

Abstract
The OTX2 homeobox-containing transcription factor gene was shown to play a key role in the development of head structures in vertebrates. In humans, OTX2 mutations result in anophthalmia/microphthalmia (A/M) often associated with systemic anomalies. We screened fifty-two unrelated individuals affected with A/M and identified disease-causing variants in four families (8%), a higher frequency than previously reported. All four mutations are predicted to result in truncation of normal OTX2 protein sequence consistent with previously reported mechanisms; three changes occurred de novo and one mutation was inherited from an affected parent. Four out of the five OTX2-positive patients in our study displayed additional systemic findings, including two novel features, Wolf-Parkinson White syndrome and an anteriorly placed anus. Analysis of the phenotypic features of OTX2-positive A/M patients in this study and those previously reported suggests the presence of pituitary anomalies and lack of genitourinary and gastrointestinal manifestations as potential distinguishing characteristics from SOX2 anophthalmia syndrome. Interestingly, pituitary anomalies seem to be more strongly associated with mutations that occur in the second half of OTX2, after the homeodomain and SGQFTP motif. OTX2 patients also show a high rate of inherited mutations (36%), often from mildly or unaffected parents, emphasizing the importance of careful parental examination/testing.
Keywords: Anophthalmia, microphthalmia, OTX2, pituitary
INTRODUCTION
OTX2 belongs to the family of homeobox-containing transcription factors related to the Drosophila gene orthodenticle (1). The Otx genes were shown to control variable developmental processes and, in particular, to be required for specification of the developing brain (2,3). Complete Otx2-deficiency in mice results in early embryonic lethality due to abnormal primitive streak formation. Otx2+/− mouse embryos show multiple abnormalities in the formation of head structures with phenotypes including anophthalmia/microphthalmia (A/M), micrognathia/agnathia, ethmocephaly, holoprosencephaly, short nose, and acephaly (2, 4). Studies in other species also support the conserved head-organizing role of Otx2 during development (5,6).
Heterozygous OTX2 mutations in humans were first identified in eight probands affected with anophthalmia/microphthalmia (A/M) along with brain malformations and short stature in some individuals (7); two of these mutations (p.P133T and p.P134A) were later determined unlikely to be pathogenic (8). Since then, thirteen additional families with point mutations in OTX2 and four patients with gene deletions were reported among individuals affected with A/M and/or pituitary abnormalities (8–15). OTX2 mutations were determined to explain at least 2–3% of A/M in the first cohorts screened (8). The ocular and other systemic anomalies observed in human patients are consistent with Otx2 activities during eye and brain development documented in animal models (2,3,16).
Anophthalmia/microphthalmia is observed in 1–3 per 10,000 births and often associated with systemic anomalies (17–19). Besides OTX2, several other genetic factors have been implicated in anophthalmia/microphthalmia (19,20) with most genes explaining only a small subset of cases (<1%), the exception being SOX2 which is mutated in 10–20% of A/M (21–24). Identification of genetic mutations in human patients allows classification of A/M into distinct subtypes based on the specific ocular and/or other systemic features associated with specific genetic causes. Genotype-phenotype correlations, if identified, can be used to target genetic testing and also to guide the diagnosis and management of patients with A/M. In this report, we present four additional OTX2 mutations identified in syndromic A/M patients and provide analysis of the new and previously reported cases to better define the OTX2 microphthalmia syndrome.
MATERIALS AND METHODS
Patients were enrolled through the A/M Clinical Registry at Albert Einstein Medical Center, Philadelphia, or the Genetic Studies of Human Developmental Disorders at the Medical College of Wisconsin, Milwaukee. This human study was approved by the Institutional Review Boards of Children's Hospital of Wisconsin and Albert Einstein Healthcare Network. Written informed consent was obtained from all participants and/or parents, as appropriate.
A total of 52 probands were screened; 40 had bilateral A/M and 12 had unilateral A/M. The control population included 96 Caucasian (European Collection of Cell Cultures, Salisbury, UK) and 96 Hispanic (Coriell Institute for Medical Research, Camden, NJ) individuals. The OTX2 coding region was examined using direct sequencing of genomic DNA, as previously described (25) and using Mutation Surveyor analysis (SoftGenetics, State College, PA); PCR products were obtained with primers: set 1 forward 5’-AAGTTAGTGCTGGAACGTGG-3’ and reverse 5’-TCAGTAAATGCCCTGGGACT-3’; set 2 forward 5’-GAAAGAAGCAGCGTGTGTCC-3’ and reverse 5’-GATTAAGTGGTGACGGGCAG-3’; set 3 forward 5’-GAGCCTCCCCAACTTTCTTAC-3’ and reverse 5’-GAAGCTGGTGATGCATAGGG-3’; set 4 forward 5’-TCAGATCCCTTGTCCACCTC-3’ and reverse 5’-TTTGTAGGCCCCTCTAAGGC-3’. Reference sequence NM_172337.1 was used.
RESULTS
Screening of OTX2 identified novel mutations in four of the 52 probands with anophthalmia or microphthalmia (Table 1 and Figure 1).
