

Abstract
In the autoimmune syndrome rheumatoid arthritis (RA), T cells and T-cell precursors have age-inappropriate shortening of telomeres and accumulate deoxyribonucleic acid (DNA) double strand breaks. Whether damaged DNA elicits DNA repair activity and how this affects T-cell function and survival is unknown. Here, we report that naïve and resting T cells from RA patients are susceptible to undergo apoptosis. In such T cells, unrepaired DNA stimulates a p53-ataxia telangiectasia mutated-independent pathway involving the non-homologous-end-joining protein DNA-protein kinase catalytic subunit (DNA-PKcs). Upregulation of DNA-PKcs transcription, protein expression and phosphorylation in RA T cells co-occurs with diminished expression of the Ku70/80 heterodimer, limiting DNA repair capacity. Inhibition of DNA-PKcs kinase activity or gene silencing of DNA-PKcs protects RA T cells from apoptosis. DNA-PKcs induces T-cell death by activating the JNK pathway and upregulating the apoptogenic BH3-only proteins Bim and Bmf. In essence, in RA, the DNA-PKcs-JNK-Bim/Bmf axis transmits genotoxic stress into shortened survival of naïve resting T cells, imposing chronic proliferative turnover of the immune system and premature immunosenescence. Therapeutic blockade of the DNA-PK-dependent cell-death machinery may rejuvenate the immune system in RA.
Keywords: apoptosis, DNA damage, DNA-PKcs, rheumatoid arthritis, T-cell
INTRODUCTION
In rheumatoid arthritis (RA), the process of T-cell ageing is accelerated (Goronzy & Weyand, 2005; Weyand et al, 2009). Telomeres of CD4 T cells are age-inappropriately shortened and clonal populations of CD28-deficient T cells accumulate (Koetz et al, 2000; Schonland et al, 2003). Premature senescence not only involves memory T cells engaged in chronic inflammatory lesions, but is most pronounced among naïve T cells unprimed by antigen. Two recent studies have identified novel mechanisms causing premature immunosenescence in RA (Fujii et al, 2009; Shao et al, 2009). Naïve RA T cells were found to be apoptosis susceptible, a defect mechanistically linked to insufficiency of the enzyme telomerase (Fujii et al, 2009). When undergoing priming, survival rates of RA T cells reached only half of those in control T cells and overexpression of hTERT, the protein component of telomerase, rescued RA T cells from death. Also, naïve RA T cells were characterized by the accumulation of deoxyribonucleic acid (DNA) double strand breaks (DSB), a defect caused by the inadequate production of the ataxia telangiectasia mutated (ATM) gene product, an enzyme critically involved in recognizing DNA DSB (Shao et al, 2009). Forced overexpression of ATM in RA T cells restored DNA repair and T-cell survival. Apoptotic susceptibility of naïve T cells determines the generation of sufficient antigen-specific clones as well as the cellular yield of homeostatic proliferation; a process that generates new T cells. Homeostatic T-cell proliferation is particularly relevant in individuals over the age of 50 years when thymic T-cell production has ceased. RA incidence rates are highest during the 7th and 8th decades of life when homeostatic proliferation is most needed for T-cell regeneration and maintenance (Doran et al, 2002; Goronzy et al, 2005).
How insufficiency of telomerase and ATM mediates apoptotic sensitivity of T cells is not understood. Proliferating naïve T cells are resistant to death ligand-death receptor-mediated apoptosis, but forced overexpression of Bcl-2 protects T cells from dying, pointing towards cell-internal signals as apoptosis initiators (Fujii et al, 2009). One of the most effective stress signals linked to apoptosis is damaged DNA, considered particularly important in senescent cells that have been chronically exposed to stressors, e.g. endogenously generated reactive oxygen species. Depending on the type of DNA damage, cells have several options to detect and repair broken DNA and similar pathways may be involved in surveillance of telomeric structures (Denchi & de Lange, 2007; Riha et al, 2006; Verdun & Karlseder, 2006). In mammalian cells, two major pathways repair DNA DSB: non-homologous end-joining (NHEJ) and homology-directed repair (HDR). Three of the DNA damage sensors involved in these pathways are PI3K-related kinases: ATM, the ataxia telangiectasia-related (ATR), and the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) (Falck et al, 2005). ATM orchestrates HDR by interacting with the DNA-damage sensing MRE11-Rad50-NBS1 (MRN) complex, phosphorylates multiple target molecules and activates cell cycle checkpoints to arrest the cell cycle and allow for fixing of the DNA (Lavin, 2007). One of the ATM targets is p53, which facilitates cell death if DNA repair fails (Lavin & Kozlov, 2007). The first step in NHEJ is the detection of fragmented DNA by Ku70/Ku80, which translocates inward from the DNA end to make the DNA termini accessible to DNA-PKcs (Lees-Miller & Meek, 2003). Cells deficient in DNA-PKcs are highly sensitive to radiation-induced cell death (Meek et al, 2008) and animals with DNA-PKcs deficiency typically have severe combined immunodeficiency (Meek et al, 2004). DNA-PKcs is not only involved in sensing/repairing DSB, but may be equally important as a repair protein in telomeric end-capping (Bailey et al, 1999; Williams et al, 2009). Proteins participating in the recognition and handling of double-stranded DNA ends, either at the telomeric end or in DSB, may be largely overlapping. RA T cells, impaired in inducing sufficient telomerase and deficient in utilizing the ATM-MRE11-p53 pathway should be a valuable model system to investigate the role of dysfunctional telomeres and unrepaired DNA in inducing DNA repair activity.
The current study was designed to identify cell-internal signals that render RA T cells apoptosis susceptible, with particular emphasis on the involvement of DNA repair mechanisms in making T-cell fate decisions. To exclude interfering mechanisms of T-cell death by external death signals, these studies were focused on naïve CD4 T cells, which are typically resistant to Fas-FasL-mediated apoptosis (Fujii et al, 2009; Krammer et al, 2007). Naïve CD4 T cells from RA patients have a spontaneous apoptosis rate twice as high as in age-matched control individuals. Resting naïve T cells progressively accumulate oxidative DNA damage and undergo apoptosis, a process accelerated in RA T cells. Apoptotic loss of resting T cells is independent from activation of the ATM-p53 pathway. Instead, RA T cells upregulate DNA-PKcs. Inhibition of DNA-PKcs activity or knockdown of the DNA-PKcs gene rescues RA T cells from death. Protection from excessive apoptosis is also achieved by inhibiting the JNK signalling pathway. Among the BH3-only proapoptotic Bcl-2 family members, Bmf and Bim transcripts and protein are selectively upregulated in RA T cells, suggesting that Bmf and Bim may be crucial in sensing and relaying stress signals. Targeting the DNA-PKcs-JNK-Bim axis may provide a novel therapeutic intervention to restore T-cell homeostasis in RA.
MATERIALS AND METHODS
Patients
The study group included 86 RA patients and 76 control subjects. RA patients fulfilled the ACR criteria, and all were positive for rheumatoid factor. Control subjects were matched for age and ethnicity; had no personal or family history of autoimmune disease. A history of cancer or chronic viral infection was considered an exclusion criterion. Demographic characteristics of RA patients and controls are summarized in Tables 1 and 2. Patients with systemic lupus erythematosus (SLE) were selected for having active disease. Their demographic characteristics are given in Supplement Table 1. The study was approved by the Institutional Review Board, and all subjects gave appropriate informed consent.
Table 1.
Demographic characteristics of study populations
| Characteristics | Controls | RA | p-Value |
|---|---|---|---|
| Number of subjects | 76 | 86 | |
| Female/malea | 62/14 | 68/18 | 0.61 |
| Age (mean ± SD years)a | 46.8 ± 10.4 | 49.5 ± 13.6 | 0.17 |
| Ethnicitya | 0.18 | ||
| African-American | 46 | 53 | |
| White | 23 | 18 | |
| Hispanic | 7 | 15 |
No significant difference.
Table 2.
Clinical characteristics of RA patients
| Characteristics | Values |
|---|---|
| Disease duration (mean ± SD years) | 7.4 ± 7.5 |
| Active diseasea | 66.2% |
| Tobacco use | 18.6% |
| Extraarticular manifestations | 36.1% |
| ESR, mm/hour | 37.6 |
| DMARD naïve | 8.2% |
| Medications | |
| Corticosteroids | 67.5% |
| Methotrexate | 67.9% |
| Hydroxychloroquine | 46.5% |
| Leflunomide | 8.2% |
| TNF inhibitors | 16.3% |
ESR, erythrocyte sedimentation rate; DMARD, disease-modifying anti-rheumatic drugs; DD, disease duration.
Active disease defined by FDA criteria [presence of three or more of the following: morning stiffness (>45 min), swollen joints (>3 min), tender joints (>6 min) and sedimentation rate (>20 mm)].
Cell purification and cell culture
Peripheral blood mononuclear cells (PBMC) were separated from whole blood with Lymphocyte Separation Medium (Mediatech Inc., Herndon, VA). CD45RO− cells were negatively selected with microbeads (Miltenyi Biotec Inc., Auburn, CA), and the naïve CD4+CD45RO− population was subsequently selected with CD4 microbeads (autoMACS, Miltenyi Biotec Inc.).
