

Abstract
BACKGROUND
Increasing evidence demonstrates that aberrantly-regulated microRNAs (miRNAs) contribute to the initiation and progression of human cancer. We previously have demonstrated that miR-125b stimulated the growth of prostate cancer (CaP) cells. In this study, we further determined the influence of miR-125b on the pathogenesis of prostate cancer.
METHODS
To evaluate the effect of miR-125b on xenograft tumor growth, male athymic mice were subcutaneously injected with PC-346C-miR-125b cells that stably overexpressed miR-125b. Potential direct target transcripts of miR-125b were identified using a bioinformatics approach and three miR-125b targeted molecules were confirmed by means of biochemical analyses.
RESULTS
Enforced expression of miR-125b promoted tumor growth in both intact and castrated male nude mice. In an effort to define the molecular mechanism(s) mediating its tumor growth properties, we found that miR-125b directly targets eight transcripts, including three key pro-apoptotic genes: p53, Puma and Bak1. Increasing the abundance of miR-125b resulted in a dramatic decrease in the levels of these three proteins in prostate cancer cells. A direct repressive effect on each of these was supported by the ability of miR-125b to significantly reduce the activity of luciferase reporters containing their 3′-untranslated regions of each gene encompassing the miR-125b-binding sites. Additionally, we found that repression of miR-125b activity was able to sensitize prostate cancer cells to different therapeutic interventions.
CONCLUSION
Data obtained in this study demonstrate that miR-125b promotes growth of prostatic xenograft tumors by down-regulating three key pro-apoptotic genes. This suggests that miR-125b is oncogenic and makes it an attractive therapeutic target in prostate cancer.
Keywords: prostate cancer, microRNA, p53, pro-apoptotic gene
INTRODUCTION
Prostate cancer (CaP) is the most frequently diagnosed malignant tumor in males and the second leading cause of cancerous deaths in American men (1). Patients with metastatic CaP are customarily treated with androgen ablative therapy (AAT). Unfortunately, failure of AAT inevitably occurs and the patient’s tumor becomes castration-resistant (CR). At present, no curative treatment is available for CR CaP (2). Although the precise mechanisms leading to castration resistance are not completely understood, considerable insight into the progression of CaP has been recently achieved. Aberrant expression of some growth/survival related signal molecules, including their modulator microRNAs (miRNAs), is thought to be one mechanism associated with the development of CR CaP (3).
Mature miRNAs are a class of 19- to 25-nucleotide RNA molecules. These endogenous small RNAs may negatively regulate up to 30% of the human genes by imperfectly binding to miRNA-binding sites located at the 3′-untranslated regions (3′-UTRs) of targeted genes, causing either degradation of mRNAs or, more commonly for mammalian miRNAs, inhibition of their translation to functional proteins. Thus, miRNAs are involved in cell proliferation, differentiation, stress response, apoptosis, immunity and transcriptional regulation in many human diseases including cancer (4). Accumulating evidence supports the potential involvement of altered expression of miRNAs in initiation and progression of a wide range of human cancers, including CaP. It was reported that a number of miRNAs exhibit distinct expression patterns in clinical prostate tumor samples compared to benign prostate tissues (5–8), suggesting that these aberrantly expressed miRNAs are likely implicated in the biology of CaP.
miR-125b is a well characterized miRNA. In humans, there are two miR-125b members: miR-125b-1 and -2, located on chromosome 11 and chromosome 21, respectively. In CaP cells, the mature miR-125b is thought to be derived primarily from miR-125b-2 (9). Dysregulation of miR-125b has been reported to occur in multiple human cancer types, including cancers of the stomach (10), colon (11), pancreases (12), bladder (13) and ovary (14), as well as in oligodendroglial tumors (15) and acute lymphoblastic leukemia (16). It was recently reported that highly expressed miR-125b confers survival advantage by suppressing apoptosis in acute lymphoblastic leukemia (17). We previously found that miR-125b stimulates androgen-independent growth of CaP cells in vitro (18). In the current study, we further explored the functional role of miR-125b in CaP, and found that enforced expression of miR-125b promoted tumor growth in intact and castrated male nude mice. We also performed a series of experiments to investigate the molecular mechanisms accounting for miR-125b-mediated growth of CaP cells. The data obtained in this study demonstrate that miR-125b has oncogenic properties through its anti-apoptotic and proliferation-promoting activities and suggest that it can be exploited as a therapeutic target in CaP.
MATERIALS AND METHODS
Reagents
Synthetic miR-125b mimic (miR-125bm), miRNA negative control (miR-NC), anti-miR-125b, anti-miR-negative control (anti-miR-NC) and the pMIR-REPORT Luciferase vector were purchased from Ambion. The miR-125bm is short double-stranded RNA oligonucleotide, and it has been chemically modified to increase the stability in vivo and the activity of miR-125b. The anti-miR-125b was single-stranded RNA oligonucleotides designed to specifically bind to endogenous miR-125b, inhibiting its activity but not downregulating its abundance. Genistein combined polysaccharide (GCP) is a nutritional supplement and was generously provided by Amino Up Chemical Company Ltd. (Sapporo, Japan) and stock solutions were made in 50% DMSO/50% EtOH. Antibodies to Bak1, p21, SMAC, CytC, proCas3 were purchased from Cell Signaling Technology (Danvers, MA) and antibodies to p53 and Puma from Calbiochem.
Cell lines
CaP cell lines used in this study include PC3, DU145, PC-346C, and LNCaP and its sublines LNCaP-cds2 (named as cds2) and LNCaP-R273H. Both cds2 and LNCaP-R273H were prepared previously in our laboratory (19,20). PC-346C cell line that was kindly provided by Dr. Adrie van Bokhoven (University of Colorado) was developed from a non-progressive prostate tumor (21). LNCaP, PC-346C, PC3 and DU145 were routinely maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. For LNCaP-R273H cells, G418 (200 µg/ml) was added to the medium. cds2 cells were cultured in RPMI 1640 medium containing 5% charcoal/dextran-stripped serum (androgen-deprived medium).
Generation of stable PC-346C-miR-125b population
A lentiviral miR-125b expression vector that expresses a ~500-base pre-miR-125b and empty lentiviral vector were purchased from System Biosciences (SBI, Mountain View, CA). Pseudovirus production and cell transduction were performed following the manufacturer’s protocol. The resulting PC-346C CaP cells, PC-346C-miR-125b that stably overexpress miR-125b, were selected through fluorescence-activated cell sorting (FACS) and were maintained in medium described above. Over-expression of miR-125b in infected PC-346C cells was detected using a quantitative RT-PCR approach with a miR-125b-specific primer (Ambion).
Tumor xenografts
Male athymic nu/nu mice (4- to 6-week-old, Harlan Laboratories) were injected subcutaneously with suspensions of 2 × 106 PC-346C-miR-125b or PC-346C-vector control cells made in 50% Matrigel basement membrane matrix (1:1 vol/vol with complete medium; Becton Dickinson). Tumor dimensions were measured once per week and volumes calculated according to the formula: 1/2 (length × width × height). Castration was performed in a subpopulation of mice when tumors reached a diameter of approximately 0.6 cm. Animal studies were performed according to protocols approved by the Animal Care and Use Committee at University of California, Davis.
Annexin V binding assay
Anti-miR-125b-induced apoptosis was analyzed using a FACS Annexin V assay kit (Trevigen, Inc. Gaithersburg, MD) following the protocol provided by the manufacturer. Briefly, cells were transfected with anti-miR-125b and incubated for 72 hours. The cells were harvested, washed once with PBS, re-suspended in 100µl of 1X Annexin V binding buffer containing 1µl of Annexin V-FITC conjugate and 10µl of propidium iodide solution and then incubated for 30 min at room temperature in the dark. Subsequently, 400 µl 1X Annexin V binding buffer was added and samples analyzed on a FACScan flow cytometer. Data analysis was performed using FACScan software (Becton Dickinson).
