2000-present; Research interests: crystallographic methods, enzyme mechanism.
Melinda Balbirnie (born Philadelphia, 20 September, 1967) B.S. in Chemistry (Virginia Tech, 1989); M.S. in Chemistry (University of Pennsylvania, 1991); Ph.D. in Biochemistry (UCLA, 2000); Research Scientist (Farmal Biomedicines, 2002-3) Postdoctoral fellowship (UCLA with David Eisenberg, 2005-present). Research interests: protein-small molecule interactions
Shilpa Sambashivan (born Bangalore, India, 5 April 1980) MSc. (Hons.) Biological Sciences (Birla Institute of Technology and Science, Pilani, India, 2001). Ph.D. in Molecular Biology (UCLA, 2006 with Prof. David Eisenberg). Research Interests: Molecular basis for amyloid diseases
Magdalena Ivanova
Anders Østergaard Madsen (born Copenhagen, 2 October 1974). Cand. Scient in Chemistry (University of Copenhagen, 2002); Graduate student in chemistry at University of Copenhagen and ESRF. Research interests: Crystallography, structural chemistry.
Christian Riekel (born Vienna, Austria, 7 August 1943). Dr. rer. nat. in Inorganic Chemistry (University of Munich, 1973), Scientist at ILL-Grenoble (1974–79), Scientist at MPI-Stuttgart (1979–81), Scientist at University of Hamburg with H.G. Zachmann (1982–86), habilitation in macromolecular chemistry (1987), constructed a synchrotron radiation beamline for polymer scattering at DESY-Hamburg, microfocus beamline (ID13) scientist and soft condensed matter group leader at ESRF-Grenoble (1986-present).
References
- 1.Sikorski P, Atkins E. Biomacromolecules. 2005;6:425–32. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- 2.Makin OS, Serpell LC. FEBS J. 2005;272:5950–61. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 3.Janowski R, Kozak M, Jankowska E, Grzonka Z, Grubb A, Abrahamson M, Jaskolski M. Nat Struct Biol. 2001;8:316–20. doi: 10.1038/86188. [DOI] [PubMed] [Google Scholar]
- 4.Kishimoto A, Hasegawa K, Suzuki H, Taguchi H, Namba K, Yoshida M. Biochem Biophys Res Commun. 2004;315:739–45. doi: 10.1016/j.bbrc.2004.01.117. [DOI] [PubMed] [Google Scholar]
- 5.Krishnan R, Lindquist SL. Nature. 2005;435:765–72. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kajava AV, Baxa U, Wickner RB, Steven AC. Proc Natl Acad Sci U S A. 2004;101:7885–90. doi: 10.1073/pnas.0402427101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kajava AV, Aebi U, Steven AC. J Mol Biol. 2005;348:247–52. doi: 10.1016/j.jmb.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. J Mol Biol. 2004;335:247–60. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 9.Ritter C, Maddelein ML, Siemer AB, Lührs T, Ernst M, Meier BH, Saupe SJ, Riek R. Nature. 2005;435:844–8. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. Proc Natl Acad Sci V S A. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. J Mol Biol. 2004;335:833–42. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 12.Westermark P. FEBS J. 2005;272:5942–9. doi: 10.1111/j.1742-4658.2005.05024.x. [DOI] [PubMed] [Google Scholar]
- 13.Astbury WT, Dickinson S, Bailey K. Biochem J. 1935;29:2351–2360. doi: 10.1042/bj0292351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eanes ED, Glenner GG. J Histochem Cytochem. 1968;16:673–7. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 15.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. J Mol Biol. 1997;273:729–39. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 16.Rochet JC, Lansbury PT., Jr Curr Opin Struct Biol. 2000;10:60–8. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 17.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid. 2002;9:197–200. doi: 10.3109/13506120209114823. [DOI] [PubMed] [Google Scholar]
- 18.Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Proc Natl Acad Sci U S A. 1998;95:4224–8. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fändrich M, Fletcher MA, Dobson CM. Nature. 2001;410:165–6. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 20.Fändrich M, Dobson CM. EMBO J. 2002;21:5682–90. doi: 10.1093/emboj/cdf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fändrich M, Forge V, Buder K, Kittler M, Dobson CM, Diekmann S. Proc Natl Acad Sci U S A. 2003;100:15463–8. doi: 10.1073/pnas.0303758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobson CM. Trends Biochem Sci. 1999;24:329–32. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 23.Wickner RB. Science. 1994;264:566–9. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 24.Patino MM, Liu JJ, Glover JR, Lindquist S. Science. 1996;273:622–6. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 25.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Science. 2000;289:1317–21. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 26.King CY, Diaz-Avalos R. Nature. 2004;428:319–23. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Nature. 2004;428:323–8. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 28.DePace AH, Santoso A, Hillner P, Weissman JS. Cell. 1998;93:1241–52. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 29.Santoso A, Chien P, Osherovich LZ, Weissman JS. Cell. 2000;100:277–88. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 30.Balbirnie M, Grothe R, Eisenberg DS. Proc Natl Acad Sci U S A. 2001;98:2375–80. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riekel C. J Synchrotron Radial. 2004;11:4–6. doi: 10.1107/s090904950302541x. [DOI] [PubMed] [Google Scholar]