CD4+CD45RO− T cells were maintained in a resting state in DMEM without mitogenic stimulation for 3 days. In selected experiments, T-cell homeostatic cytokines were supplemented. To test for the role of signalling networks in mediating apoptosis, the following inhibitors were added to the cultures at day 0: DNA-PKcs inhibitor IC86621 (Sigma–Aldrich, St. Louis, MO), JNK inhibitor II (Calbiochem, La Jolla, CA), p38 inhibitor (Alexis Biochemicals, Plymouth Meeting, PA, SB203580), CaM inhibitor (Calbiochem), NF-κB inhibitor (Calbiochem) and AKT inhibitor (Calbiochem).
To induce DNA damage, naïve CD4 T cells were incubated in DMEM containing 0, 56, 112, 224 µM H2O2 for 1 h. After a brief rinse in medium, cells were collected for DNA damage analysis or kept for an additional 3 h before DNA-PKcs transcripts were quantified.
Comet assay
Deoxyribonucleic acid damage was quantified with CometAssay™ kits (R&D Systems, Minneapolis, MN), modified as previously described (Shao et al, 2009). Cells (1 × 105/ml) mixed with molten LMAgarose (1:10 at 37°C) were immediately transferred onto a CometSlide, kept in the dark for 10 min in a flat position, immersed in cold lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris base, 1% sodium lauryl sarcosinate, 1% Triton-100, 10% DMSO) for 60 min and treated with freshly prepared alkaline solution (300 mM NaOH, 1 mM EDTA, pH > 13) for 20 min at room temperature. After washing twice in 1× TBE buffer for 5 min, slides were transferred to a horizontal electrophoresis chamber and aligned at equidistance from the electrodes. One volt/cm was applied for 10 min. Stained slides (20 µg/ml ethidium bromide) were analyzed by fluorescence microscopy. A minimum of 50 cells were evaluated in each sample using the CometScore™ software (TriTek Corp., Morrisville, VA).
Deoxyribonucleic acid damage was quantified by the tail moment (TM) calculated as percentage of DNA in the tail × tail length (Hellman et al, 1995).
Flow cytometry
To quantify T-cell apoptosis, CD4+CD45RO− cells were washed and suspended in 500 µl fluorescence-activated cell sorting (FACS) buffer supplemented with EasyComp Blank Particles (Spherotech, Lake Forest, IL). Uptake of propidium iodide (PI) (Sigma–Aldrich) was determined by flow cytometry. Apoptotic cells were detected with PE-Annexin V (BD Biosciences, San Jose, CA) and 7-AAD (BD Biosciences).
For intracellular staining, cells were fixed directly with 1.5% formaldehyde for 10 min at room temperature and resuspended in 90% methanol. After overnight incubation, 107 cells/ml were stained with fluorescein isothiocyanate (FITC)-pATM (Rockland Immunochemicals, Gilbertsville, PA), Alexa Fluor 488-pp53, FITC-Bcl-2, FITC-pJNK, Alexa Fluor 488-Bim, PE-Bmf (Cell Signalling Technology, Danvers, MA), Alexa Fluro 488-Ku70 (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies, or FITC-conjugated 8-oxoguanine probe (OxyDNA Assay Kit; Calbiochem). Proteins or 8-oxoguanine expression were detected by an LSRII flow cytometer (BD Biosciences). Data were analyzed by FlowJo software (Tree Star Inc., Ashland, OR).
DNA isolation and quantitative PCR
Total ribonucleic acid (RNA) was extracted from 1.0 × 105 cells, and complementary DNA (cDNA) was synthesized with AMV-reverse transcriptase and random hexamer primers. Polymerase chain reaction (PCR) was conducted as previously described. Primer sequences are shown in Table 3.
Table 3.
Primer sequences
| 18S ribosomal RNA | 5′-AGGAATTCCCAGTAAGTGCG-3′ |
| 5′-GCCTCACTAAACCATCCAA-3′ | |
| DNA-PKcs | 5′-CTTTGTCGTGTGGAGGGAA-3′ |
| 5′-CACAACGGGGTTCAGAAGTT-3′ | |
| Puma | 5′-GGACGACCTCAACGCACAGTA-3′ |
| 5′-GGCAGGAGTCCCATGATGAGA-3′ | |
| Noxa | 5′-CCT GGG AAG AAG GCG CG-3′ |
| 5′-TCA GGT TCC TGT GCA GAA G-3′ | |
| Bim | 5′-ACG CTT ACT ATG CAA GGA GGG-3′ |
| 5′-GGT CTT CGG CTG CTT GGT AAT-3′ | |
| Bmf | 5′-CTGCACAACCTTGCTTTGAA-3′ |
| 5′-CAAGGGCCTGACAGAGAAAG-3′ |
Quantitative reverse transcription PCRs were completed in triplicate following previously described protocols (Niessner et al, 2006). Expression levels were determined by interpolation with a standard curve. cDNA copies were adjusted for 1 × 108 ribosomal RNA copies.
Western blot
Whole cell lysates were prepared in RIPA buffer (Cell Signalling Technology) plus phenylmethylsulphonyl fluoride (PMSF) and a protease inhibitor cocktail (Sigma). For each sample, equal amounts of total protein were electrophoresed, transferred to a nitrocellulose membrane (Amersham, Piscataway, NJ) and blocked with 5% non-fat milk. Antibodies specific for DNA-PKcs, pDNA-PKcs, JNK (1:1000, Santa Cruz) and pJNK (Cell Signalling Technology) were added overnight at 4°C, followed by washing with washing buffer. Membranes were subsequently incubated with secondary antibodies (1:6000; Santa Cruz) for 1 h at room temperature and developed with a chemiluminescent detection system (GE Healthcare, Buckinghamshire, UK). To ensure equal loading, membranes were stripped and reprobed for β-actin using goat anti-human actin antibodies (1:4000; Santa Cruz).
siRNA transfection
DNA-dependent protein kinase siRNA (sc-35200) was purchased from Santa Cruz, CA. Six micrograms of siRNA oligonucleotides were transfected into resting CD4 T cells using the Amaxa Nucleofector® system and Human T-cell Nucleofector® kits (Amaxa, Fremont, CA) as previously described (Shao et al, 2009). AllStars Negative Control siRNA (Qiagen) served as a control. Twenty-four hours after transfection, knockdown efficiencies were monitored by qPCR and Western blotting.
Detection of 53BP1 foci
CD4+CD45RO− T cells were maintained in DMEM without mitogenic stimulation for 72 h. Immunofluorescence staining was performed as previously published (Song et al, 2007). T cells were incubated in cytoskeleton buffer (10 mM PIPES at pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100) for 5 min on ice, followed by incubation in cytoskeleton stripping buffer (10 mM Tris–HCl at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1% Tween 40, 0.5% sodium deoxycholate) for 5 min on ice and then cells were fixed with fixation buffer (3.7% formaldehyde in 1× PBS). The fixed cells were blocked with 1% bovine serum albumin (BSA) in PBS for 30 min at 37°C and incubated with a rabbit polyclonal antibody against 53BP1 for 1 h at room temperature. The cells were washed three times in PBS, incubated with secondary antibodies for 1 h at room temperature, washed and stained with DAPI. Images were acquired with a confocal laser-scanning inverted microscope (LSM 510 META Axiovert 200; Carl Zeiss, Inc.).
Statistical analysis
Groups were compared using parametric tests for independent or paired samples as appropriate. Results are expressed as the mean ± SEM. p Values of less than 0.05 were considered significant.
RESULTS
In rheumatoid arthritis, resting CD4 T cells are prone to apoptosis
In vivo the vast majority of CD4 T cells are in a resting state and, accordingly, are not undergoing apoptosis. In normal healthy donors, only 2.6% of freshly isolated naïve CD4 T cells expressed Annexin V. In RA patients, the rate of spontaneous apoptosis was significantly higher (3.53% Annexin V+ cells, p = 0.05) (Fig 1A and B). When removed from their natural resources and kept ex vivo, human T cells progressively die. By 72 h, 15% of T cells from control donors displayed apoptotic features (Fig 1A and B). The susceptibility to undergo apoptosis was significantly higher in RA T cells with death rates of 25–30% (p = 0.01).
Figure 1. Apoptotic susceptibility and DNA damage in naïve CD4 T cells from RA patients.