Table 1.
Summary of OTX2 mutations in human patients reported to date. The reference sequence NM_172337.1 was used.
| Reference | Nucleotide Mutation* |
Amino Acid Mutation |
Eye Anomalies | Other Structural Brain Anomaly |
Abnormal Pituitary Structure/ Function |
SS | DD/ MR |
Other features | Family history | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AN | MI | ONH/A | ASD | Other | |||||||||
| Ragge et al. (7) | c.81delC | p.S28PfsX23 | - | B | B | - | - | U | U | U | - | - | Unaffected mother mosaic; affected sibling |
| Ragge et al. (7) | c.81delC | p.S28PfsX23 | - | B | U | - | - | Y | U | U | U | Pregnancy terminated | Sibling of above |
| Wyatt et al. 2008 | c.93C>G | p.Y31X | - | L | U | - | R | U | U | U | U | U | Parents DNA normal |
| Wyatt et al. 2008 | c.106dupC | p.R36PfsX52 | - | R | U | - | - | U | U | - | - | - | Affected sibling (below), Parents DNA normal; Presumed mosaicism |
| Wyatt et al. 2008 | c.106dupC | p.R36PfsX52 | R | - | U | - | L | U | U | - | - | - | Brother of above |
| Ragge et al. (7) | c.117_118delCC | p.R40GfsX47 | B | - | B | - | - | Y | NS | Y | - | - | Unaffected mother carries mutation |
| Ragge et al. (7) | c.117_118delCC | p.R40GfsX47 | - | - | - | - | - | U | U | - | - | - | Mother of above |
| Patient 1A, this study | c.136dupA | p.T46NfsX42 | - | B | B | - | - | - | NS | - | - | Microcephaly, anteriorly placed anus, sacral dimple | Affected father carries mutation |
| Patient 1B, this study | c.136dupA | p.T46NfsX42 | - | L | L | L | B | U | U | - | - | - | Father of above |
| Dateki et al. 2010 | c.214_217delGCACinsCA | p.A72HfsX15 | - | B | U | U | U | U | - | - | U | - | Parents' DNA normal |
| Dateki et al. 2010 | c.221_236del | p.K74SfsX30 | R | L | U | U | U | - | Y | Y | Y | Retractile testis | Parents' DNA normal |
| Ragge et al. 2005 | c.265C>G | p.R89G | - | B | B | - | B | - | NS | - | - | - | Mother had previous stillbirth; Parents' DNA normal |
| Wyatt et al. 2008 | c.289C>T | p.Q97X | - | B | U | - | - | U | U | U | U | U | Dizygotic twin and father with mutation |
| Wyatt et al. 2008 | c.289C>T | p.Q97X | - | - | U | - | B | U | U | U | U | U | Twin of above |
| Wyatt et al. 2008 | c.289C>T | p.Q97X | U | U | U | U | 1 | U | U | U | U | 'Reduced vision" one eye | Father of above |
| Ragge et al. (7) | c.295C>T | p.Q99X | B | - | B | - | - | Y | U | U | Y | Seizures | Unaffected father carries mutation |
| Ragge et al. (7) | c.295C>T | p.Q99X | - | - | - | - | - | U | U | - | - | - | Father of above |
| Patient 2, this study | c.313C>T | p.Q105X | B | - | B | - | - | - | NS | - | Y | Wolf-Parkinson White syndrome, feeding difficulties | Parents' DNA normal |
| Wyatt et al. (8) | c.371_372delAG | p.S125Wf sX11 | B | - | U | - | - | U | U | U | Y | U | Unaffected mother carries mutation |
| Wyatt et al. (8) | c.371_372delAG | p.S125Wf sX11 | - | - | - | - | - | U | U | - | - | - | Mother of above |
| Dateki et al. (11) | c.402_403insC | p.S135LfsX2 | B | - | B | - | - | - | Y | Y | Y | Cleft palate | Parents' DNA normal |
| Tajima et al. 2009 | c.405_406insCT | p.S136LfsX43 | B | - | B | - | - | Y | Y | Y | Y | Microcephaly, small penis, cryptorchidism | Parents' DNA normal |
| Henderson et al. (10) | c.413C>G | p.S138X | - | - | - | - | B | - | Y | Y | U | Feeding difficulties | Parents' DNA normal |