Computational identification of miR-125b target transcripts
In order to gain insight into the mechanism(s) through which miR-125b influences growth and survival of CaP cells, an integrative bioinformatics strategy was developed to identify the target transcripts specifically relevant within the context of our models. This target profiling strategy entailed four steps: 1) defining a miR-125b-regulated gene expression profile; 2) generating a comprehensive miR-125b target site database by compilation of predicted targets (~943 separate sites) sourced from various publicly-available databases (22,23); 3) using this database as an index for mining and extracting of the miR-125b targets present in our list of differentially-expressed genes; and 4) selecting candidate genes based upon gene ontology and pathway analyses. For the first component, the list of miR-125b-regulated genes in LNCaP cells was obtained by overexpression of miR-125b followed by Affymetrix gene expression profiling and comparison analysis for differentially-expressed genes with DNA-Chip analyzer (dChip) software (24). Extraction of predicted miR-125b target transcripts from the expression profile was accomplished with an Excel add-in “parsing” tool from the Macros for Microarray (MfM) software (UC Davis Cancer Center Genomics and Expression Resource). Since miRNAs can negatively regulate the genes via accelerated decay, the resulting miR-125b target profile was limited to the down-regulated transcripts. Using the GO terms appended to each entity, the genes were then filtered based upon function in biological processes including apoptosis, tumor suppression and metastasis.
Western blot analysis
Cells were grown to 70–80% confluence and lysed using the cell lysis buffer (Cell Signaling Technology, Cat#: 9803). Denatured protein samples (20 to 100 µg) were resolved on 10% SDS-polyacrylamide gels, and then transferred to Immobilon PVDF membrane (Millipore). After blocking with 5% nonfat dry milk in Tris-buffered saline/0.05% Tween 20 (TBST), the membrane was incubated with a specific primary antibody, followed by the horseradixh peroxidase-conjugated secondary antibody. Proteins were visualized using ECL reagents.
Reporter constructs and luciferase assay
To construct reporter plasmids containing the 3′-DNA regions of p53 and PUMA, a 0.5-kb DNA fragment of the p53 3′-UTR and a 0.85-kb DNA fragment of the PUMA 3′-UTR that contain the putative miR-125 binding sites were prepared. Negative control plasmids containing 0.4-kb p53 and 0.7-kb PUMA 3′-UTR fragments that lack the miR-125b binding sites were also prepared. DNA fragments were cloned into the pMIR-REPORT Luciferase vector downstream of the reporter gene. The sequences and cloning direction of these PCR products were validated by DNA sequencing. For luciferase assay, cells (4×104 per well) were seeded into 24-well plates and cultured for 24 hours. The cells were then transfected with reporter plasmids. The pRL-SV40 Renilla luciferase plasmid (Promega) was used as an internal control. Two days later, cells were harvested and lysed with passive lysis buffer (Promega). Luciferase activity was measured using a dual luciferase reporter assay (Promega). The results were expressed as relative luciferase activity (Luc/Renilla).
Evaluation of miR-125b antagonism on growth with WST-1 assay
Cells (4.5 × 103 per well) were plated in 96-well plates in androgen-deprived medium. After being cultured for 24 hours, cells were transfected with anti-miR-125b or anti-miR-NC at a concentration of 50 nM using lipofectamine 2000 (Invitrigen). Five hours later, cells were treated with fresh medium or, if necessary, fresh medium containing anti-CaP drug in optimized concentration. The transfection protocol was optimized using a fluorescent pEGFP-N1 vector (Clontech), and resulted in the transfection of >90% of cds2 cells. Tetrazolium-based cell proliferation assay (WST-1; Promega) was then carried out at various days after transfection according to the manufacturer’s protocol.
RESULTS
miR-125b promotes tumor growth in intact and castrated male nude mice
Our previous in vitro experiments demonstrated that miR-125b stimulated androgen-independent growth (18). In this study, we determined whether miR-125b was able to enhance tumorigenicity. For this purpose, we generated a stable PC-346C-miR-125b pool by infection with a miR-125b-expressing lentiviral vector. Quantitative RT-PCR analysis of miR-125b abundance exhibited a 19-fold increase in miR-125b levels in miR-125b-infected cells compared to that in control cells (data not shown). PC-346C cell line was selected since it expresses wild-type p53 and wild-type AR (25) and is androgen-dependent/sensitive (26). Furthermore, this cell line has a limited ability to form tumors in intact nude mice and fails to grow in castrated mice (27). Two in vivo experiments were performed using the lentiviral-transduced PC-346C-miR-125b cells. First, we tested whether miR-125b determined tumorigenicity with respect to tumor formation rate and growth kinetics. Intact male nude mice (five mice per group) were injected subcutaneously with PC-346C-miR-125b or PC-346C-vector control cells. Tumors arose in all mice injected with PC-346C-miR-125b and in four of five control mice. However, PC-346C-miR-125b cells produced palpable tumors much faster than control cells with tumors appearing at 7 to 10 days vs. at approximately 21 days, respectively. Furthermore, PC-346C-miR-125b tumor volumes at 5 weeks were markedly larger than control tumors (Fig. 1A), indicating that in intact mice, elevated miR-125b expression accelerates tumor growth kinetics. We next tested whether miR-125b could support tumor growth in the absence of androgens. For this, male mice were injected with PC-346C-miR-125b cells. When tumors reached ~0.6 cm in the diameter, a subpopulation of mice bearing miR-125b tumors were castrated and tumor growth monitored. Castration resulted in a temporary growth regression of miR-125b tumors, followed by rapid growth (Fig. 1B). In summary, these data indicate that miR-125b increases the tumorigenicity of and promotes CR growth of PC-346C cells.
Fig. 1.
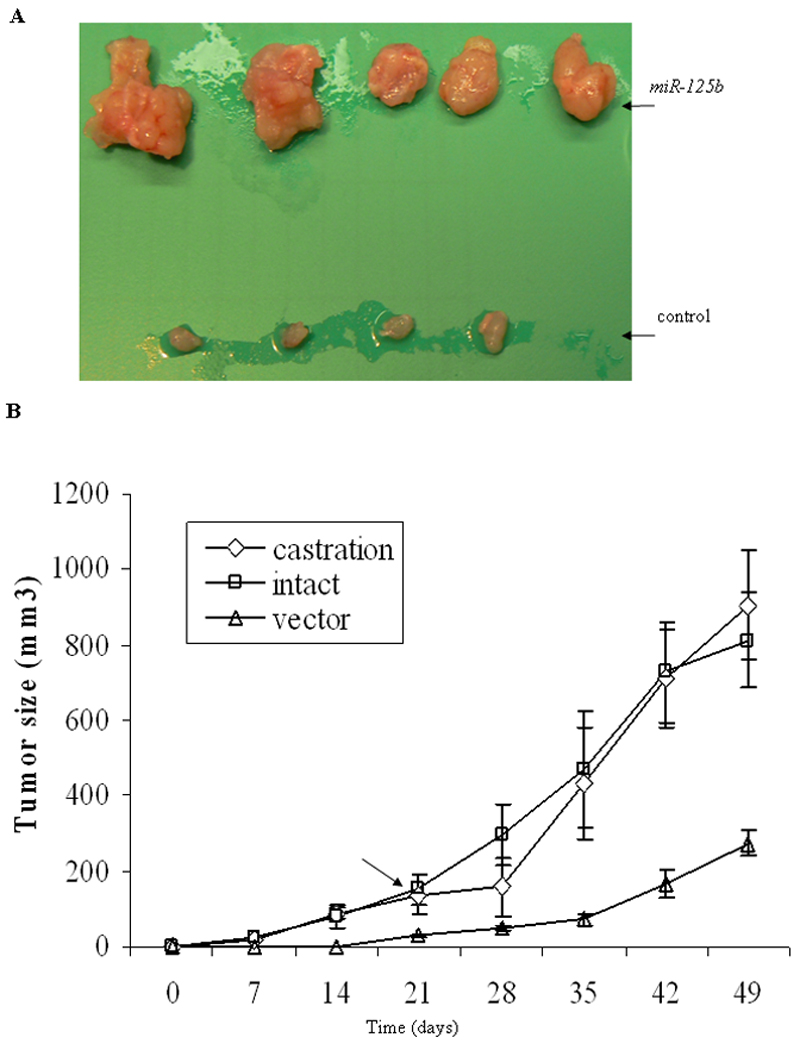
miR-125b promotes androgen-dependent and castration-resistant growth in vivo. A) Intact male nude mice (5 per group) each were injected subcutaneously with 2×106 PC-346C-miR-125b cells or PC-346C-vector control cells. Tumors were dissected from miR-125b mice (top) and control mice (bottom) five weeks after inoculation. B) Sixteen intact nude mice each were injected subcutaneously with 2×106 PC-346C-miR-125b cells which overexpress of miR-125b. When their tumor size reached approximately 0.125 cm3 (~0.6 cm in the diameter), eight of these 16 mice were castrated. The arrow indicates the castration time. Eight intact nude mice were injected subcutaneously with 2×106 PC-346C-vector control cells (vector). Each time point represents mean ± SD of 8 independent values.