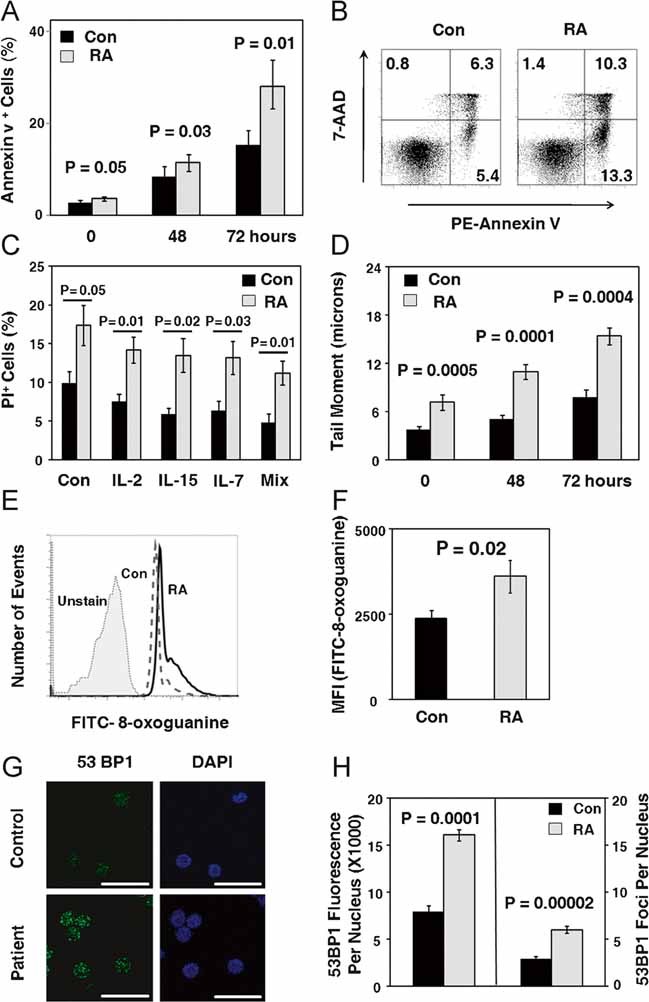
CD4+CD45RO− T cells were isolated from the peripheral blood of RA patients and controls. Cells were maintained in culture without mitogenic stimulation for 72 h.
- T cells undergoing apoptosis were identified by Annexin V and 7-AAD staining in patient-derived and control samples. Data are given as mean ± SEM from six patients and six controls.
- Representative data from one patient and one control collected after 72 h are shown.
- Cells were maintained in culture with IL-2 (20 U/ml), IL-7 (20 ng/ml), IL-15 (10 ng/ml) or a mixture of the three (Mix) for 72 h. T cells undergoing apoptosis were identified by PI staining. Data are given as mean ± SEM from seven patients and five controls.
- DNA breaks were quantified by comet assay after 72 h. Data are given as mean ± SEM of seven patients and six controls with a minimum of 90 individual cells analyzed in each sample.
- Flow cytometry analysis of 8-oxoguanine levels in control (dashed line) and RA T cells (solid line). Unstained control is shown as light grey.
- The level of 8-oxoguanine in five RA and five control samples is given as mean fluorescence intensity (MFI) of FITC-8-oxoguanine ± SEM.
- 53BP1 foci were determined by immunofluorescence staining and confocal laser microscopy after 72 h. Bar, 20 µM.
- The levels of 53BP1 foci in three RA and three control samples is given as total 53BP1 intensity per nucleus ± SEM and 53BP1 foci per nucleus ± SEM.
T-cell survival may depend on the availability of growth-promoting cytokines (interleukin-2, IL-2; interleukin-7, IL-7; interleukin-15, IL-15) (Ma et al, 2006; Surh & Sprent, 2008). Therefore, T cells were supplemented with IL-2, IL-7, IL-15 or a mixture of all three cytokines. Optimal doses were determined in pilot experiments (data not shown). IL-2, IL-7 and IL-15 reduced apoptosis rates with about equal potency, but prevented only one-third of the T-cell attrition (Fig 1C). Anti-apoptotic effects of the cytokines were similar in control and RA T cells and could not abolish the difference in apoptosis between patients and controls.
To identify death-inducing signals, different from cytokine withdrawal, the load of damaged DNA was determined. In the absence of mitogenic or antigenic stimulation, levels of oxidatively damaged DNA detected as 8-oxoguanine bases by flow cytometry were low in almost all control T cells (Fig 1D). RA T cells contained significantly higher levels of 8-oxoguanine DNA lesions, often displaying a biphasic flow cytometry pattern indicative of a cell subpopulation with markedly elevated signals for 8-oxoguanine sites. In naïve CD4 T cells from RA patients, fluorescence intensities marking oxidized DNA were 1.5-fold higher than in controls (Fig 1E, p = 0.02).
To search for DNA DSB, comet assays were employed to examine purified CD4+CD45RO− T-cell populations immediately after isolation and 48+ and 72 h later (Fig 1F). TMs were low in fresh T cells, but continuously increased over the 72 h observation period. The load of DNA breaks was almost twice as high in RA T cells (p < 0.001, p = 0.0001, p < 0.001), with a steeper slope of accumulation over the 3-day culture.
Accumulation of DNA DSB was confirmed by immunostaining for 53BP1 foci in the nuclei of RA and control T cells. Quantification of immunofluorescence staining showed significant higher anti-53BP1 binding in the nucleus of RA T cells (Fig 1G) and a higher number of 53BP1 foci per nucleus.
In essence, spontaneous apoptosis in naïve CD4 T cells was closely correlated with the accrual of damaged DNA.
RA T cells die independently from the ATM-p53 pathway
T cells with fragmented DNA are culled from the pool of DNA damage sensing and repair mechanisms fail to restore genomic intactness. The most lethal DNA lesions are DSB which upon recognition by the DNA repair machinery elicit cell cycle arrest to allow for repair. One of the major downstream targets of ATM is p53, which facilitates cell death in case repair is insufficient. Given the increased prevalence of DSB and oxidized DNA lesions in RA T cells, we analyzed the expression of pATM and pp53 in paired samples of control and RA naïve CD4 T cells (Fig 2). Among control T cells, essentially all cells with activated caspase-3 expressed pATM, suggesting that the ATM-p53 pathway controls apoptosis of most healthy CD4 T cells. In contrast, patient-derived CD4 T cells undergoing apoptosis and expressing activated caspase-3 lacked pATM expression. Decreased expression of pATM and pp53 was confirmed in a comparative analysis of control and patient-derived samples demonstrating decreased expression of both pATM and pp53 in the RA T cells (Fig 2B, p = 0.01 and Fig 2C, p = 0.05).
Figure 2. Apoptosis in RA CD4 T cells is ATM and p53 independent.
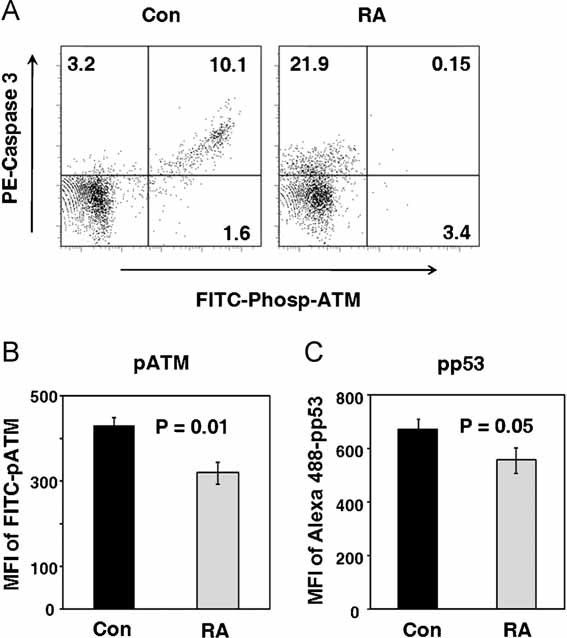
CD4+CD45RO− T cells were isolated from six controls and six RA patients. Cells were maintained in culture without stimulation and collected after 72 h.
- T cells undergoing apoptosis were stained with PE-caspase-3 and phosphorylated ATM was detected by FITC-Phosp-ATM. Representative cytometric data from one patient and one control are shown.
- Expression levels of pATM were quantified by flow cytometry in five RA and five control samples and are given as mean MFI of FITC-pATM ± SEM.
- Expression levels of pp53 were quantified by flow cytometry in six RA and six control samples and are given as mean MFI of Alexa Fluor 488-pp53 ± SEM.
These data strongly suggested an ATM-p53-independent pathway mediating T-cell apoptosis in RA.
Spontaneous upregulation of the DNA damage sensing enzyme DNA-PKcs
Besides ATM and ATR, the PI3K-related enzyme DNA-PKcs is an important player amongst the DNA damage sensors. To implicate this enzyme in regulating the fate of resting naïve T cells, DNA-PKcs transcript and protein levels were measured in naïve CD4 T cells from healthy control donors and RA patients (Fig 3). Expression of DNA-PKcs specific sequences in T-cell extracts were increased 1.4-fold in the freshly isolated RA samples (Fig 3A, p = 0.04). Spontaneous upregulation was a disease-specific finding. CD4 T cells isolated from patients with active SLE had significant lower transcript levels for DNA-PKcs than controls (Supplement Figure 1, p = 0.04). Also, RA T cells contained significantly higher concentrations of DNA-PKcs protein (Fig 3A and B). Western blotting confirmed that RA T cells contained higher amounts of phosphorylated DNA-PKcs (Fig 3B and C). The kinase is only one of the core elements of the NHEJ repair complex and needs to partner with Ku70/80, a heterodimer critically involved in detecting DSB. Quantification of Ku70 and Ku80 protein revealed a statistically significant reduction in RA T cells compared to control T cells to about half of the levels (Fig 3C; p = 0.05 for Ku70 and p = 0.02 for Ku80).
Figure 3. RA CD4 T cells express increased levels of DNA-PKcs but decreased levels of Ku70 and Ku80.