| Patient 3, this study | c.456_457delGAinsAT | p.W152X | R | L | B | - | - | - | Y | Y | Y | Microcephaly, feeding difficulties | Parents' DNA normal |
| Ragge et al. (7) | c.464_465dup | p.S156LfsX23 | R | L | B | - | L | Y | NS | - | Y | Microcephaly | Parents' DNA normal |
| Ragge et al. 2005 | c.537T>A | p.Y179X | - | B | U | B | B | U | U | Y | Y | Seizures | Mother mosaic with pigmentary retinopathy; Affected brother and niece |
| Ragge et al. 2005 | c.537T>A | p.Y179X | - | B | - | B | B | - | NS | Y | Y | - | Brother of above, affected daughter (below) |
| Henderson et al. 2007 | c.537T>A | p.Y179X | - | R | U | B | B | Y | U | U | Y | Microcephaly, NF type 1; Mutations in PAX6 and NF1 | Daughter of above; mother with NF1 and PAX6 mutations |
| Patient 4, this study | c.553_556dupTATA | p.S186IfsX2 | - | B | B | - | R | - | Y | Y | Y | Microcephaly, hypotonia, hypoplastic labia minora | Parents' DNA normal |
| Dateki et al. 2010 | c.562G>T | p.G188X | - | B | U | U | U | - | Y | Y | Y | - | Parental testing not reported |
| Dateki et al. 2010 | c.562G>T | p.G188X | - | B | U | U | U | U | - | - | Y | Seizures | Unrelated to above; Parental testing not reported |
| Diaczok et al. 2008 | c.674A>G | p.N225S | U | U | - | U | U | - | Y | U | U | Feeding difficulties | Parental testing not reported |
| Diaczok et al. 2008 | c.674A>G | p.N225S | U | U | - | U | U | - | Y | U | U | Seizures, respiratory distress | Parental testing not reported |
| Dateki et al. 2010 | 2.86 Mb deletion incl OTX2 | whole gene deletion | L | R | U | U | U | - | Y | Y | Y | - | Parents' DNA normal |
| Wyatt et al. 2008 | 3.07 Mb deletion incl. OTX2 | whole gene deletion | - | B | - | - | - | - | NS | - | - | - | Parents' DNA normal |
| Wyatt et al. 2008 | 1.28 Mb deletion incl. OTX2 | whole gene deletion | B | - | U | - | - | U | U | U | Y | Skull fractures, Feeding difficulties | Parents' DNA normal |
| Nolen et al. (9) | 9.66 Mb deletion incl. OTX2 | whole gene deletion | B | - | B | - | - | Y | Y | Y | Y | Microcephaly, hypotonia, MCA | Parents' DNA normal |
AN=anophthalmia, MI=microphthalmia, ONH/A=optic nerve hypoplasia/aplaisa, ASD=anterior segment dysgenesis, SS=short stature, DD/MR=developmental delay/mental retardation, B=bilateral, R=right eye only, L=left eye only, U=unknown/not reported, 1=one eye affected, Y=yes, - = feature not present, NS=normal structure (pituitary), MCA=multiple congenital anomalies
Other' eye anomalies include cataract, coloboma, retinal anomalies, nystagmus, retinal dystrophy, posterior vitreous opacities, persistent pupillary membrane,
Mutations were numbered based upon OTX2 cDNA sequence NM_172337.1 transcript variant 2, which is the canonical sequence according to UniProt (http://www.uniprot.org/), with +1 as the A of the ATG start site to allow direct comparison;
Two mutations reported by Ragge et al. 2005, p.P133T and p.P134A, were excluded from this table due to their later assertion in Wyatt et al. 2008 (8) that these mutations were unlikely to be pathogenic. Two mutations reported by Dateki et al. 2010, p.T178S and p.A245V, were also excluded due to the authors assertion that they were likely to be rare polymorphisms (15).
Figure 1.
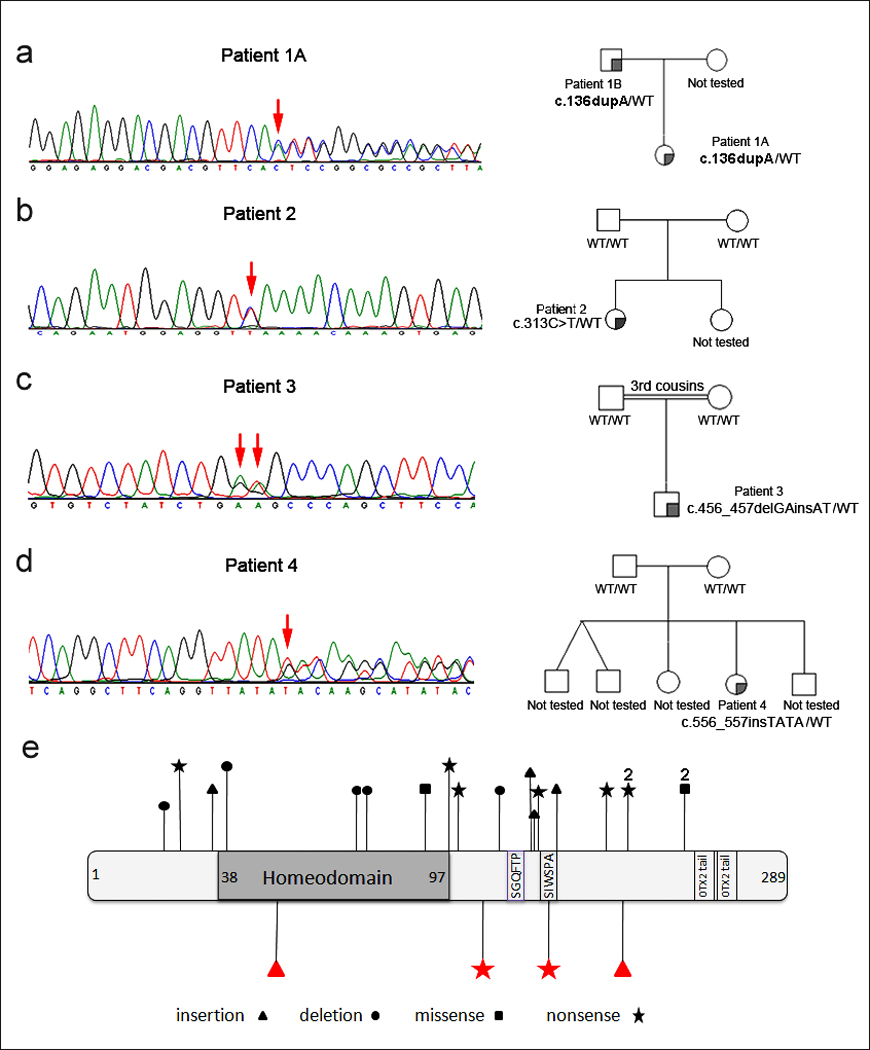