Inactivation of miR-125b induces apoptosis of CaP cells
Reduced apoptosis can figure prominently in the CR growth of CaP (28). Based upon the results above, we wanted to test the hypothesis that downregulation of miR-125b activity would result in increased apoptotic cell death. To address this, the activity of miR-125b in PC-346C cells was antagonized by transfection with anti-miR-125b (50 nM). Cells were then cultured in androgen-deprived medium for four days and apoptotic cells detected and quantitated by Annexin V binding assay. We observed that in the absence of androgen anti-miR-125b induced 30.2% of PC-346C cells to undergo apoptotic cell death (Fig. 2A). The comparative figure for the anti-miR-NC cells was 5.4% (p<0.01). To provide biochemical evidence for the occurrence of apoptosis, we determined whether downregulation of miR-125b increases the release of mitochondrial cytochrome c, SMAC and activated caspase 3. As expected, treatment of PC-346C cells with anti-miR-125b induced an enhancement of cytochrome c and SMAC, and a reduction of procaspase 3 (Fig. 2B). Therefore, miR-125b-mediated suppression of apoptosis signaling contributes to the growth of CaP cells.
Fig. 2.
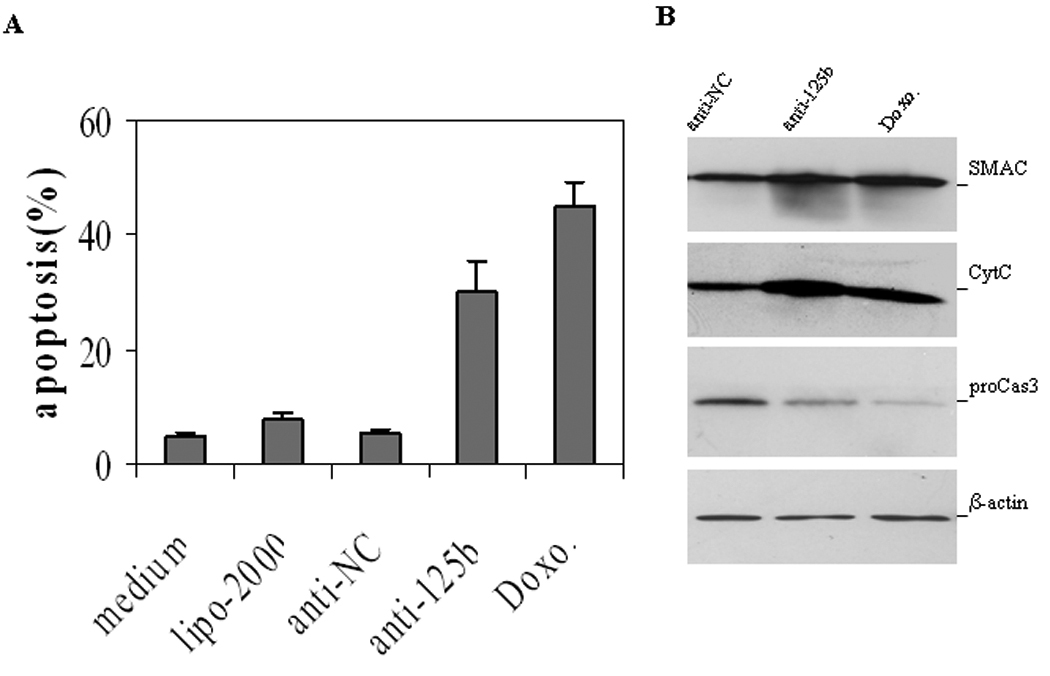
Downregulation of miR-125b activity induces apoptosis. A) Annexin V assay of apoptosis. PC-346C cells grown in charcoal-deprived medium were treated with 50 nM anti-miR-125b (anti-125b) or anti-miRNA negative control (anti-NC) for four days and stained with Annexin V and propidium iodide. Both early apoptotic cells (%) and late apoptotic/necrotic cells (%) are combined. Data are means ± SD from 3 independent experiments. Charcoal-deprived medium (medium), transfection reagent lipofectamine 2000 (lipo-2000) and doxorubicin (Doxo) were the controls. B) Wastern blot analysis of SMAC, cytochrome c (CytC) and pro-caspases 3 (proCas3). PC-346C cells were treated with 50 nM anti-miR-125b (anti-125b) or anti-miRNA negative control (anti-NC) for four days. The expression levels of proteins were analyzed with specific antibodies. Significant reduction of the pro-caspase 3 band indicates the activation of caspase-3 (49).
Identification of miR-125b targets
In order to better define the molecular mechanisms by which miR-125b promotes CaP cell growth and modulates apoptosis, we utilized a bioinformatics approach to identify its potential targets. For this, we compiled a database of the computationally-predicted miR-125b target sites (from MSKCC) and then used it as an index to mine a gene expression profile composed of transcripts that were differentially-expressed as a result of transient overexpression of miR-125b in CaP cells. In this manner, candidate direct targets are extracted from the entire dataset, which more than likely also contained transcripts regulated as a secondary response. The resulting putative, CaP-relevant miR-125b targets were then classified based upon Gene Ontology terms in order to determine the mRNAs whose suppression was most likely responsible for the alteration in one or more biological processes, which was described in Materials and Methods. In addition to Bak1 which we previously confirmed and characterized as a miR-125b target, seven additional genes were identified that contain miR-125b-binding sites in their 3′UTRs (Table I). Of these, three pro-apoptotic molecules (p53, Puma and Bak1) that are functionally inactivated either by mutation and/or dysregulation, have been reported to play important roles in the initiation and progression of CaP. The other five genes are potential tumor or metastasis suppressors in other cancer types. Myotubularin related protein 3 (MTMR3) induces the expression of p27 (29); BRCA1 associated protein-1 (BAP1) interacts with BRCA1 and suppresses cancer cell growth (30); sphingosine-1-phosphate lyase 1 (SGPL1) may play a role in cancer-surveillance pathways (31); aryl-hydrocarbon receptor repressor (AHRR) is an angiogenic factor (32); and sialidase 1 (lysosomal sialidase) (NEU1) inhibits tumor metastasis by decreasing integrin β4 signaling and down-regulating MMP-7 (33). However, their involvement in CaP remains largely unknown.
Table I.