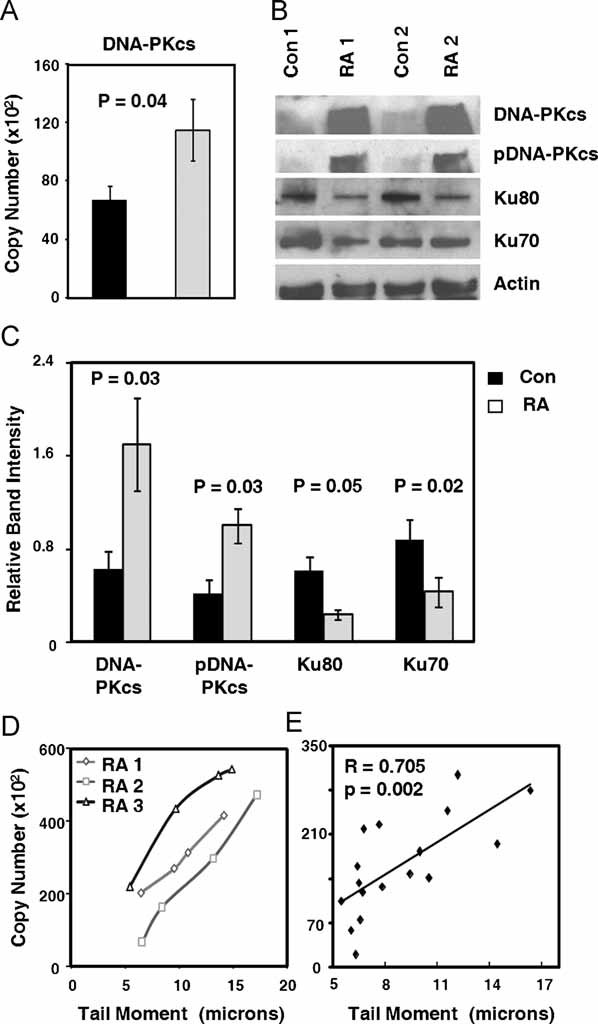
CD4+CD45RO− T cells were purified from RA patients and age-matched controls. Cells were analyzed immediately after isolation and after 72 h of culture without stimulation.
- DNA-PKcs transcript levels were measured by RT-PCR in control (black bar) and RA T cells (light grey bar). Results are presented as mean ± SEM from 15 controls and 16 RA patients.
- Quantification of DNA-PKcs, pDNA-PKcs, Ku70 and Ku80 protein levels by Western blotting in cells harvested after 72 h.
- Relative expression levels of the four proteins were quantified by measuring band intensities adjusted by β-actin. The results are shown as mean ± SEM for six RA patients and six control donors.
- Correlation of DNA-PKcs expression with DNA damage. CD4+CD45RO− T cells from three RA donors were treated with the indicated doses of H2O2 for 1 h, and DNA damage was assayed by comet assay. DNA-PKcs mRNA expression was quantified by qPCR.
- Correlation of DNA-PKcs expression with DNA damage. Transcript levels of DNA-PKcs from 16 RA samples were quantified by qPCR and standardized by 18S ribosomal RNA; DNA damage was assayed by comet assay.
To link increased DNA-PKcs expression with the cellular burden of damaged DNA, TMs and DNA-PKcs transcript concentrations were compared in individual T-cell samples (Fig 3D). The higher the T cells’ TMs, the more intense the induction of DNA-PKcs transcription. In an alternative approach, T cells were exposed to exogenous genotoxic stress and induction of DNA-PKcs was monitored. TMs tripled as the T cells were exposed to increasing concentrations of H2O2. Transcript levels of DNA-PKcs increased linearly, suggesting swift reactivity of this enzyme system to DNA fragmentation in T cells (Fig 3E).
In essence, naïve RA T cells are characterized by a dysbalance of the core elements of the NHEJ complex, with overexpression and activation of DNA-PKcs and underrepresentation of Ku70 and Ku80.
Inhibition of DNA-PKcs activity and silencing of DNA-PKcs restores apoptotic resistance in RA T cells
To implicate DNA-PKcs directly in rendering RA T cells apoptosis susceptible, control and RA T cells were treated with the DNA-PKcs inhibitor IC86621, and their apoptotic response pattern was quantified by flow cytometry measurement of PI-positive T cells (Fig 4A and B). The DNA-PKcs inhibitor essentially did not affect survival in control T cells. After 72 h of ex vivo culture 12–14% of the control T cells were apoptotic, independent from treatment with the DNA-PKcs inhibitor. In contrast, inhibition of the enzymatic activity of DNA-PKcs improved survival rates among RA T cells. Doses of 100 nM were sufficient to normalize apoptotic susceptibility and reduced the proportion of PI-positive cells to levels seen in the control T cells. The protection of RA T cells from apoptosis was dose-dependent (Fig 4B), suggesting that DNA-PKcs-dependent signalling pathways are dominant in regulating life-death decisions in these T cells. DNA-PKcs appears to be less relevant in normal T cells as DNA-PKcs inhibition had essentially no effect.
Figure 4. Inhibition of DNA-PKcs activity protects RA T cells from apoptosis.
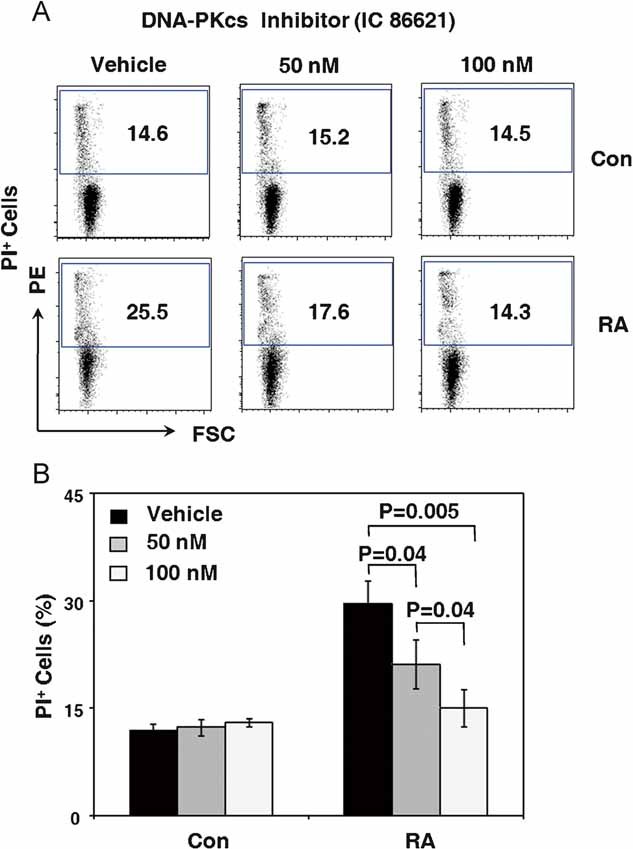
CD4+CD45RO− T cells were isolated from six control donors and seven RA patients. Cells were maintained in culture without stimulation for 48 h and then were treated with the DNA-PKcs inhibitor IC86621 (50 nM, 100 nM) or vehicle for 24 h.
- T-cell apoptosis was measured by flow cytometry staining for PI. Representative cytometric data from one patient and one control are shown.
- Frequencies of PI-positive T cells in the absence and presence of the DNA-PKcs inhibitor from six controls and seven RA patients are presented as mean ± SEM.
In an alternative approach, the function of DNA-PKcs was inhibited by siRNA interference. By transfecting RA T cells with DNA-PKcs specific-interfering sequences, transcript levels for the gene were reduced by about 70% (Fig 5A). Western blotting confirmed that RNA interference was able to depress protein levels markedly (Fig 5B). Reduction of DNA-PKcs levels had immediate consequences for the propensity of naïve CD4 T cells to undergo apoptosis (Fig 5C and D). The rates of PI-positive T cells diminished from 15.76% in populations with intact DNA-PKcs to 9.27% in T cells with silenced DNA-PKcs (Fig 5D, p = 0.03).
Figure 5. Silencing of the DNA-PKcs gene reduces apoptosis in RA T cells.
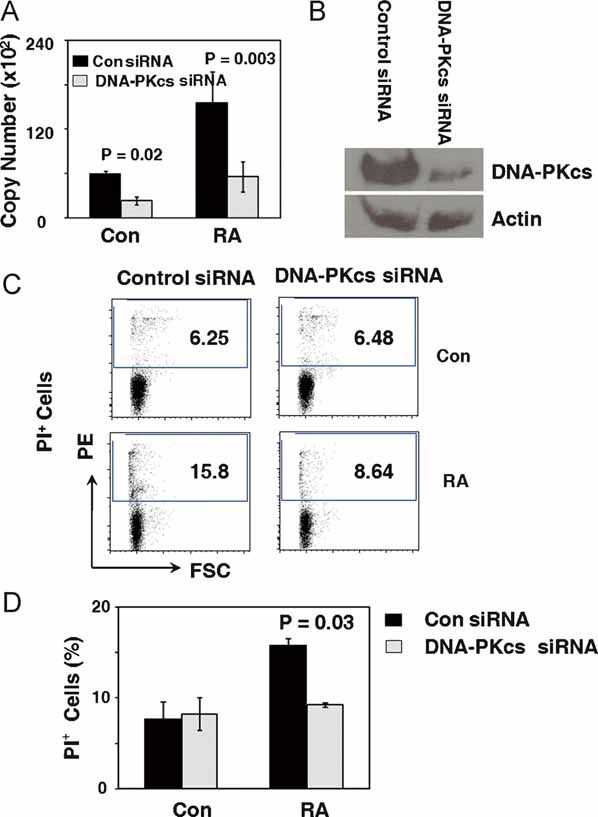
CD4+CD45RO− T cells were purified from three control donors and three RA patients and transfected with control or DNA-PKcs specific siRNA oligonucleotides by nucleofection.