A–D. Pedigrees and chromatograms corresponding to affected OTX2 coding sequence for Patients 1 (A and B) through 4. Positions of mutations are indicated with red arrows. OTX2 genotypes corresponding to tested pedigrees members are shown. E. OTX2 diagram showing positions of mutations reported in this manuscript (bottom) as well as the previously reported changes (top); all mutations are unique except for p.N225S and p.G188X, each observed in two unrelated patients (indicated with 2).
Patient 1A is a 25-month-old Caucasian female born at full term with no further details available regarding birth history. She has bilateral microphthalmia and head CT at birth showed very severe optic nerve hypoplasia but normal midline structures and cerebral parenchyma. On physical exam she was noted to have a sacral dimple and an anteriorly placed anus. Her height was normal at the 10th centile, weight was <3rd centile (at the 50th centile for a 13-month-old), and head circumference was at the 3rd centile. Pituitary function studies were not performed. Development was normal with no formal assessment deemed necessary. The patient was found to have a heterozygous duplication mutation, c.136dupA (p.T46NfsX84), resulting in frameshift (Figure 1A). This mutation was not seen in Caucasian controls. Family history revealed that the patient’s father (Patient 1B) was affected with unilateral microphthalmia, cataract, optic nerve aplasia, and anterior segment dysgenesis (iris pseudocoloboma and posterior synechiae), lack of vascularization in the retinal periphery and absent papilla of the left eye. His right eye was affected with pallor of the papilla and vascular paucity in the retinal periphery only. His growth and development were normal; head imaging studies were not available. He was found to carry the same mutation seen in his daughter.
Patient 2 is a 5½-year-old Caucasian female born at full term with a birth weight of 3.6 kg (50th–75th centile), length of 50 cm (50th–75th centile), and head circumference of 36 cm (75th centile). She was diagnosed with bilateral clinical anophthalmia. Head CT in the neonatal period identified bilateral dysplastic globes, optic nerve aplasia, and otherwise apparently normal brain structures. The patient’s growth was normal, with a height of 110 cm (25th centile), weight of 18.5 kg (25th centile), and head circumference of 49 cm (10th centile). Early developmental milestones were within the normal range: she sat at 6 months, walked independently at 18 months, and spoke three words together at 18 months-2 years. However, at the age of 4 years old she was diagnosed with an autism spectrum disorder by a neurologist and developmental pediatrician due to sensory/tactile issues, echolalia, and lack of functional language. She has severe feeding difficulties, felt to be related to her sensory issues, consisting of choking on solid foods despite normal oral motor cavity and swallowing function. A diagnosis of latent Wolf-Parkinson-White syndrome was made by a pediatric cardiologist due to a history of cold sweats and racing pulse throughout her life. ECG at rest and echocardiogram were normal and there is no family history of similar cardiac events. She was found to have a heterozygous nonsense mutation in exon 5, c.313C>T (p.Q105X), creating a premature stop codon (Figure 1B). This mutation was not seen in Caucasian or Hispanic controls. Family history was unremarkable; clinical testing of buccal samples did not identify the mutation in either unaffected parent.