Genes directly targeted by miR-125b in LNCaP cells
| Name | Fold change* |
Function |
miR-125b binding site at 3’UTR |
|---|---|---|---|
| p53 | −1.3 | Tumor suppressor | 733–739 |
| Puma | −2.5 | Pro-apoptosis | 24–30 |
| Bak1 | −2.0 | Pro-apoptosis | 630–637 |
| Mtmr3 | −1.4 | Tumor suppressor | 941–947 |
| Bap1 | −1.6 | Tumor suppressor | 580–586 |
| Sgpl1 | −1.9 | Tumor suppressor | 1586–1592 |
| Ahrr | −1.5 | Tumor suppressor | 1275–1281 |
| Neu1 | −1.7 | Metastasis suppressor | 331–337 |
Fold change: signal in miR-125 treated cells/that in miR-NC-treated cells.
miR-125b down-regulates pro-apoptotic molecules
Several approaches were then used to confirm that these three pro-apoptotic genes are bona fide targets of miR-125b. First, Western blot analysis of lentiviral-transduced PC-346C-miR-125b tumor cell lysates demonstrated that miR-125b overexpression markedly reduced the protein levels of p53, Puma and Bak1 (left panel in Fig. 3A). Similar results were observed (right panel in Fig. 3A) in cds2 cells that express 5-fold greater miR-125b than parental LNCaP cells do (18). Next, we determined whether elevating miR-125b levels ectopically with a chemically-modified miR-125b mimic (miR-125bm) would down-regulate these pro-apoptotic molecules in untreated or irradiated LNCaP cells. As expected, irradiation significantly enhanced the cellular level of p53 and its two direct effectors, p21 and Puma. Treatment with miR-125bm resulted in a marked decrease in both basal and irradiation-induced p53 levels (Fig. 3B). Moreover, downregulation of p53 by miR-125bm also reduced the expression of its effectors p21 and Puma. Consistent with our previous observation (18), miR-125bm targets Bak1. Additionally, we performed immunocytochemistry using enzymatic approache to stain both p53 and Puma in LNCaP cells that had been transfected with miR-125bm and then irradiated. Similar to the data obtained from Western blot analysis, ectopic expression of miR-125bm induced a marked downregulation of p53, Puma and Bak1 (Fig. 3 C).
Fig. 3.
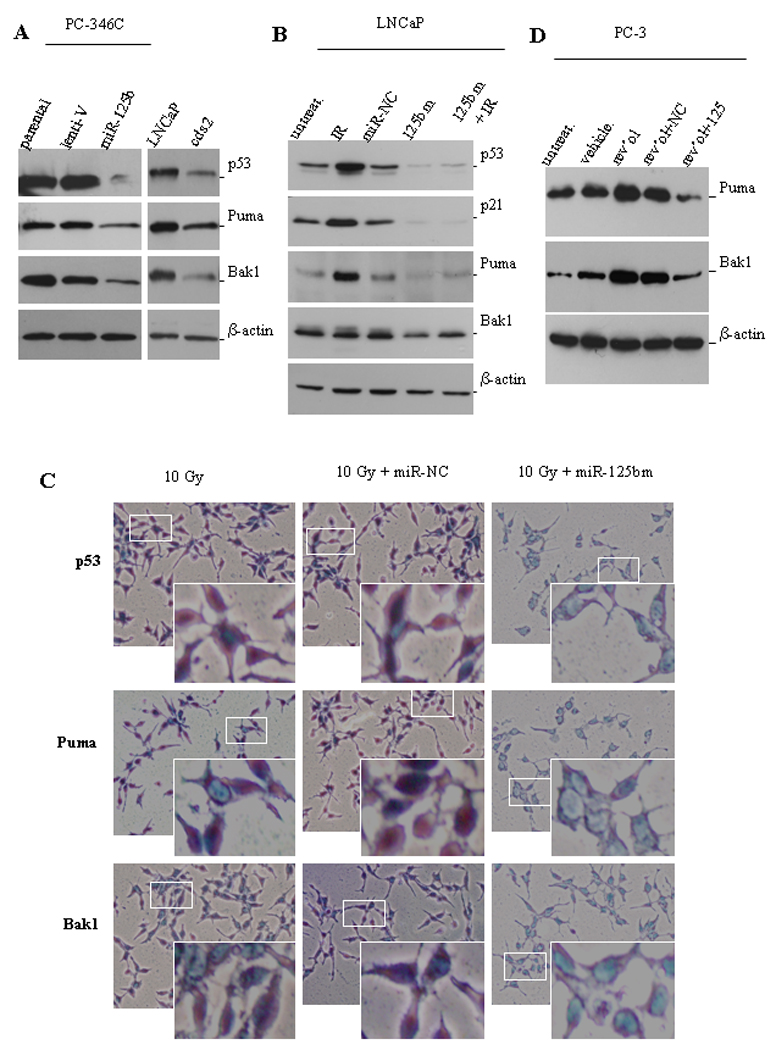
miR-125b targets pro-apoptotic genes. A) Western blot analyses of the expression levels of p53, Puma and Bak1 in lenti-miR-125b infected PC-346C cells (left panel), and in LNCaP cells grown in 10% FBS medium and in cds2 cells grown in androgen-deprived medium (right panel). Lent-V, lentiviral vector. B) Western blot analysis of the expression of p53, p21, Puma and Bak1 in miR-125bm-treated LNCaP cells. Cells were first transfected with 50 nM of chemically-modified miR-125b mimic (125bm) and 24 hours later irradiated with 10 Gys (IR). Eight hours after irradiation, cells were lysed and protein was extracted for Western blot analysis of p53, p21, Puma and Bak1. The controls include untreated cells (untreat.) and miRNA negative control (miR-NC)-treated cells. C) Detection of the expression levels of p53, Puma and Bak1 in LNCaP cells by immunostaining. LNCaP cells were grown for 24 hours on sterile slides in 100-mm Petri dishes in 10% FBS medium. Cells were first transfected with 50 nM of chemically-modified miR-125bm and 24 hours later irradiated with 10 Gys. Eight hours later, cells were fixed. p53, Puma or Bak1 were stained using specific 1st antibodies followed by a HRP-labeled 2nd antibody. Then, addition of the substrate (DAB) led to generate the brown color. The enzymatic approach shows that p53 locates mainly in nuclei, and Puma and Bak1 in cytoplasm. The white squares indicate the areas of magnified images. D) PC3 cells were transfected with 50 nM of miR-125bm (125bm) and 24 hours later treated with 100 µM resveratrol. Cells were lysed next day and protein was isolated for Western blot analysis of Puma and Bak1. Untreated cells (untreat.) and vehicle-treated cells are used as controls. β-actin is a loading control.
Mechanistic analysis of miR-125b as a direct regulator of Puma expression in the context of p53-expressing CaP cells is complicated by the fact that Puma is a transcriptional target of p53, which is also a target of miR-125b. Thus, it is difficult to discern whether the downregulation of Puma is due to a direct effect of miR-125b or to the decrease of p53 caused by miR-125b. To validate that miR-125b directly targets Puma, p53-null PC3 cells were treated with miR-125bm followed by treatment with resveratrol (100 µM), which was previously reported to upregulate Puma and Bak1 in PC3 cells (34). Western blot analysis confirmed that transfection with miR-125bm resulted in obvious reductions in resveratrol-stimulated Puma and Bak1 expression, respectively (Fig. 3D). Taken together, these data strongly support that miR-125b down-regulates the expression of p53, Puma and Bak1 in CaP cells.
Androgen downregulates pro-apoptosis molecules
As presented in our previous reporter (18), androgen upregulates the expression of miR-125b and consequently resulted in a reduction of Bak1 levels, we thus tested whether androgen treatment decreases the levels of p53 and Puma as well. To address this issue, LNCaP cells were treated with the synthetic androgen R1881 and Western blot analysis was performed to determine the levels of p53 and Puma, as well as Bak1 as a positive control. Similar to the previous observation, Bak1 expression was down-regulated by androgen and up-regulated by anti-miR-125b. When compared to untreated and vehicle-treated cells, R1881 induced a marked reduction of basal p53 and Puma levels (Fig. 4A). LNCaP cells were also treated with anti-miR-125b to decrease the activity of miR-125b. We found that ectopic expression of anti-miR-125 increased the expression of p53 and Puma proteins by approximately 2.0-fold (Fig. 4A), which was quantitatively measured using a Molecular Imager. In addition, when LNCaP cells were pre-treated with R1881 for 48 hours prior to irradiation with 10 Gys to stimulate increased expression of p53, it was found that R1881 treatment resulted in a 71% and 62% reduction of irradiation-stimulated up-regulation of p53 and Puma, respectively (Fig. 4B).