A,B. Twenty-four hours after transfection, DNA-PKcs transcript levels were quantified by qPCR, and DNA-PKcs protein levels were detected by Western blotting.
C. T-cell apoptosis was analyzed by staining for PI. Representative data from one patient and one control are shown.
D. Cell apoptosis from three control and three RA samples are presented as mean ± SEM.
These experiments directly involved the DNA-damage sensing enzyme DNA-PKcs in setting the apoptotic threshold in RA T cells.
Inhibition of the JNK pathway protects RA T cells from cell death
Since levels of phosphorylated p53 are decreased in RA T cells (see Fig 2), it was important to understand how DNA-PKcs activates the cell death machinery. A screening approach with a panel of signal pathway inhibitors was applied to pinpoint which of these pathways has a role in transmitting death signals (Fig 6A). As expected, inhibiting survival signals transmitted by the NF-κB and the AKT pathway increased T-cell death rates. Only one of the inhibitors, blocking the functional activity of JNK was able to protect RA T cells from dying. Dose–response experiments demonstrated that inhibiting JNK activity could normalize the death rate and that 10 µM JNK inhibitor II were more effective than 5 µM (Fig 6B and C).
Figure 6. Spontaneous apoptosis of RA CD4+ T cells is mediated through the JNK pathway and JNK is upregulated by DNA-PKcs.
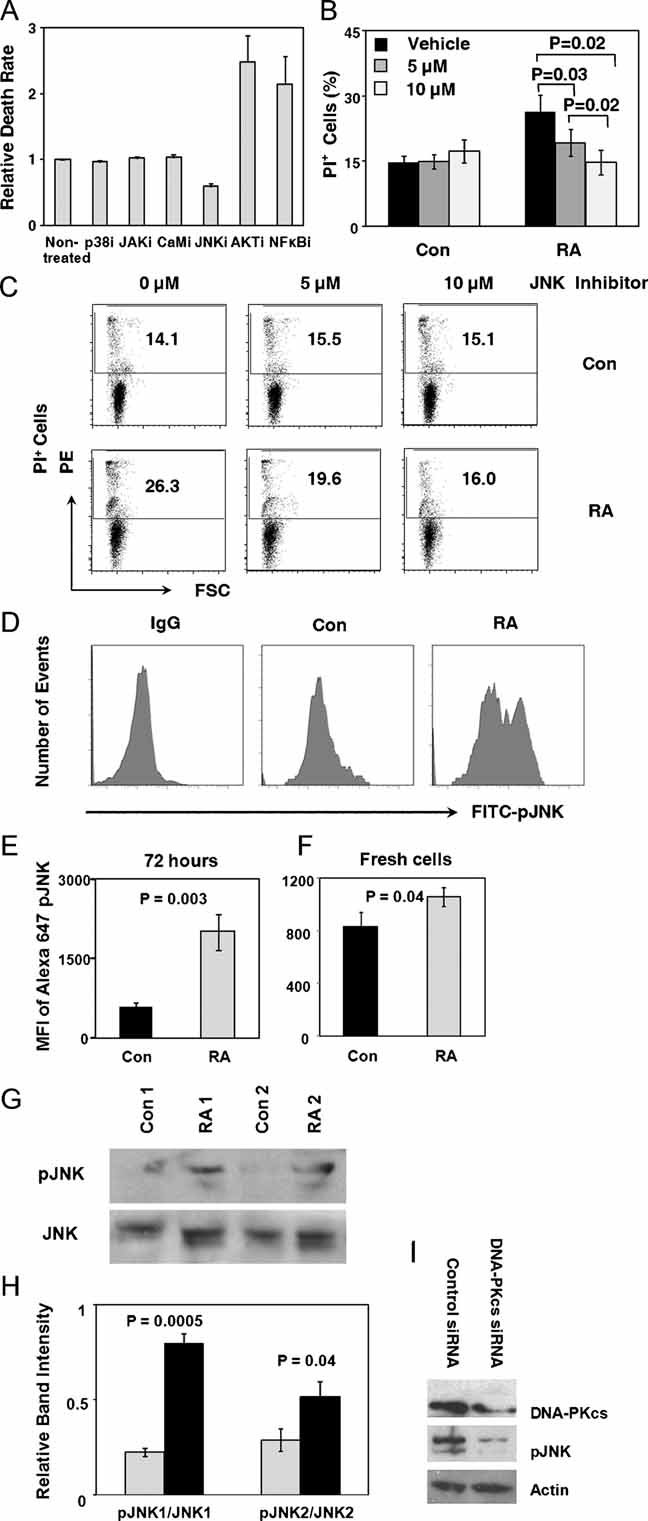
- CD4+CD45RO− T cells were isolated and cultured in the presence of the indicated enzyme inhibitors for 24 h. T-cell apoptosis was quantified through flow cytometry staining for PI.
- Cells were maintained in culture without stimulation for 48 h and then were treated with the JNK inhibitor. PI-positive T cells were measured after 24 h of treatment with the JNK inhibitor II (5 or 10 µM) or vehicle. Frequencies of apoptotic T cells from six controls and seven RA patients are presented as mean ± SEM.
- Representative flow cytometry results from one control and one patient.
- Flow cytometry analysis of phosphorylated JNK levels in control and RA T cells after culturing without mitogenic stimulation for 72 h. Representative data from one patient and one control are presented.
- Expression of pJNK protein in n = 6 RA and n = 6 control samples at 72 h is given as MFI of FITC-pJNK.
- Flow cytometry analysis of phosphorylated JNK levels in freshly isolated control and RA naïve CD4 T cells. Expression of pJNK protein in n = 7 RA and n = 7 control samples at day 0 is given as MFI of FITC-pJNK.
- Quantification of JNK and pJNK protein expression at 72 h by Western blotting.
- Relative expression levels of pJNK were quantified by measuring band intensities adjusted by total JNK in six RA patients and six control donors. Data are presented as mean ± SEM.
- CD4+CD45RO− T cells were purified from RA donors and transfected with control or DNA-PKcs-specific siRNA oligonucleotides by nucleofection. Twenty-four hours after transfection, DNA-PKcs and pJNK protein levels were detected by Western blotting. A representative blot from three independent experiments is shown.
To confirm overactivation of the JNK signalling pathway, naïve CD4 T cells from RA patients were isolated and immediately following isolation analyzed for the spontaneous expression of pJNK. Cytometric analysis confirmed higher expression of pJNK in freshly isolated CD4 T cells from RA patients compared to control T cells. Sustained and spontaneous signalling activity of the JNK pathway was confirmed after 72 h of maintaining T cells in culture, a stressor that upregulates T-cell death rate significantly (Fig 6D and E). FACS analysis demonstrated strong expression of pJNK in a subset of T cells, forming a shoulder in the histograms (Fig 6D). Mean fluorescence intensity (MFI) of pJNK was more than doubled in the patient-derived T cells (Fig 6E). Western blotting of cell protein extracts confirmed that pJNK was expressed at significantly higher concentrations in RA T cells (Fig 6G and H). In patient-derived T cells, the band intensities for both pJNK1 and pJNK2 were consistently higher (p = 0.0005 and 0.04), supporting the concept that the JNK signalling pathway is spontaneously activated in RA patients.
To establish a link between the induction of the DNA damage sensing enzyme DNA-PKcs and the increased baseline activation of JNK signalling, DNA-PKcs was knocked down by siRNA interference in RA T cells. Forced reduction in DNA-PKcs (Fig 6I) was accompanied by a marked loss in JNK phosphorylation.
Activation of the DNA-PKcs-JNK pathway in RA T cells induces the proapoptotic BH3-only proteins Bim and Bmf
Spontaneous upregulation of the DNA-PKcs-JNK pathway in RA T cells indicated continuous DNA repair activity that activates cell-internal stress kinases and jeopardizes T-cell survival. Stress-induced apoptosis mostly involves triggering of the intrinsic cell death machinery, employing BH3-only members of the Bcl-2 protein family as essential initiators of apoptotic cell death. BH3-only proteins, including Bim, Bmf, Noxa, Puma, Bad and Bid are thought to unleash Bax/Bak-like proteins from their sequestration by prosurvival Bcl-2 family members to eventually facilitate the release of apoptogenic molecules, such as cytochrome c and downstream activation of caspases.
To understand how activation of DNA-PKcs and JNK renders resting RA T cells susceptible to apoptosis, production of Puma, Noxa, Bim and Bmf was assessed (Fig 7A). In freshly isolated T cells from RA patients and controls, transcript levels of Puma and Noxa were indistinguishable, but levels of Bim and Bmf specific sequences were almost doubled in the patient-derived cells. These data were confirmed by flow cytometry analysis of Bim and Bmf protein (Fig 7B and C). Expression of the proapoptotic protein Bim as well as Bmf was significantly higher in RA T cells. There was a tendency for RA T cells to contain lower levels of Bcl-2 protein (Fig 7D), which did not reach statistical significance.
Figure 7. The DNA-PKcs-JNK pathway upregulates the apoptogenic molecules Bim and Bmf in RA CD4+ T cells.