Patient 3 is a 20-month-old Caucasian male born at 41 weeks’ gestation with a birth weight of 4.1 kg (75th–90th centile), length of 51.5 cm (50th–75th centile), and head circumference of 36 cm (50th centile). He was diagnosed with right clinical anophthalmia and left microphthalmia. Cranial magnetic resonance imaging (MRI) in the neonatal period showed an extremely tiny globe to the right, left severe microphthalmia, bilateral optic nerve hypoplasia, and absent optic chiasms on the right, left severe microphthalmia, bilateral optic nerve hypoplasia, and absent optic chiasms. The patient was diagnosed with hypopituitarism at 9 months of age (treated with thyroxine, hydrocortisone, and growth hormone). Clinical Genetics evaluation at 20 months of age revealed feeding difficulties and failure to thrive, with a eight of 71.5 cm (<3rd centile, 50th centile for a 9-month-old), weight of 8.0 kg (<3rd centile, 50th centile for a 6-month old), and microcephaly with a head circumference of 43 cm (<3rd centile, 50th centile for a 5-month-old). He also had global developmental delay: he was able to sit unsupported and had <10 single words, but was not walking or putting two words together. He was found to have a heterozygous c.456_457delGAinsAT mutation which is predicted to create a premature stop codon (p.W152X) (Figure 1C). This mutation was not seen in Caucasian or Hispanic controls. The family history was unremarkable for similar features, but the parents are 3rd cousins; testing of buccal samples from both parents did not identify this mutation.
Patient 4 is a 4½-year-old Hispanic female born at 41-weeks gestation with a birth weight of 3.2 kg (25th– 50th centile), length of 48 cm (25th centile), and head circumference of 34 cm (25th– 50th centile). Ophthalmology evaluation identified left severe microphthalmia with minimal ocular remnants and right mild microphthalmia with maldevelopment of the retina, choroid, and optic nerve. Cranial MRI at 4 years of age noted bilateral microphthalmia, hypoplastic optic nerves, and a small optic chiasm, along with a small anterior pituitary and absent posterior pituitary glands. Clinical genetics evaluation at this time revealed hypoplastic labia minora, hypotonia, failure to thrive with a height of 91 cm (3rd centile, 50th centile for a 30-month-old) and weight of 13.1 kg (<3rd centile, 50th centile for a 31-month-old), and microcephaly with head circumference of 43 cm (<3rd centile, 50th centile for a 7-month old). She also had significant developmental delay: she sat alone at 8 months, is not yet crawling or walking, and has no meaningful words. The patient was found to have a heterozygous insertion mutation, c.556_557insTATA, resulting in frameshift (p.S186IfsX187) (Figure 1D). This mutation was not seen in Hispanic or Caucasian controls. Family history is non-contributory; clinical testing of buccal samples did not identify this mutation in either parent.
DISCUSSION
In this report, we present four additional families with mutations in OTX2 which increases the total number of families with OTX2 alterations to twenty-seven (Table 1). The frequency of OTX2 mutations in our sample (~8%) is somewhat higher than the previously reported rate (2–3%) (8), which may be due to the inclusion of ocular anomalies other than A/M, such as coloboma, within the initial cohorts. This higher frequency further underscores the importance of clinical testing for OTX2 mutations in A/M patients. OTX2 represents the second most common cause of anophthalmia/microphthalmia after SOX2, which explains up to 20% of this condition (23–24).
The main phenotype seen in OTX2-positive patients in our study is anophthalmia/microphthalmia associated with additional ocular and non-ocular defects such as optic nerve hypoplasia, brain malformations and pituitary abnormalities; short stature, developmental delay/mental retardation, and feeding difficulties were also observed. Previously unreported features include Wolf-Parkinson White syndrome in one patient and an anteriorly placed anus in another affected individual. These characteristics may represent coincidental findings; it is also possible that they may be low-penetrance features associated with OTX2 deficiency but support from additional mutation screens and/or expression studies is needed (OTX2 transcripts were reported in human gastrointestinal tract tissues (Genome Browser: http://genome.ucsc.edu)). All families in our study displayed inheritance consistent with a dominant pattern with three de novo alterations and one mutation inherited from an affected parent.
The OTX2 protein contains a paired type homeodomain (amino acids 38–97); two regions, SGQFTP (128–133) and SIWSPA (150–155), which are highly conserved within OTX family members; and tandemly repeated conserved OTX tail motifs (255–263 and 273–281) which were shown to be required for the transactivation activity of the protein (1,26). The four novel OTX2 mutations are predicted to result in truncation of normal OTX2 protein at 16% (p.T46NfsX42), 36% (p.Q105X), 53% (p.W152X) and 64% (p.S186IfsX2) of its sequence (Figure 1). Only the first mutation, p.T46NfsX42, is expected to be subject to nonsense medicated decay (27,28). The three remaining mutant proteins are predicted to lack the OTX tail domains; in addition, p.W152X is predicted to be short of SIWSPA motif and p.Q105X is expected to lack the SIWSPA and SGQFTP motifs.
Overall, the newly identified mutations are consistent with the previously reported disease-causing variants, the majority of which result in truncation or lack of protein product. To date, twenty-five different disease-causing variants have been reported with only two missense mutations. Of the remaining variants, eight were nonsense mutations, eleven resulted in frameshift and four were whole gene deletions. Only two recurrent mutations have been reported: a missense mutation, p.N225S, seen in two unrelated patients with combined pituitary hormone deficiency (12) and a nonsense mutation, p.G188X, seen in two unrelated patients with microphthalmia and pituitary abnormalities in one (15) (Table 1). In 35% of families tested, the proband inherited the mutation from a parent. Family 1 is the third case of an OTX2 mutation inherited from an affected non-mosaic parent; the majority (~2/3) of parents carrying OTX2 mutations to date have been unaffected due to mosaicism or incomplete penetrence (Table 1). The frequency of unaffected carriers indicates the importance of parental testing even in cases with apparently de novo phenotypes.