Fig. 4.
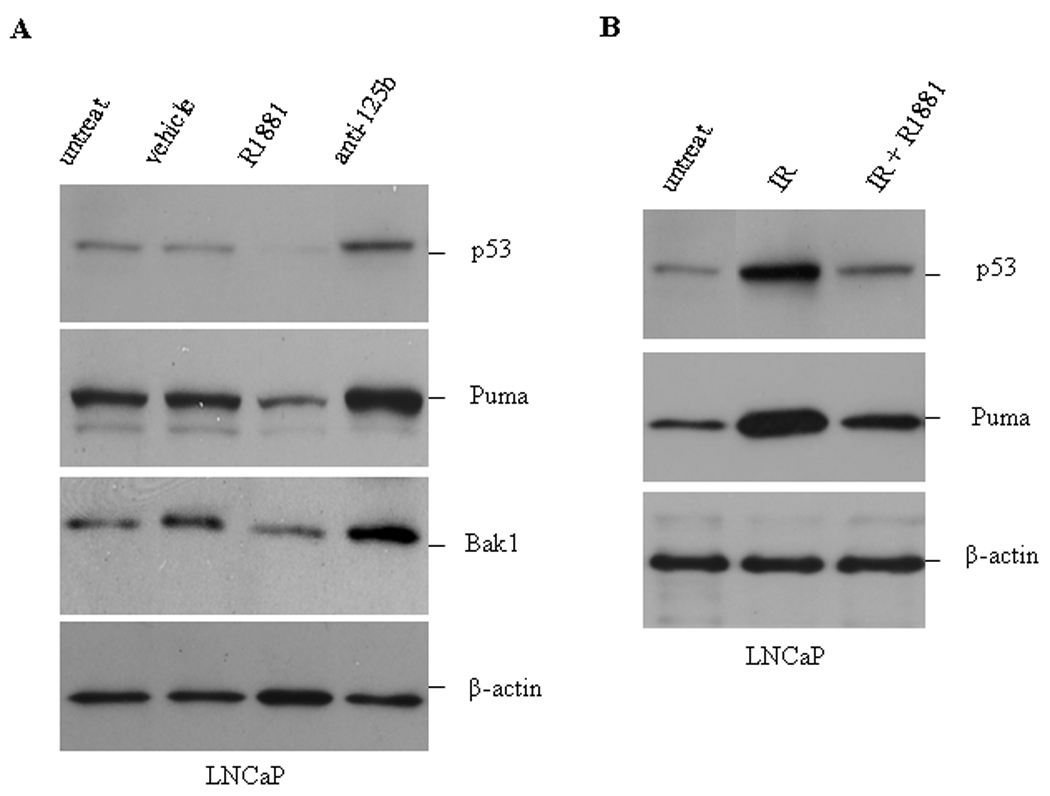
Effect of androgen on the expression of p53, Puma and Bak1. A) LNCaP cells were treated with 5.0 nM of R1881 or 100 nM anti-miR-125b (anti-125b) for 48 hours. Subsequently cells were harvested for Western blot analysis of p53, Puma and Bak1. B) LNCaP cells were treated with 5.0 nM R1881 for 48 hours followed by 10 Gys of irradiation (IR). Eight hours after irradiation, the cellular levels of p53 and Puma were analyzed by Western blotting. Controls include untreated cells (untreat.) and ethanol vehicle. β-actin is a loading control.
miR-125b binds to the 3’-UTRs of p53 and PUMA
Mature miRNAs negatively regulate gene expression by binding to the 3'-UTRs of target transcripts mediated by the complementarity of their 5'-end seed sequences (i.e., nucleotides 2–8 or 2–7) with miRNA-binding site sequences (35,36). We previously reported that miR-125b can bind to the 3′ UTR of BAK1 transcript that contains a binding site for miR-125b (18). Using the TargetScan program, we identified the miR-125b binding sites in the 3′-UTR regions of both p53 and PUMA. To determine whether these putative miR-125b binding sites are responsible for the negative post-transcriptional regulation of p53 and PUMA by miR-125b, pMIR-REPORT luciferase reporter constructs containing the 3′-UTR fragments of p53 or PUMA were co-transfected with miR-125bm into the miR-125b-null DU145 cells. As shown in Fig. 5A, cotransfection resulted in a 57% and 45% reduction of the reporter activity for p53 and PUMA, respectively. As a complimentary approach, we compared the androgen-regulated expression of p53 protein in the parental LNCaP cell line and its LNCaP-R273H subline. The latter ectopically expresses a p53R273H mutant allele from a cDNA expression cassette that does not contain a miR-125b binding site due to its lack of the p53 3′-UTR. Consistent with the previous observation, LNCaP-R273H cells expressed markedly increased p53 compared to the parental LNCaP (20). R1881 treatment induced an 78% reduction of p53 in the parental LNCaP cells, but only 40% reduction in LNCaP-R273H cells (Fig. 5B), suggesting that R1881-induced miR-125b only targeted endogenous wild-type p53 while ectopically-expressed p53R273H evaded regulation. Taken together, these results provide experimental validation that the 3′ UTRs of p53 and PUMA are targets of miR-125b.
Fig. 5.
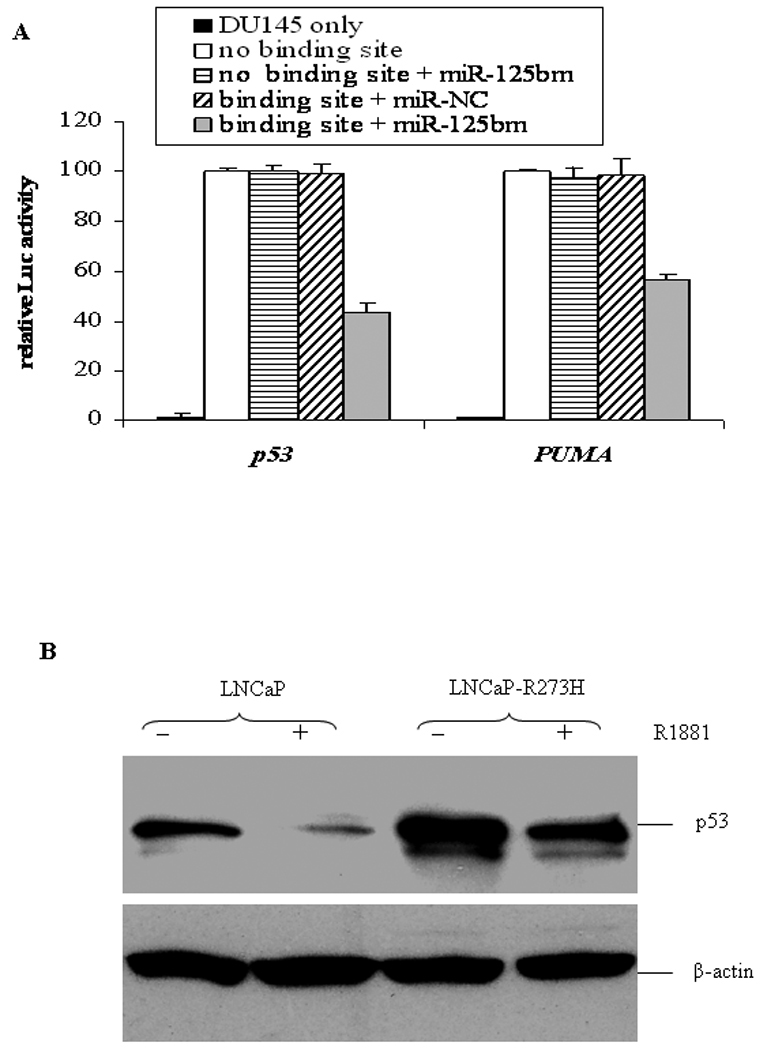
MiR-125b binds to the 3’UTRs of p53 and PUMA. A) Luciferase analysis of the 3’UTRs of p53 and PUMA in DU145 cells. Individual truncated 3’UTRs of p53 and PUMA were used as controls (no binding site) in these experiments. The assay was repeated three times with each assay being performed in three wells and similar results were obtained each time. The representative results are shown as M ± SD (n = 3). B) Western blot analysis of p53 protein in 5.0nM R1881-treated LNCaP cells and LNCaP-R273H cells.