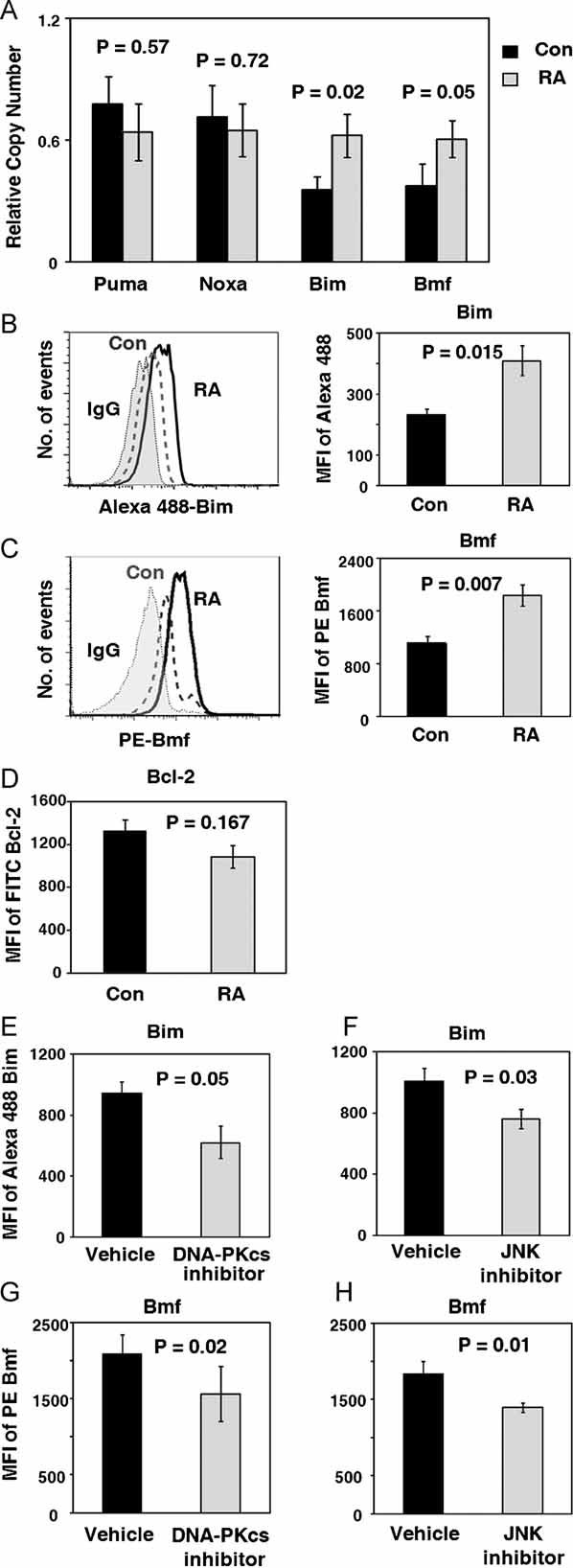
CD4+CD45RO− T cells were isolated from the peripheral blood of controls and RA patients. Cells were analyzed immediately after isolation or maintained in culture without mitogenic stimulation for 72 h. DNA-PKcs or JNK inhibitors were added for the final 24 h of culture.
- Puma, Noxa, Bim and Bmf transcript levels in freshly isolated cells were quantified by qPCR. Data are presented as mean ± SEM.
- Flow cytometry analysis of Bim levels in control and RA T cells after 72 h of culture. Expression of Bim protein in n = 5 RA and n = 5 controls is given as MFI of Alexa Fluor 488 Bim.
- Flow cytometry analysis of Bmf levels in control and RA T cells after 72 h of culture. Expression of Bmf protein in n = 6 RA and n = 4 controls is given as MFI of PE Bmf.
- Bcl-2 protein levels were analyzed by flow cytometry after 72 h of culture. Expression of Bcl-2 protein in n = 6 RA and n = 6 control samples is given as MFI of FITC Bcl-2.
- Bim protein levels in the absence or presence of the DNA-PKcs inhibitor IC86621 (100 nM) were analyzed by flow cytometry in samples from four RA patients. Results are presented as MFI ± SEM.
- Bim levels in the absence or presence of the JNK inhibitor II (10 µM) were analyzed in naïve T cells from six RA patients. Results are presented as MFI ± SEM.
- Flow cytometry analysis of Bmf protein expression in T cells from three RA patients in the absence or presence of the DNA-PKcs inhibitor II. Results are presented as MFI ± SEM.
- Flow cytometry analysis of Bmf protein expression in T cells from three RA patients in the absence or presence of the JNK inhibitor II. Results are presented as MFI ± SEM.
To examine whether the overexpression of Bim protein was mechanistically connected to the upregulation of DNA-PKcs and JNK, RA T cells were treated with the DNA-PKcs inhibitor IC 86621 or the JNK inhibitor II. Bim and Bmf protein concentrations were compared in treated and untreated T cells by flow cytometry (Fig 7E–H). Pharmacological blockade of DNA-PKcs activity or inhibition of the JNK pathway both reduced intracellular Bim and Bmf levels, supporting a direct connection between DNA-PKcs-JNK upregulation and BH3 protein-mediated apoptotic susceptibility.
These data suggest that Puma and Noxa are uninvolved in the shortened survival of RA T cells; rather, the internal apoptosis machinery responds to the cell's failure to repair DNA by upregulating Bim, shifting the Bim:Bcl-2 ratio and rendering T cells apoptosis susceptible.
DISCUSSION
In RA, the process of immune ageing is accelerated. RA patients accumulate CD4 T cells that have lost expression of CD28 and utilize alternate co-stimulatory molecules to sustain pro-inflammatory functions in peripheral tissues (Park et al, 1997; Schmidt et al, 1996; Weyand & Goronzy, 1997, 2006). Molecularly, RA T cells are distinct in that they have an intrinsic deficiency of two enzymes implicated in maintaining genomic stability; the telomere-elongating enzyme telomerase and ATM, a kinase critically involved in recognizing and repairing DNA DSB (Fujii et al, 2009; Shao et al, 2009). The current study has examined whether chronic DNA damage activates alternative pathways in DNA surveillance and repair activity and how deficiencies in DNA repair and telomere repair affect T-cell survival. The study focused on naïve and resting CD4 T cells as they represent the reserve pool of the immune system and their survival critically affects the process of immune ageing. Such naïve CD4 T cells were found to be apoptosis susceptible, to accumulate damaged DNA and to upregulate DNA-PKcs phosphorylation. Remarkably, the induction of DNA-PKcs in RA T cells was combined with a reduction in Ku70 and Ku80 protein, causing a dysbalance of core components of the NHEJ complex. Nevertheless, the overexpression of DNA-PKcs had profound functional consequences; inhibition of enzyme activity protected T cells from apoptotic death. DNA-PKcs-dependent apoptosis was mediated through the stress kinase signalling pathway, specifically involving JNK. RA T cells expressed increased amounts of the apoptogenic BH3-only proteins Bim and Bmf and blockade of either DNA-PKcs or JNK activity normalized T-cell death rates.
Mechanisms of T-cell apoptosis have been implicated in the formation of the T-cell repertoire and in T-cell autoimmunity. Yet, studies have concentrated on antigen-experienced memory T cells and antigen-induced clonal expansion. How T-cell death is regulated in the pool of peripheral naïve T cells, the host's lymphocyte reserve, remains unclear. Antigen-naïve T cells are Fas resistant and need to be restimulated and acquire memory T-cell features before they can die through the Fas-FasL pathway (Miyawaki et al, 1992). The current study implicates the sensing of damaged DNA in the regulation of T-cell survival. T cells are long-lived cells and thus have ample opportunity to accumulate DNA breaks. Under physiological conditions, DNA-PKcs-dependent repair does not seem to be important as inhibition of the enzyme in healthy T cells did not increase T-cell death. In RA T cells, DNA-PKcs becomes an important regulator of T-cell longevity. Apoptotic loss of T cells was normalized when enzyme activity was blocked.
T cells require growth factors to stay alive and to repopulate the T-cell pool through homeostatic proliferation. Naïve T cells require IL-7 and, in humans, respond to IL-15 as a homeostatic cytokine. The pool of naïve T cells persistently expresses the low-affinity receptor for IL-2 (Ma et al, 2006; Surh & Sprent, 2008). T cells closely monitor the availability of such growth and survival factors and cytokine deficiency activates intracellular sensors, such as FOXO3A (Dijkers et al, 2000). However, supplementation of growth-promoting cytokines, including the homeostatic cytokines IL-2, IL-7 and IL-15, could not rescue naïve human T cells from dying whereas inhibition of DNA-PKcs as well as JNK were able to reset the apoptotic threshold. These findings implicate the DNA-PKcs-JNK pathway in the regulation of T-cell homeostasis and the long-term health of the immune system.