- 32.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. Proc Natl Acad Sci U S A. 2004;101:711–6. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burkoth TS, Benzinger TLS, Urban V, Morgan DM, Gregory DM, Thiyagarajan P, Botto RE, Meredith SC, Lynn DG. J Am Chem Soc. 2000;122:7883–9. [Google Scholar]
- 35.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. Proc Natl Acad Sci V S A. 2002;99:16742–7. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paravastu AK, Petkova AT, Tycko R. Biopnys J. 2006;90:4618–29. doi: 10.1529/biophysj.105.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siemer AB, Ritter C, Steinmetz MO, Ernst M, Riek R, Meier BH. J Biomol NMR. 2006;34:75–87. doi: 10.1007/s10858-005-5582-7. [DOI] [PubMed] [Google Scholar]
- 38.Serag AA, Altenbach C, Gingery M, Hubbell WL, Yeates TO. Nat Struct Biol. 2002;9:734–9. doi: 10.1038/nsb838. [DOI] [PubMed] [Google Scholar]
- 39.Török M, Milton S, Kayed R, Wu P, Mclntire T, Glabe CG, Langen R. J Biol Chem. 2002;277:40810–5. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 40.Hoshino M, Katou H, Hagihara Y, Hasegawa K, Naiki H, Goto Y. Nat Struct Biol. 2002;9:332–6. doi: 10.1038/nsb792. [DOI] [PubMed] [Google Scholar]
- 41.Thakur AK, Wetzel R. Proc Natl Acad Sci U S A. 2002;99:17014–9. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiménez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR. EMBO J. 1999;18:815–21. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiménez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, Saibil HR. Proc Natl Acad Sci U S A. 2002;99:9196–201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P, Muller SA, Kistler J, Cooper GJ, Aebi U. J Struct Biol. 2000;130:352–62. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 45.McParland VJ, Kalverda AP, Homans SW, Radford SE. Nat Struct Biol. 2002;9:326–31. doi: 10.1038/nsb791. [DOI] [PubMed] [Google Scholar]
- 46.Lopez de la Paz M, Serrano L. Proc Natl Acad Sci V S A. 2004;101:87–92. doi: 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson R, Eisenberg D. Adv Protein Chem. 2006 doi: 10.1016/S0065-3233(06)73008-X. In press. [DOI] [PubMed] [Google Scholar]
- 48.Lawrence MC, Colman PM. J Mol Biol. 1993;234:946–50. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 49.Perutz MF, Johnson T, Suzuki M, Finch JT. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perutz MF, Finch JT, Berriman J, Lesk A. Proc Natl Acad Sci U S A. 2002;99:5591–5. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varley P, Gronenborn AM, Christensen H, Wingfield PT, Pain RH, Ciore GM. Science. 1993;260:1110–3. doi: 10.1126/science.8493553. [DOI] [PubMed] [Google Scholar]
- 52.Sivaraman T, Kumar TK, Chang DK, Lin WY, Yu C. J Biol Chem. 1998;275:10181–9. doi: 10.1074/jbc.273.17.10181. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Gotte G, Libonati M, Eisenberg D. Nat Struct Biol. 2001;8:211–4. doi: 10.1038/84941. [DOI] [PubMed] [Google Scholar]
- 54.Crestfield AM, Stein WH, Moore S. J Biol Chem. 1963;238:2421–8. [PubMed] [Google Scholar]
- 55.Gotte G, Bertoldi M, Libonati M. Eur J Biochem. 1999;265:680–7. doi: 10.1046/j.1432-1327.1999.00761.x. [DOI] [PubMed] [Google Scholar]
- 56.Sambashivan S, Liu Y, Sawaya MR, Gingery M, Eisenberg D. Nature. 2005;457:266–9. doi: 10.1038/nature03916. [DOI] [PubMed] [Google Scholar]
- 57.Baxa U, Cheng N, Winkler DC, Chiu TK, Davies DR, Sharma D, Inouye H, Kirschner DA, Wickner RB, Steven AC. J Struct Biol. 2005;150:170–9. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 58.Bousset L, Briki F, Doucet J, Melki R. J Struct Biol. 2003;141:132–42. doi: 10.1016/s1047-8477(02)00606-8. [DOI] [PubMed] [Google Scholar]
- 59.Bousset L, Redeker V, Decottignies P, Dubois S, Le Marechal P, Melki R. Biochemistry. 2004;43:5022–32. doi: 10.1021/bi049828e. [DOI] [PubMed] [Google Scholar]
- 60.Eakin CM, Attenello FJ, Morgan CJ, Miranker AD. Biochemistry. 2004;45:7808–15. doi: 10.1021/bi049792q. [DOI] [PubMed] [Google Scholar]
- 61.Baxa U, Taylor KL, Wall JS, Simon MN, Cheng N, Wickner RB, Steven A. C J Biol Chem. 2003;278:43717–27. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- 62.Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D. Proc Natl Acad Sci U S A. 2006;103:4074–8. doi: 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ivanova MI, Sawaya MR, Gingery M, Attinger A, Eisenberg D. Proc Natl Acad Sci U S A. 2004;101:10584–9. doi: 10.1073/pnas.0403756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ivanova MI, Thompson MJ, Eisenberg D. Proc Natl Acad Sci U S A. 2006;103:4079–82. doi: 10.1073/pnas.0511298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Staniforth RA, Giannini S, Higgins LD, Conroy MJ, Hounslow AM, Jerala R, Craven CJ, Waltho JP. EMBO J. 2001;20:4774–81. doi: 10.1093/emboj/20.17.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]