Down-regulation of miR-125b sensitizes CaP cells to anti-cancer drugs
Our data presented above and in previous report (18) demonstrated that downregulation of miR-125b abundance inhibits the growth of CaP cells. We thus asked whether this miRNA is a potential therapeutic target that would improve the efficiency of existing CaP therapy. To address this issue, we evaluated the effect of a combination of anti-miR-125b and an anti-CaP drug on the growth inhibition or apoptosis of CaP cells. We have previously reported that GCP, an isoflavone-enriched nutriceutical, lowers the threshold at which CaP cells undergo apoptosis; hence, it was used here. To verify that the combination would increase the growth inhibition, GCP and anti-miR-125b were used to treat androgen-independent cds2 cells that overexpress natural miR-125b (18). We observed that the combined treatment resulted in a significant inhibition (p<0.05) of the CR growth compared to treatment with a single chemical (Fig. 6A). Additionally, we also tested cisplatin as it is a commonly-used chemotherapy in the treatment of solid malignancies including CaP. We used this combinational strategy to treat PC-346C cells that were cultured in medium containing 10% FBS and evaluated the extent to which they underwent apoptosis. It was found that anti-miR-125b plus 5 µM cisplatin increased the number of CaP cells that entered apoptosis in the presence of androgens (p<0.05, Fig 6B). The results suggest that down-regulation of miR-125b level can increase the sensitivity of CaP cells to different treatments.
Fig. 6.
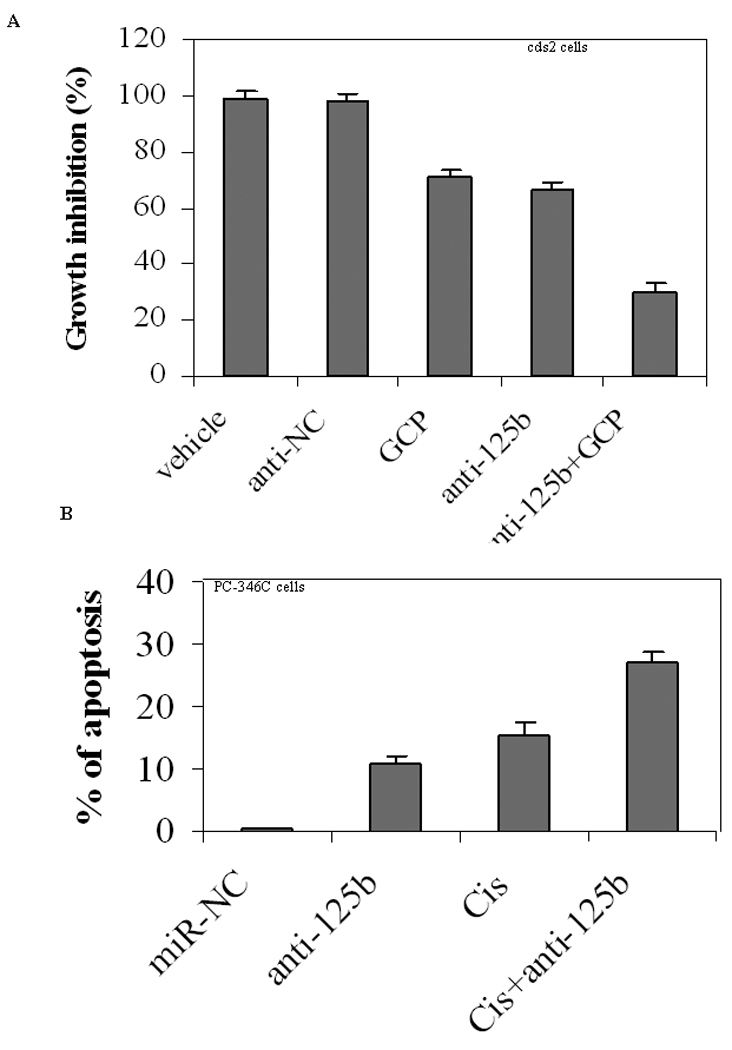
Effect of down-regulation of miR-125b on anti-cancer drugs. A) WST-1 assay of growth inhibition. Castration-resistant cds2 cells were grown in androgen-deprived medium and treated for 5 days using GCP (75µg/ml) and anti-miR-125b (anti-125b, 50 nM) in alone or in a combination. The controls include both vehicle and anti-miRNA-negative control (anti-NC). WST-1 analysis was used to measure the cell growth. The results are expressed as the growth inhibition relative to that treated with vehicle (100%), and shown as M ± SD (n=4). B) Annexin V assay of apoptosis. PC-346C cells cultured in medium containing 10% FBS were treated using Cisplatin (Cis, 5 µM) and anti-miR-125b (anti-125b, 50 nM) for two days in alone or in a combination. Cells were harvested and analyzed on the FACScan flow cytometer. Both early apoptotic cells (%) and late apoptotic/necrotic cells (%) are combined. Data are means ± SD from 3 independent experiments.
DISCUSSION
This study extends our previous observation that miR-125b stimulates CR growth in vitro (18). In the present study, we demonstrated that miR-125b enhances tumorigenicity of CaP cells in intact and castrated mice. Furthermore, we found that miR-125b mediates a broad attenuation of the intrinsic apoptosis pathway by targeting p53, Puma, and Bak1. Although an early investigation reported that miR-125b was down-regulated in 5 of 9 clinical CaP tissues related to 4 benign prostate samples (6), several later studies demonstrated that clinical CaP samples over-expressed miR-125b. Prueitt et al. (8) detected a 4.29-fold increase in miR-125b in locally-invasive tumors compared to those without local invasion. Mitchell et al. (37) observed a 6.35-fold increase in circulating miR-125b in metastatic CaP compared to that found in sera from healthy men. We previously reported that prostate tumors with high Gleason scores overexpressed miR-125b (18). Our experimental results obtained in the present study, together with a majority of previous clinical data, strongly suggest that aberrantly-expressed miR-125b has attributes of an oncogene by virtue of promoting the development and progression of CaP. Moreover, we found that androgen withdrawal by lowering miR-125b leads to an increase in p53 expression, which is consistent with Agus’s observation that androgen withdrawal induced expression of p53 (38). Our finding provides a molecular rationale for why androgen withdrawal may improve the outcome for CaP patients who undergo radiation therapy. Additionally, we found that repression of miR-125b activity sensitizes CaP cells to undergo apoptosis after treatment with various therapies, suggesting that miR-125b may be exploited as a target for CaP therapy.
While miR-125b has been reported to be highly expressed in many types of human cancers, including CaP, it remains to be determined how miR-125b affects the growth of CaP cells. At the initiation of the study, like most of miRNAs, the precise physiologic targets of miR-125b had not been validated experimentally. We reasoned that for miR-125b to be a driver miRNA, it would need to satisfy the criteria of targeting relevant and potent effector molecules in the context of CaP cells. In our earlier report, Bak1 was identified as a target of miR-125b. In the present study, p53 and Puma were further validated to be direct targets of miR-125b, which is consistent with a recent finding that miR-125b is an important negative regulator of p53 (39). Since p53, Puma and Bak1 are pro-apoptotic molecules, our results show that miR-125b is involved in the p53 network and plays a role in how it regulates apoptosis. Conversely, the data provide a mechanistic explanation for why suppression of miR-125b can inhibit tumor growth. In addition to these three pro-apoptotic molecules, miR-125b also targets five additional tumor suppressor mRNAs. The precise roles of these putative targets in CaP are unknown. It is possible that decreased expression of these tumor suppressors, together with down-regulated pro-apoptotic molecules, contributes to tumorigenesis and progression of CaP by providing the cells a survival advantage in an androgen-sufficient or androgen-deprived environment. In the ongoing studies, we will conduct in vitro and in vivo experiments to evaluate the effects of individual molecules on the growth of CaP cells. Elucidation of their contribution to CaP will shed light on understanding mechanism underlying the tumorigenesis and progression of CaP, which might result in identify new molecular targets that would be used to develop potential treatments for this disease.