Interestingly, both ATM and DNA-PKcs have a role in regulating T-cell fate. Resting healthy T cells died predominantly through activating ATM (Fig 2) whereas RA T cells lack availability of that mechanism and seem to resort to alternative means of DNA surveillance. Besides the repression of ATM transcription, RA T cells also produce low amounts of p53 (Shao et al, 2009). The tumour repressor p53 is well known for its role in cellular responses to DNA breaks, with double-strand breaks being the most powerful lesion to activate p53-dependent apoptosis. Broken DNA initiates recruitment of ATM which rapidly phosphorylates p53, leading to its stabilization, induction of downstream transcriptional targets and cell-cycle arrest or apoptosis (Shiloh, 2003; Vousden & Lu, 2002). p53 is so important that p53-deficient cells fail to respond to this cell cycle checkpoint (Brown & Attardi, 2005; Maclean et al, 2008). Yet, data presented here strongly support the notion that under conditions of p53 deficiency T cells are able to employ DNA-PKcs-mediated activation of stress kinases to access the endogenous cell death machinery. Knockdown of DNA-PKcs promptly reduced JNK phosphorylation, establishing a mechanistic link between the two signalling networks (Fig 6). How and where DNA-PKcs communicates with the JNK pathway is unknown and needs to be explored. Also, the reduction in ATM and the overexpression of DNA-PKcs in RA T cells suggests as one possibility that the two enzymes involved in DNA repair activity may cross-regulate each other.
An intriguing result of the current study is the dysbalance of DNA-PKcs and Ku70/80. Under physiological conditions, DNA-PKcs is only recruited to DNA ends after the Ku70/80 heterodimer has bound DNA ends (Smith et al, 1999). DNA-PK synapses the ends and removes the overhanging 3′ and 5′ ends to allow for rejoining by DNA ligase (DeFazio et al, 2002; Lees-Miller & Meek, 2003; Spagnolo et al, 2006). An interesting scenario emerges in RA T cells. Only the catalytic subunit of the enzyme is activated, whereas Ku70/Ku80 remain low, undermining the cell's options to proceed with DNA end joining. Under these conditions chronically induced catalytic subunit of DNA-PK ultimately harms the cell and mediates T-cell apoptosis. Thus, coordinated regulation of both DNA-PKcs and Ku70/Ku80 may be necessary to facilitate successful repair of DNA lesions. DNA-PKcs alone, however, may be sufficient to trigger other signalling pathways, e.g. the JNK pathway to regulate cellular functions (Damrot et al, 2009; Fritz & Kaina, 2006). These findings suggest that in human T cells DNA-PK activation is possible, even when expression of the Ku70/Ku80 heterodimer is low, broadening the role of the enzyme in cellular regulation.
Data collected in this study provide detailed information that sensing of damaged DNA by DNA-PK initiates an integrated pathway, with activation of the JNK signalling network and eventually, engagement of the intrinsic apoptosis machinery. Expression studies indicated selectivity in this process as Bim and Bmf were expressed at higher levels in apoptosis-sensitive RA T cells and their levels normalized when the DNA-PKcs-JNK axis was blocked (Fig 7). Proteins of the Bcl-2 family, including the prosurvival members Bcl-2, Bcl-xL, Bcl-w and Mcl-10 and the prodeath Bax/Bak-like proteins ultimately control cell survival and response patterns to apoptotic stressors. The BH3-only proteins Bad, Bik, Bid, Bim, Bmf, Puma and Noxa are essential apoptotic initiators (Danial & Korsmeyer, 2004), yet each of them may respond to particular cellular stressors. Bim has emerged as critical in apoptosis following withdrawal of cytokines and is necessary in the deletion of autoreactive murine T cells (Bouillet et al, 2002; Enders et al, 2003). Bim has also been implicated in regulating the downregulation of T-cell responses as immune activation subsides (Hildeman et al, 2002; Pellegrini et al, 2003). Puma and Noxa are recognized as p53-induced apoptosis regulators and have been connected to both cytokine-deprivation and DNA damage-induced apoptosis (Ekert et al, 2006; Jeffers et al, 2003; Shibue et al, 2003; Villunger et al, 2003). Studies of Bid deficiency have produced controversial results (Kaufmann et al, 2007; Zinkel et al, 2003) with a recent report that could not confirm increased genomic instability and leukemogenesis upon mitomycin treatment (Kaufmann et al, 2007) removing Bid as a candidate for monitoring DNA damage in healthy and RA T cells. Gene expression screening focused attention onto Bim and Bmf, compatible with the p53 independence of the apoptosis.
Bim and Bmf share subcellular localization and activation mechanisms. Under non-apoptotic conditions, Bim and Bmf are sequestered via dynein light chains to the actin and tubulin cytoskeleton, respectively, which keeps them from activating Bak and Bax (Puthalakath et al, 1999, 2001). Cell stress signals activate Bim and Bmf through phosphorylation within their dynein binding sites, mobilizing both proteins from the cytoskeleton and making them available to cause conformational changes within Bak and Bax, enabling them to homooligomerize and forming pores in the outer mitochondrial membrane (Antonsson et al, 2001; Wei et al, 2000). JNK has been proposed to mediate Bim and Bmf phosphorylation (Lei & Davis, 2003; Tianhu et al, 2010), possibly releasing them from their cytoskeletal sequestration. Pharmacological inhibition of either JNK or of DNA-PKcs normalized the levels of Bim as well as Bmf in the patient-derived T cells, localizing activation of DNA-PKcs and JNK upstream of the intrinsic apoptosis machinery.
The paper explained
PROBLEM:
Rheumatoid arthritis is an autoimmune syndrome that causes chronic destructive inflammation of joints and shortens life expectancy by rendering patients susceptible to cardiovascular disease. Cells of the adaptive immune system, including T cells and B cells, have been implicated in abnormal immune responses in RA and are targeted by non-specific immunosuppressive therapy.
Within the last decade it has become clear that patients with RA have premature ageing of the immune system. They accumulate aged T cells that are prone to inflammatory responses. In RA patients even naïve T cells, not involved in inflammatory reactions, are prematurely aged. Such naïve T cells have telomeric dysfunction with shortening of chromosomal ends by 1500 kb, a telomere loss associated with 25–30 years of life time progression. Mechanisms underlying premature immunosenescence in RA and the functional implications for the disease process are not understood.
RESULTS:
To explore whether DNA instability contributes to accelerated immune ageing we have screened T cells from a cohort of RA patients for DNA double strand beaks and apoptotic susceptibility. RA T cells carried a high load of DNA DSB and were prone to undergo apoptosis. DNA-PKcs, an enzyme involved in DNA repair, was chronically activated but failed to promote DNA repair due to the deficiency of essential components of the repair complex, specifically the DNA-binding proteins Ku70 and Ku80. However, chronic DNA-PKcs activation had marked functional consequences for RA T cells. Downstream of DNA-PKcs activation the JNK stress kinase pathway and the apoptogenic proteins Bim and Bmf were upregulated. Inhibition of DNA-PKcs normalized JNK activation and protected RA T cells from apoptotic death.
IMPACT:
Deoxyribonucleic acid instability renders RA T cells susceptible to apoptosis, causing a continuous leak from the T-cell pool. In humans older than 45 years of age, as most RA patients are, T cells cannot be replenished by thymic production but only by homeostatic proliferation. Continuous T-cell loss imposes proliferative stress and exhausts the immune system's proliferative reserve. As T-cell loss in RA results from a deficiency to properly repair fragmented DNA, the process is potentially amendable to therapeutic interventions.
A most interesting aspect of the present work is how it can impact the understanding of the autoimmune syndrome RA and current therapeutic approaches. Chronic T-cell loss, particularly in the naïve compartment, will elicit homeostatic control mechanisms; restoring T-cell numbers by enforced autoproliferation. Thus, the T-cell pool in RA patients is under high turnover, eventually jeopardizing the proliferative reserve needed for immune competence. So far, RA has not been regarded as a syndrome of chronic lymphopenia (Weyand et al, 2003). Yet, constant leaking of T cells and widespread DNA damage in surviving T cells must have consequences for the composition of the T-cell repertoire and the functional competency of individual T cells. Ideally, damaged DNA could be repaired and nuclear stability maintained to ensure optimal survival time and prevention of cellular senescence. Enhancing production of the DNA-PK subunits Ku70/Ku80 may have beneficial effects, although it may make DNA-PKcs activation even more efficient and accelerate cell loss. Experimental data presented in this study suggest that inhibiting chronically activated DNA-PKcs protects RA T cells from apoptosis and thus should be considered as a therapeutic intervention to counteract T-cell loss in RA. However, considerations of utilizing DNA-PKcs inhibition to rescue RA T cells from apoptosis, reduce the proliferative stress in the immune system and slow down immune ageing require a careful weighing of potential benefits and harm. In the current study, protection from the induction of the apoptogenic mediators Bim and Bmf could be achieved with either inhibiting DNA-PKcs activity or blocking JNK (Fig 7). Thus, either intervention could be explored as a new strategy to prolonging the survival of naïve CD4 T cells. Downregulation of JNK activity may have additional anti-inflammatory effects (Bennett et al, 2001). Targeting DNA-PKcs, however, would exploit the most upstream component of the DNA-PK-JNK axis.