We examined the 37 reported cases with mutations in OTX2 (Tables 1 and 2) in order to determine if any specific traits can distinguish OTX2-positive anophthalmia/microphthalmia from A/M caused by other genetic factors. The spectrum of features associated with the major A/M gene, SOX2, has been described and includes genitourinary (micropenis, cryptorchidism, kidney defects), gastrointestinal (tracheoesophageal fistula, esophageal atresia, feeding disorder), endocrine (hypopituitarism, abnormal pituitary structure, hypogonadotropic hypogonadism), and brain defects (cavum/absent septum pellucidum, hypoplastic/agenesis corpus callosum, hippocampal abnormalities, hypothalamic hamartoma), developmental delay, especially gross motor, and anophthalmia/microphthalmia (22,24,29). Review of the anomalies linked with OTX2 mutations (Table 2) demonstrates overlap with features reported in SOX2 patients, especially ocular, brain, and endocrine abnormalities, while genitourinary and gastrointestinal structural anomalies seem to be more specific to SOX2 mutations (Tables 1 and 2). The observed similarities in ocular, brain and endocrine defects may be due to the involvement of OTX2 and SOX2 in related developmental processes; recent reports have demonstrated functional interactions between SOX2/Sox2 and OTX2/Otx2 in directing tissue development and regulation of downstream target genes (20,30).
Table 2.
Frequency of features described in patients with mutations in/deletions of OTX2. Only features present in two or more individuals were included.
| Feature | Percentage | Frequency/all cases |
|---|---|---|
| Anophthalmia/microphthalmia | 78% | 29/37 |
| Optic nerve hypoplasia | 35% | 13/37 |
| Anterior segment defects | 11% | 4/37 |
| Other eye defects | 32% | 12/37 |
| No eye defects | 8% | 3/37 |
| Abnormal pituitary structure/function | 30% | 11/37 |
| Other structural brain anomalies | 19% | 7/37 |
| Short stature | 32% | 12/37 |
| DD/MR | 46% | 17/37 |
| Microcephaly | 19% | 7/37 |
| Feeding difficulties | 14% | 5/37 |
| Seizures | 11% | 4/37 |
| Hypotonia | 5% | 2/37 |
Note: not all reported cases were assessed for each feature
The most common extra-ocular feature associated with mutations in OTX2 is abnormal pituitary structure and/or function. Out of 37 total reported OTX2-positive patients (from twenty-seven unrelated families), pituitary structure/function was reported as abnormal in eleven, pituitary structure was reported as normal in nine, and the remaining 17 patients had unknown status (head imaging/endocrine evaluation was not reported). While reported pituitary dysfunction was typically associated with short stature, three additional patients were noted to have short stature; specific endocrine evaluation was not reported for these patients, but two of the three were noted to have normal pituitary structure (Table 1). Given these data, there appears to be a strong association between OTX2 mutations and pituitary abnormalities with more than half of patients with reported pituitary assessment displaying functional/structural pituitary defects and/or short stature (38% of all patients). This is similar to the frequency seen in patients with SOX2 mutations (29% with pituitary abnormalities and an additional 10% with short stature) (24).
Interestingly, mutations in the second half of the OTX2 gene (from amino acid 135) appear more likely to be associated with pituitary dysfunction (or short stature) (p≈0.001 based on chi-squared analysis). Data on pituitary structure/function or stature are available for twelve of the thirteen patients with mutations affecting amino acids 135–225; eight have known pituitary abnormalities, two additional patients have short stature, and only two are reported to have normal stature. Data on pituitary structure/function or stature are available for twelve of the twenty patients with mutations earlier in the protein; only one has a confirmed pituitary dysfunction, one has short stature with normal pituitary structure, and ten have reported normal stature (Table 1). Based on location, all of the mutations in the second portion and at least half of the mutations in the first portion of the gene are not expected to be subject to nonsense mediated decay (NMD) (27,28) making this an unlikely explanation for the observed phenotypic differences. One feature that seems to better differentiate the groups is that mutant proteins associated with mutations in the second half of the gene retain the homeodomain and SGQFTP motif while earlier mutations disrupt one or both of these regions. It may be that retention of these two regions in the absence of other functional domains of OTX2 produces proteins with dominant-negative effects. These dominant-negative mutant proteins may further decrease the functional dosage of OTX2 through interference with wild-type protein activities leading to abnormal pituitary development. While the presence of pituitary anomalies in one patient with a whole gene deletion would argue against a dominant negative explanation, there are several other genes within the deleted region which are also important in pituitary development, suggesting the possibility of an alternate explanation for this patient’s pituitary anomalies (9,31). Several studies have investigated the activities of human OTX2 mutations (11–13, 15, 26) and shown decreased transactivation by the mutant proteins. In terms of dominant-negative effect, although one study identified a dominant-negative effect for the p.N225S mutation associated with pituitary anomalies in two patients (12), most reports describe a lack of dominant-negative effects for studied mutants, including the p.S136LfsX43, p.S135LfsX2 mutations that are associated with pituitary defects (11, 13, 15, 26). The analysis is further complicated by the fact that one mutation, p.G188X, has been found to be associated with a discordant pituitary phenotype in two unrelated patients (15). Identification and study of additional OTX2 mutations are needed to provide further support for or dispute the association of pituitary defects with mutations in the latter portion of the gene and to determine the explanation for this phenomenon.