Data obtained from this study show that miR-125b is involved in the governance of proliferation and apoptosis in CaP. Under normal conditions, pro-apoptotic molecules induce mitochondrial outer membrane permeability, causing the release of apoptotic molecules (e.g., cytochrome c), subsequent caspase activation, and ultimately leading to apoptotic cell death. In contrast, anti-apoptotic molecules promote survival by antagonizing apoptotic mediators at one or more levels. In CaP cells, particularly in CR tumors, growth is determined by the relative balance between cell proliferation and cell death (apoptosis). Aberrantly-expressed miR-125b down-regulates p53, Puma and Bak1. As critical regulators of apoptosis, these molecules have been shown to play a crucial role in suppression of the malignant phenotype of CaP cells (40–42). Therefore, aberrant miR-125b expression re-sets the balance of apoptosis by directly targeting these molecules and indirectly affecting other members of the Bcl-2 family, conferring a survival advantage to CaP cells. Additionally, it is well known that the AR plays a dominant role in both the initiation and progression of CaP. Studies have demonstrated that wild-type p53 can repress AR activity (43). We observed that the AR signaling up-regulates miR-125b and miR-125b in turn down-regulates wild-type p53. We thus speculate that this is a self sustaining loop in CaP cells, which positively regulates the growth of CaP cells.
We, as well as others (39), validated that miR-125b is a direct, negative regulator of wild-type p53 by suppressing its basal expression and up-regulation in response to pro-apoptotic stimuli. Here, we asked how aberrant miR-125b expression affects apoptosis in CaP cells harboring a p53 mutation. Addressing this issue is of clinical importance, since mutation of p53 occurs in more than 40% of CaPs (44–46). At present, the exact answer to this question has not been determined. Data obtained from this present study provides some insight regarding repression of apoptosis by miR-125b in p53-inactivated CaP cells, which may involve targeting Puma and Bak1. In addition to p53 that directly regulates Puma, some of the other cell death and survival signals have been reported to upregulate Puma in a p53-independent fashion (47,48). Consistent with a previous report (34), we also observed that resveratrol induces an increase in Puma and Bak1 in p53-null PC3 CaP cells. Furthermore, increased miR-125b was able to significantly reduce resveratrol-stimulated Puma and Bak1. Therefore, miR-125b-mediated downregulation of Puma and Bak1 may play a key role in the p53-inactivated cases. Indeed, one study demonstrated that low Puma expression was associated with a rapid biochemical recurrence of CaP (41).
Given the complexity implicated in the castration resistance of CaP, treatment of this disease may rely on multimodal combination regimens. Until recently, however, chemotherapy has demonstrated only a small survival benefit in patients with CR CaP. The current strategy regarding the treatment of this disease is to prevent occurrence of castration resistance. The fact that one miRNA may target a large number of coding genes involving diverse signaling pathways make the non-encoding small RNA attractive in the design of combination therapy regimens. Indeed, recent study showed that molecular alterations elicited by targeting single miRNAs can provide growth advantages to tumor cells (17). Aberrant expression of miR-125b occurs in at least a subset of CaP cases. This miRNA targets various molecules related to the progression and therapy-resistance of CaP, which implicates it as an emerging alternative treatment option. In this study, we presented evidence that miR-125b has oncogenic function. It stimulates CaP growth in xenografts through an anti-apoptotic pathway, very importantly in both the presence and absence of androgens. Therefore, beyond CR CaP, this miRNA also is important in CaP progression. The anti-apoptotic phenotype was not an artifact of miR-125b overexpression in CaP cells. Its relevance to CaP cells is suggested by the sensitization to apoptosis induced by GCP after knockdown of the endogenous miR-125b in CaP cells. Furthermore, we validated that miR-125b directly targets p53, Puma and Bak1, which provide new insights into the molecular basis of CaP progression. There is little doubt that both p53 and AR play important roles in the progression of CaP. Here, we demonstrate that miR-125b links these two important pathways. Thus, it would appear to be an excellent target to pursue in our attempts to discover improved treatments for patients with prostate cancer.
CONCLUSION
The present results demonstrate that overexpression of miR-125b promoted tumor growth in intact and castrated male nude mice. A series of experiments was performed to investigate the molecular mechanisms accounting for miR-125b-mediated growth of CaP cells. It was found that miR-125b down-regulates three key pro-apoptotic molecules: p53, Puma and Bak1. Furthermore, reduction of miR-125b activity by using anti-miR- 125b oligonucleotides induced apoptosis of CaP cells and sensitizes CaP cells to anti-cancer drugs. These results suggest that miR-125b is oncogenic and makes it an attractive therapeutic target in prostate cancer.
Acknowledgments
Supported by NCI grant CA92069 and DOD grant PC080488
Footnotes
Conflict of Interest. The authors have declared no conflict of interest.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Lassi K, Dawson NA. Emerging therapies in castrate-resistant prostate cancer. Curr Opin Oncol. 2009;21(3):260–265. doi: 10.1097/CCO.0b013e32832a1868. [DOI] [PubMed] [Google Scholar]
- 3.Shi XB, Tepper CG, White RW. MicroRNAs and prostate cancer. J Cell Mol Med. 2008;12(5A):1456–1465. doi: 10.1111/j.1582-4934.2008.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowland JB, Hother C, Gronbaek K. MicroRNAs and cancer. APMIS. 2007;115(10):1090–1106. doi: 10.1111/j.1600-0463.2007.apm_775.xml.x. [DOI] [PubMed] [Google Scholar]
- 5.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67(13):6130–6135. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 7.Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27(12):1788–1793. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- 8.Prueitt RL, Yi M, Hudson RS, Wallace TA, Howe TM, Yfantis HG, Lee DH, Stephens RM, Liu CG, Calin GA, Croce CM, Ambs S. Expression of microRNAs and protein-coding genes associated with perineural invasion in prostate cancer. Prostate. 2008;68(11):1152–1164. doi: 10.1002/pros.20786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee YS, Kim HK, Chung S, Kim KS, Dutta A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem. 2005;280(17):16635–16641. doi: 10.1074/jbc.M412247200. [DOI] [PubMed] [Google Scholar]
- 10.Ueda T, Volinia S, Okumura H, Shimizu M, Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, Yoshida K, Sasaki H, Nomura S, Seto Y, Kaminishi M, Calin GA, Croce CM. Relation between microRNA expression and progression and prognosis of gastric cancer: a microRNA expression analysis. Lancet Oncol. 11(2):136–146. doi: 10.1016/S1470-2045(09)70343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baffa R, Fassan M, Volinia S, O'Hara B, Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM, Rosenberg A. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J Pathol. 2009;219(2):214–221. doi: 10.1002/path.2586. [DOI] [PubMed] [Google Scholar]
- 12.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C, Croce CM. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297(17):1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 13.Veerla S, Lindgren D, Kvist A, Frigyesi A, Staaf J, Persson H, Liedberg F, Chebil G, Gudjonsson S, Borg A, Mansson W, Rovira C, Hoglund M. MiRNA expression in urothelial carcinomas: important roles of miR-10a, miR-222, miR-125b, miR-7 and miR-452 for tumor stage and metastasis, and frequent homozygous losses of miR-31. Int J Cancer. 2009;124(9):2236–2242. doi: 10.1002/ijc.24183. [DOI] [PubMed] [Google Scholar]