The DNA-PKcs inhibitor used in the current study displays high specificity for this kinase. In previous reports concentrations of up to 100 µM have shown neither activity against distantly related protein kinases (protein kinase A, protein kinase C, checkpoint protein kinase 1, cyclin-dependent kinase 2, Rous sarcoma tyrosine kinase, aurora-related kinase 2 and casein kinase 1) nor against the closely related protein kinases ATR and ATM (Kashishian et al, 2003). Here, T-cell survival was improved at doses 1000-fold lower (50–100 nM). Another important consideration is the role of DNA-PK as a component of the DNA DSB recombination apparatus which is critically involved in the end joining of V(D)J recombination (Schlissel, 2002). Thus, blocking of DNA-PKcs could lead to immunodeficiency. However, there exists an alternative end-joining pathway that generates chromosomal translocations and it is possible that this mechanism would dominate if the DNA-PK pathway of NHEJ were shut down. In previous studies doses of the DNA-PKcs inhibitor IC 86621 that were needed to prevent DNA end joining were as high as 25–100 µM (Allen et al, 2003), again 1000-fold higher than the dose range employed to protect apoptosis-sensitive T cells. The potential side effects of DNA-PKcs inhibition are of particular relevance for RA patients as the disease is associated with an increased risk to develop lymphoma (Weyand et al, 2006). In essence, complete blockade of DNA-PKcs should certainly not be pursued as a therapeutic goal but bringing DNA-PKcs activity into a normal range may be all that is necessary to avoid the negative consequences of chronic DNA repair activity. Ultimately, the mechanisms underlying DNA instability in RA T cells need to be unraveled as protection from DNA damage and reconstitution of proper DNA repair would open entirely new approaches to the conceptional understanding and the clinical management of this autoimmune syndrome.
Acknowledgments
This work was supported by the National Institutes of Health grants R01 AI44142, R01 AR42527 and U19 AI57266.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Abbreviations
- ACR
American College of Rheumatology
- AKT
v-akt murine thymoma viral oncogene homolog
- AMV
avian myeloblastosis virus
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia related
- CaM
calcium modulated protein
- CD28
cluster of differentiation 28
- CD4
cluster of differentiation 4
- DMEM
Dulbecco's modified Eagle medium
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DSB
double strand breaks
- EGTA
ethyleneglycol-bis-(β-aminoethylether)-N,N,N′,N′-tetraacetic acid
- Fas
tumor necrosis factor receptor superfamily, member 6
- FasL
Fas ligand
- FOXO3A
forkhead box O3A
- HDR
homology-directed repair
- JNK
c-Jun NH2-terminal kinase
- pJNK
phosphorylated c-Jun–NH2-terminal kinase
- MRN
Mre11-Rad50-NBS1 complex
- NF-κB
nuclear factor κB
- NHEJ
non-homologous end-joining
- PBS
phosphate buffered saline
- PI
propidium iodide
- PI3K
phosphatidylinositol-3′-kinase
- PIPES
piperazine-N,N′-bis(2-ethanesulfonic acid)
- qPCR
quantitative polymerase chain reaction
- RIPA
radioimmunoprecipitation assay
- siRNA
small (or silencing), interfering RNA.
Author contributions
L. S. designed and performed experiments, analyzed data and contributed to manuscript writing. J. J. G. designed the project, supervised experiments and data analysis and was responsible for patient recruitment and clinical analysis. C. M. W. conceived the project idea, designed the experimental approach, supervised every stage of the project and wrote the manuscript.
For more information
Institute for Immunity, Transplantation and Infection, at Standford University
Rheumatoid Arthritis
http://www.nature.com/nrrheum/journal/v5/n10/full/nrrheum.2009.180.html
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Allen C, Halbrook J, Nickoloff JA. Interactive competition between homologous recombination and non-homologous end joining. Mol Cancer Res. 2003;1:913–920. [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci USA. 1999;96:14899–14904. doi: 10.1073/pnas.96.26.14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- Damrot J, Helbig L, Roos WP, Barrantes SQ, Kaina B, Fritz G. DNA replication arrest in response to genotoxic stress provokes early activation of stress-activated protein kinases (SAPK/JNK) J Mol Biol. 2009;385:1409–1421. doi: 10.1016/j.jmb.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002;21:3192–3200. doi: 10.1093/emboj/cdf299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- Doran MF, Pond GR, Crowson CS, O'Fallon WM, Gabriel SE. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 2002;46:625–631. doi: 10.1002/art.509. [DOI] [PubMed] [Google Scholar]
- Ekert PG, Jabbour AM, Manoharan A, Heraud JE, Yu J, Pakusch M, Michalak EM, Kelly PN, Callus B, Kiefer T, et al. Cell death provoked by loss of interleukin-3 signaling is independent of Bad, Bim, and PI3 kinase, but depends in part on Puma. Blood. 2006;108:1461–1468. doi: 10.1182/blood-2006-03-014209. [DOI] [PubMed] [Google Scholar]
- Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J Exp Med. 2003;198:1119–1126. doi: 10.1084/jem.20030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Fritz G, Kaina B. Late activation of stress kinases (SAPK/JNK) by genotoxins requires the DNA repair proteins DNA-PKcs and CSB. Mol Biol Cell. 2006;17:851–861. doi: 10.1091/mbc.E05-07-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA. 2009;106:4360–4365. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, Henel G, Sawai H, Singh K, Lee EB, Pryshchep S, Weyand CM. Costimulatory pathways in rheumatoid synovitis and T-cell senescence. Ann N Y Acad Sci. 2005;1062:182–194. doi: 10.1196/annals.1358.022. [DOI] [PubMed] [Google Scholar]
- Hellman B, Vaghef H, Bostrom B. The concepts of tail moment and tail inertia in the single cell gel electrophoresis assay. Mutat Res. 1995;336:123–131. doi: 10.1016/0921-8777(94)00047-a. [DOI] [PubMed] [Google Scholar]
- Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T-cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Kashishian A, Douangpanya H, Clark D, Schlachter ST, Eary CT, Schiro JG, Huang H, Burgess LE, Kesicki EA, Halbrook J. DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Mol Cancer Ther. 2003;2:1257–1264. [PubMed] [Google Scholar]
- Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, Johnstone RW, Dixit VM, Strasser A. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–433. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T-cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7:532–542. doi: 10.1038/nri2115. [DOI] [PubMed] [Google Scholar]
- Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–7758. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Meek K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie. 2003;85:1161–1173. doi: 10.1016/j.biochi.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek K, Gupta S, Ramsden DA, Lees-Miller SP. The DNA-dependent protein kinase: the director at the end. Immunol Rev. 2004;200:132–141. doi: 10.1111/j.0105-2896.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends. Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- Miyawaki T, Uehara T, Nibu R, Tsuji T, Yachie A, Yonehara S, Taniguchi N. Differential expression of apoptosis-related Fas antigen on lymphocyte subpopulations in human peripheral blood. J Immunol. 1992;149:3753–3758. [PubMed] [Google Scholar]
- Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation. 2006;114:2482–2489. doi: 10.1161/CIRCULATIONAHA.106.642801. [DOI] [PubMed] [Google Scholar]
- Park W, Weyand CM, Schmidt D, Goronzy JJ. Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+ CD28− T cells. Eur J Immunol. 1997;27:1082–1090. doi: 10.1002/eji.1830270507. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T-cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci USA. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–1832. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- Riha K, Heacock ML, Shippen DE. The role of the nonhomologous end-joining DNA double-strand break repair pathway in telomere biology. Annu Rev Genet. 2006;40:237–277. doi: 10.1146/annurev.genet.39.110304.095755. [DOI] [PubMed] [Google Scholar]
- Schlissel MS. Does artemis end the hunt for the hairpin-opening activity in V(D)J recombination. Cell. 2002;109:1–4. doi: 10.1016/s0092-8674(02)00694-3. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7− CD28− T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, Weyand CM. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci USA. 2003;100:13471–13476. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. 2009;206:1435–1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Smith GC, Divecha N, Lakin ND, Jackson SP. DNA-dependent protein kinase and related proteins. Biochem Soc Symp. 1999;64:91–104. [PubMed] [Google Scholar]
- Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9:573–580. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- Spagnolo L, Rivera-Calzada A, Pearl LH, Llorca O. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol Cell. 2006;22:511–519. doi: 10.1016/j.molcel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Tianhu Z, Shiguang Z, Xinghan L. Bmf is upregulated by PS-341-mediated cell death of glioma cells through JNK phosphorylation. Mol Biol Rep. 2010;37:1211-1219. doi: 10.1007/s11033-009-9491-9. [DOI] [PubMed] [Google Scholar]
- Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–720. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ. Pathogenesis of rheumatoid arthritis. Med Clin North Am. 1997;81:29–55. doi: 10.1016/s0025-7125(05)70504-6. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ. T-cell-targeted therapies in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2:201–210. doi: 10.1038/ncprheum0142. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Fulbright JW, Goronzy JJ. Immunosenescence, autoimmunity, and rheumatoid arthritis. Exp Gerontol. 2003;38:833–841. doi: 10.1016/s0531-5565(03)00090-1. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ, Kurtin PJ. Lymphoma in rheumatoid arthritis: an immune system set up for failure. Arthritis Rheum. 2006;54:685–689. doi: 10.1002/art.21674. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Fujii H, Shao L, Goronzy JJ. Rejuvenating the immune system in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:583–588. doi: 10.1038/nrrheum.2009.180. [DOI] [PubMed] [Google Scholar]
- Williams ES, Klingler R, Ponnaiya B, Hardt T, Schrock E, Lees-Miller SP, Meek K, Ullrich RL, Bailey SM. Telomere dysfunction and DNA-PKcs deficiency: characterization and consequence. Cancer Res. 2009;69:2100–2107. doi: 10.1158/0008-5472.CAN-08-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, Kutok JL, Alt FW, Korsmeyer SJ. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. 2003;17:229–239. doi: 10.1101/gad.1045603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.