Despite the wide overlap, differences between the A/M syndromic phenotypes associated with OTX2 versus SOX2 mutations can be seen. Based on the presented analysis, A/M patients with genitourinary and gastrointestinal defects in combination with pituitary anomalies are more likely to have SOX2 mutations while individuals lacking these associated features and displaying pituitary anomalies are more likely to harbor alterations in OTX2. Other associated features do not seem to provide significant aid in discriminating between OTX2- and SOX2- phenotypes. Ideally, screening of both genes should be performed in a stepwise manner, with the more likely candidate screened first, followed by screening of the second if no mutation is identified. Given the high frequency of inherited mutations in OTX2, careful parental examination and testing when a mutation in OTX2 is detected is critical for accurate counseling.
Acknowledgements
We would like to thank the families for their participation in our studies. Funding was provided by NIH grant EY015518 and Children’s Hospital of Wisconsin Foundation (EVS), Albert B. Millett Memorial Fund, A Mellon Mid-Atlantic Charitable Trust, Rae S. Uber Trust, A Mellon Mid-Atlantic Charitable Trust and Gustavus and Louis Pfeiffer Research Foundation (AS and TB).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Simeone A, Acampora D, Mallamaci A, Stornaiuolo A, D'Apice MR, Nigro V, Boncinelli E. A vertebrate gene related to orthodenticle contains a homeodomain of the bicoid class and demarcates anterior neuroectoderm in the gastrulating mouse embryo. EMBO J. 1993;12(7):2735–2747. doi: 10.1002/j.1460-2075.1993.tb05935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acampora D, Simeone A. The TINS Lecture. Understanding the roles of Otx1 and Otx2 in the control of brain morphogenesis. Trends Neurosci. 1999;22(3):116–122. doi: 10.1016/s0166-2236(98)01387-3. [DOI] [PubMed] [Google Scholar]
- 3.Simeone A, Puelles E, Acampora D. The Otx family. Curr Opin Genet Dev. 2002;12(4):409–415. doi: 10.1016/s0959-437x(02)00318-0. [DOI] [PubMed] [Google Scholar]
- 4.Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev. 1995;9(21):2646–2658. doi: 10.1101/gad.9.21.2646. [DOI] [PubMed] [Google Scholar]
- 5.Pannese M, Polo C, Andreazzoli M, Vignali R, Kablar B, Barsacchi G, Boncinelli E. The Xenopus homologue of Otx2 is a maternal homeobox gene that demarcates and specifies anterior body regions. Development. 1995;121(3):707–720. doi: 10.1242/dev.121.3.707. [DOI] [PubMed] [Google Scholar]
- 6.Andreazzoli M, Pannese M, Boncinelli E. Activating and repressing signals in head development: the role of Xotx1 and Xotx2. Development. 1997;124(9):1733–1743. doi: 10.1242/dev.124.9.1733. [DOI] [PubMed] [Google Scholar]
- 7.Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, Ruddle P, Hurst J, Collin JR, Salt A, Cooper ST, Thompson PJ, Sisodiya SM, Williamson KA, Fitzpatrick DR, van Heyningen V, Hanson IM. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. 2005;76(6):1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wyatt A, Bakrania P, Bunyan DJ, Osborne RJ, Crolla JA, Salt A, Ayuso C, Newbury-Ecob R, Abou-Rayyah Y, Collin JR, Robinson D, Ragge N. Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia, microphthalmia and coloboma. Hum Mutat. 2008;29(11):E278–E283. doi: 10.1002/humu.20869. [DOI] [PubMed] [Google Scholar]
- 9.Nolen LD, Amor D, Haywood A, St Heaps L, Willcock C, Mihelec M, Tam P, Billson F, Grigg J, Peters G, Jamieson RV. Deletion at 14q22–23 indicates a contiguous gene syndrome comprising anophthalmia, pituitary hypoplasia, and ear anomalies. Am J Med Genet A. 2006;140(16):1711–1718. doi: 10.1002/ajmg.a.31335. [DOI] [PubMed] [Google Scholar]
- 10.Henderson RA, Williamson K, Cumming S, Clarke MP, Lynch SA, Hanson IM, FitzPatrick DR, Sisodiya S, van Heyningen V. Inherited PAX6, NF1 and OTX2 mutations in a child with microphthalmia and aniridia. Eur J Hum Genet. 2007;15(8):898–901. doi: 10.1038/sj.ejhg.5201826. [DOI] [PubMed] [Google Scholar]
- 11.Dateki S, Fukami M, Sato N, Muroya K, Adachi M, Ogata T. OTX2 mutation in a patient with anophthalmia, short stature, and partial growth hormone deficiency: functional studies using the IRBP, HESX1, and POU1F1 promoters. J Clin Endocrinol Metab. 2008;93(10):3697–3702. doi: 10.1210/jc.2008-0720. [DOI] [PubMed] [Google Scholar]