- 14.Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol Oncol. 2008;111(3):478–486. doi: 10.1016/j.ygyno.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 15.Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA. 2006;12(2):187–191. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonoki T, Iwanaga E, Mitsuya H, Asou N. Insertion of microRNA-125b-1, a human homologue of lin-4, into a rearranged immunoglobulin heavy chain gene locus in a patient with precursor B-cell acute lymphoblastic leukemia. Leukemia. 2005;19(11):2009–2010. doi: 10.1038/sj.leu.2403938. [DOI] [PubMed] [Google Scholar]
- 17.Gefen N, Binder V, Zaliova M, Linka Y, Morrow M, Novosel A, Edry L, Hertzberg L, Shomron N, Williams O, Trka J, Borkhardt A, Izraeli S. Hsa-mir-125b-2 is highly expressed in childhood ETV6/RUNX1 (TEL/AML1) leukemias and confers survival advantage to growth inhibitory signals independent of p53. Leukemia. 24(1):89–96. doi: 10.1038/leu.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi XB, Xue L, Yang J, Ma AH, Zhao J, Xu M, Tepper CG, Evans CP, Kung HJ, deVere White RW. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A. 2007;104(50):19983–19988. doi: 10.1073/pnas.0706641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi XB, Ma AH, Tepper CG, Xia L, Gregg JP, Gandour-Edwards R, Mack PC, Kung HJ, deVere White RW. Molecular alterations associated with LNCaP cell progression to androgen independence. Prostate. 2004;60(3):257–271. doi: 10.1002/pros.20039. [DOI] [PubMed] [Google Scholar]
- 20.Nesslinger NJ, Shi XB, deVere White RW. Androgen-independent growth of LNCaP prostate cancer cells is mediated by gain-of-function mutant p53. Cancer Res. 2003;63(9):2228–2233. [PubMed] [Google Scholar]
- 21.van Weerden WM, de Ridder CM, Verdaasdonk CL, Romijn JC, van der Kwast TH, Schroder FH, van Steenbrugge GJ. Development of seven new human prostate tumor xenograft models and their histopathological characterization. Am J Pathol. 1996;149(3):1055–1062. [PMC free article] [PubMed] [Google Scholar]
- 22.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36((Database issue)):D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2(11):e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98(1):31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57(3):205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 26.Limpens J, Schroder FH, de Ridder CM, Bolder CA, Wildhagen MF, Obermuller-Jevic UC, Kramer K, van Weerden WM. Combined lycopene and vitamin E treatment suppresses the growth of PC-346C human prostate cancer cells in nude mice. J Nutr. 2006;136(5):1287–1293. doi: 10.1093/jn/136.5.1287. [DOI] [PubMed] [Google Scholar]
- 27.Marques RB, Erkens-Schulze S, de Ridder CM, Hermans KG, Waltering K, Visakorpi T, Trapman J, Romijn JC, van Weerden WM, Jenster G. Androgen receptor modifications in prostate cancer cells upon long-termandrogen ablation and antiandrogen treatment. Int J Cancer. 2005;117(2):221–229. doi: 10.1002/ijc.21201. [DOI] [PubMed] [Google Scholar]
- 28.Cohen MB, Rokhlin OW. Mechanisms of prostate cancer cell survival after inhibition of AR expression. J Cell Biochem. 2009;106(3):363–371. doi: 10.1002/jcb.22022. [DOI] [PubMed] [Google Scholar]
- 29.Yoo YD, Cho SM, Kim JS, Chang YS, Ahn CM, Kim HJ. The human myotubularin-related protein suppresses the growth of lung carcinoma cells. Oncol Rep. 2004;12(3):667–671. [PubMed] [Google Scholar]
- 30.Jensen DE, Rauscher FJ., 3rd BAP1, a candidate tumor suppressor protein that interacts with BRCA1. Ann N Y Acad Sci. 1999;886:191–194. doi: 10.1111/j.1749-6632.1999.tb09414.x. [DOI] [PubMed] [Google Scholar]
- 31.Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends Mol Med. 2007;13(5):210–217. doi: 10.1016/j.molmed.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, Martinez A, Narayan G, Kirsch I, Franklin W, Hirsch F, Birrer M, Cuttitta F. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118(2):640–650. doi: 10.1172/JCI30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uemura T, Shiozaki K, Yamaguchi K, Miyazaki S, Satomi S, Kato K, Sakuraba H, Miyagi T. Contribution of sialidase NEU1 to suppression of metastasis of human colon cancer cells through desialylation of integrin beta4. Oncogene. 2009;28(9):1218–1229. doi: 10.1038/onc.2008.471. [DOI] [PubMed] [Google Scholar]
- 34.Shankar S, Siddiqui I, Srivastava RK. Molecular mechanisms of resveratrol (3,4,5-trihydroxy-trans-stilbene) and its interaction with TNF-related apoptosis inducing ligand (TRAIL) in androgen-insensitive prostate cancer cells. Mol Cell Biochem. 2007;304(1–2):273–285. doi: 10.1007/s11010-007-9510-x. [DOI] [PubMed] [Google Scholar]
- 35.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 36.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agus DB, Cordon-Cardo C, Fox W, Drobnjak M, Koff A, Golde DW, Scher HI. Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst. 1999;91(21):1869–1876. doi: 10.1093/jnci/91.21.1869. [DOI] [PubMed] [Google Scholar]
- 39.Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, Lodish HF, Lim B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23(7):862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Isaacs WB, Carter BS, Ewing CM. Wild-type p53 suppresses growth of human prostate cancer cells containing mutant p53 alleles. Cancer Res. 1991;51(17):4716–4720. [PubMed] [Google Scholar]
- 41.Diallo JS, Aldejmah A, Mouhim AF, Peant B, Fahmy MA, Koumakpayi IH, Sircar K, Begin LR, Mes-Masson AM, Saad F. NOXA and PUMA expression add to clinical markers in predicting biochemical recurrence of prostate cancer patients in a survival tree model. Clin Cancer Res. 2007;13(23):7044–7052. doi: 10.1158/1078-0432.CCR-07-1224. [DOI] [PubMed] [Google Scholar]
- 42.Choi S, Singh SV. Bax and Bak are required for apoptosis induction by sulforaphane, a cruciferous vegetable-derived cancer chemopreventive agent. Cancer Res. 2005;65(5):2035–2043. doi: 10.1158/0008-5472.CAN-04-3616. [DOI] [PubMed] [Google Scholar]
- 43.Shenk JL, Fisher CJ, Chen SY, Zhou XF, Tillman K, Shemshedini L. p53 represses androgen-induced transactivation of prostate-specific antigen by disrupting hAR amino- to carboxyl-terminal interaction. J Biol Chem. 2001;276(42):38472–38479. doi: 10.1074/jbc.M103652200. [DOI] [PubMed] [Google Scholar]
- 44.Shi XB, Gandour-Edwards R, Beckett LA, Deitch AD, de Vere White RW. A modified yeast assay used on archival samples of localized prostate cancer tissue improves the detection of p53 abnormalities and increases their predictive value. BJU Int. 2004;94(7):996–1002. doi: 10.1111/j.1464-410X.2004.05093.x. [DOI] [PubMed] [Google Scholar]
- 45.Gumerlock PH, Chi SG, Shi XB, Voeller HJ, Jacobson JW, Gelmann EP, deVere White RW. p53 abnormalities in primary prostate cancer: single-strand conformation polymorphism analysis of complementary DNA in comparison with genomic DNA. The Cooperative Prostate Network. J Natl Cancer Inst. 1997;89(1):66–71. doi: 10.1093/jnci/89.1.66. [DOI] [PubMed] [Google Scholar]
- 46.Chi SG, deVere White RW, Meyers FJ, Siders DB, Lee F, Gumerlock PH. p53 in prostate cancer: frequent expressed transition mutations. J Natl Cancer Inst. 1994;86(12):926–933. doi: 10.1093/jnci/86.12.926. [DOI] [PubMed] [Google Scholar]
- 47.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7(3):673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 48.You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203(7):1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcelli M, Cunningham GR, Walkup M, He Z, Sturgis L, Kagan C, Mannucci R, Nicoletti I, Teng B, Denner L. Signaling pathway activated during apoptosis of the prostate cancer cell line LNCaP: overexpression of caspase-7 as a new gene therapy strategy for prostate cancer. Cancer Res. 1999;59(2):382–390. [PubMed] [Google Scholar]