- 12.Diaczok D, Romero C, Zunich J, Marshall I, Radovick S. A novel dominant negative mutation of OTX2 associated with combined pituitary hormone deficiency. J Clin Endocrinol Metab. 2008;93(11):4351–4359. doi: 10.1210/jc.2008-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K, Fujieda K. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocrinol Metab. 2009;94(1):314–319. doi: 10.1210/jc.2008-1219. [DOI] [PubMed] [Google Scholar]
- 14.Henderson RH, Williamson KA, Kennedy JS, Webster AR, Holder GE, Robson AG, FitzPatrick DR, van Heyningen V, Moore AT. A rare de novo nonsense mutation in OTX2 causes early onset retinal dystrophy and pituitary dysfunction. Mol Vis. 2009;15:2442–2447. [PMC free article] [PubMed] [Google Scholar]
- 15.Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, Muroya K, Adachi M, Tajima T, Motomura K, Kinoshita E, Moriuchi H, Sato N, Fukami M, Ogata T. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab. 2010;95(2):756–764. doi: 10.1210/jc.2009-1334. [DOI] [PubMed] [Google Scholar]
- 16.Acampora D, Gulisano M, Broccoli V, Simeone A. Otx genes in brain morphogenesis. Prog Neurobiol. 2001;64(1):69–95. doi: 10.1016/s0301-0082(00)00042-3. [DOI] [PubMed] [Google Scholar]
- 17.Kallen B, Robert E, Harris J. The descriptive epidemiology of anophthalmia and microphthalmia. Int J Epidemiol. 1996;25(5):1009–1016. doi: 10.1093/ije/25.5.1009. [DOI] [PubMed] [Google Scholar]
- 18.Forrester MB, Merz RD. Descriptive epidemiology of anophthalmia and microphthalmia, Hawaii, 1986–2001. Birth Defects Res A Clin Mol Teratol. 2006;76(3):187–192. doi: 10.1002/bdra.20237. [DOI] [PubMed] [Google Scholar]
- 19.Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007;2:47. doi: 10.1186/1750-1172-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hever AM, Williamson KA, van Heyningen V. Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin Genet. 2006;69(6):459–470. doi: 10.1111/j.1399-0004.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- 21.Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33(4):461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 22.Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JR, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, Fitzpatrick DR. SOX2 anophthalmia syndrome. Am J Med Genet A. 2005;135(1):1–7. doi: 10.1002/ajmg.a.30642. discussion 8. [DOI] [PubMed] [Google Scholar]
- 23.Bakrania P, Robinson DO, Bunyan DJ, Salt A, Martin A, Crolla JA, Wyatt A, Fielder A, Ainsworth J, Moore A, Read S, Uddin J, Laws D, Pascuel-Salcedo D, Ayuso C, Allen L, Collin JR, Ragge NK. SOX2 anophthalmia syndrome: 12 new cases demonstrating broader phenotype and high frequency of large gene deletions. Br J Ophthalmol. 2007;91(11):1471–1476. doi: 10.1136/bjo.2007.117929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider A, Bardakjian T, Reis LM, Tyler RC, Semina EV. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am J Med Genet A. 2009;149A(12):2706–2715. doi: 10.1002/ajmg.a.33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reis LM, Tyler RC, Abdul-Rahman O, Trapane P, Wallerstein R, Broome D, Hoffman J, Khan A, Paradiso C, Ron N, Bergner A, Semina EV. Mutation analysis of B3GALTL in Peters Plus syndrome. Am J Med Genet A. 2008;146A(20):2603–2610. doi: 10.1002/ajmg.a.32498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatelain G, Fossat N, Brun G, Lamonerie T. Molecular dissection reveals decreased activity and not dominant negative effect in human OTX2 mutants. J Mol Med. 2006;84(7):604–615. doi: 10.1007/s00109-006-0048-2. [DOI] [PubMed] [Google Scholar]
- 27.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36(8):801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 28.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14(10):1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 29.Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116(9):2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danno H, Michiue T, Hitachi K, Yukita A, Ishiura S, Asashima M. Molecular links among the causative genes for ocular malformation: Otx2 and Sox2 coregulate Rax expression. Proc Natl Acad Sci U S A. 2008;105(14):5408–5413. doi: 10.1073/pnas.0710954105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu X, Lin CR, Prefontaine GG, Tollkuhn J, Rosenfeld MG. Genetic control of pituitary development and hypopituitarism. Curr Opin Genet Dev. 2005;15(3):332–340. doi: 10.1016/j.gde.2005.04.011. [DOI] [PubMed] [Google Scholar]