

Abstract
Circulating insulin-like growth factor I (IGF-1) levels are closely associated with cardiac performance although the role of IGF-1 in alcoholic cardiac dysfunction is unknown. This study was designed to evaluate the impact of severe liver IGF-1 deficiency (LID) on chronic alcohol-induced cardiomyocyte contractile and intracellular Ca2+ dysfunction. Adult male C57 and LID mice were placed on a 4% alcohol diet for 15 weeks. Cardiomyocyte contractile and intracellular Ca2+ properties were evaluated including peak shortening (PS), maximal velocity of shortening/relengthening (±dL/dt), time-to-relengthening (TR90), change in fura-fluorescence intensity (ΔFFI) and intracellular Ca2+ decay. Levels of apoptotic regulators caspase-3, Bcl-2 and c-Jun NH2-terminal kinase (JNK), the ethanol metabolizing enzyme mitochondrial aldehyde dehydrogenase (ALDH2), as well as the cellular fuel gauge AMP-activated protein kinase (AMPK) were evaluated. Chronic alcohol intake enlarged myocyte cross-sectional area, reduced PS, ± dL/dt and ΔFFI as well as prolonged TR90 and intracellular Ca2+ decay, the effect of which was greatly attenuated by IGF-1 deficiency. The beneficial effect of LID against alcoholic cardiac mechanical defect was ablated by IGF-1 replenishment. Alcohol intake increased caspase-3 activity/expression although it down-regulated Bcl-2, ALDH2 and pAMPK without affecting JNK and AMPK. IGF-1 deficiency attenuated alcoholism-induced responses in all these proteins with the exception of Bcl-2. In addition, the AMPK agonist 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside abrogated short-term ethanol incubation-elicited cardiac mechanical dysfunction. Taken together, these data suggested that IGF-1 deficiency may reduce the sensitivity to ethanol-induced myocardial mechanical dysfunction. Our data further depicted a likely role of Caspase-3, ALDH2 and AMPK activation in IGF-1 deficiency induced ‘desensitization’ of alcoholic cardiomyopathy.
Keywords: alcoholism, IGF-1, cardiomyocytes, contractile function, AMPK
Introduction
Insulin-like growth factor 1 (IGF-1) plays a pivotal role in the maintenance of cardiac architecture and function in the settings of both healthy and failing hearts [1]. Deficiency in IGF-1 is known to contribute to altered body composition, neuroendocrine activation, cardiac atrophy and compromised cardiac function [2, 3]. Paradoxically, recent evidence from our lab and others has demonstrated that reduced IGF-1 secretion and circulating levels may retard myocardial injury in response to oxidative stress and aging [4–6]. Data from our group using the liver IGF-1 deficiency (LID) murine model revealed enhanced resistance against the pro-oxidant paraquat-induced cardiomyocyte apoptosis and contractile dysfunction [5]. This finding is in consistence with the prolonged lifespan with genetic mutation of IGF-1 or its receptor such as the Ames Dwarf and igf-1r+/− mice (IGF-1 receptor knockout) [7, 8]. Nonetheless, the precise role of IGF-1 in the maintenance of myocardial architecture and function under various pathological conditions remains ambiguous. Evidence from both clinical and experimental studies has demonstrated that chronic alcohol abuse markedly and irreversibly reduces IGF-1 bioavailability and alters IGF-1 signalling [9, 10], indicating a role of IGF-1 in the pathogenesis of alcoholic cardiomyopathy characterized by cardiac hypertrophy and ventricular dysfunction [11, 12].
Furthermore, alcohol intoxication-induced inhibition of protein synthesis may be associated with alcohol-induced IGF-1 resistance, the effect of which is ameliorated by deficiency of mitochondrial branched-chain aminotransferase [13]. On the other hand, alcohol is capable of disrupting reproductive function in adolescent human beings through interrupted IGF-1 signalling [14], further substantiating complex interactions between alcohol and IGF-1 in both physiological and pathophysiological processes. To better elucidate the role of IGF-1 in chronic alcohol intake-induced myocardial morphological and contractile defect, we hereby took advantage of the LID mouse model, which possesses only ∼25% of normal circulating IGF-1 levels [15], to examine the impact of severe LID on alcohol-induced cardiomyocyte morphological and functional alterations. To further consolidate the role of IGF-1 in alcoholic cardiac injury, recombinant IGF-1 was replenished exogenously to the IGF-1 deficient cardiomyocytes prior to mechanical evaluation. Expression of apoptotic proteins, the ethanol metabolizing enzyme mitochondrial aldehyde dehydrogenase (ALDH2) and the cellular fuel gauge molecule AMP-activated protein kinase (AMPK) was evaluated, in an effort to better understand the mechanism of action behind IGF-1 deficiency-induced effect on chronic alcohol intake-elicited cardiomyocyte mechanical dysfunction.
Materials and methods
Experimental animals and chronic alcohol feeding
The experimental procedure was approved by our Institutional Animal Use and Care Committee at the University of Wyoming (Laramie, WY, USA). All animal procedures were in accordance with the NIH standard. Generation of the LID mice was described in detail previously using the Cre/loxP system [15]. To determine the presence of the IGF-1/loxP transgene, genomic DNA was isolated from tail clips using a Quick extraction and amplification kit (BioPioneer, Inc., San Diego, CA, USA). To test the presence of Cre transgene (i.e. liver-specific IGF-1 gene knockout), primers Cre-5′ and Cre-3′ were used, which yielded a 0.6 kb band for the Cre transgene. Mice homozygous or heterozygous for IGF-1/loxP carrying the albumin-Cre transgene were crossed. The homozygous offspring along with negative controls were used for experiment. The mouse genotyping was executed using a double PCR strategy. To identify the genotype of IGF-1/loxP, primers of insulinoma-associated 2 (IA6), insulinoma-associated 8 (IA8) and inhibitor of DNA binding 3 (ID3) were used in PCR reaction. Mice which yield one 0.4 kb band are negative for IGF-1/loxP whereas those with one 0.2 kb band are positive. Presence of both 0.4 kb and 0.2 kb bands indicates for heterozygous IGF-I/loxP. To determine the presence of the Cre transgene, primers Cre-5′ and Cre-3′ were used which yield a 0.6 kb band. Mice positive for both IGF-1/loxP and Cre transgene were deemed as the LID mice, while the IGF-I/loxP– mice with or without Cre transgene were used as LID– (C57) mice. Ten-month-old aged-matched adult male C57 and LID mice were placed on a nutritionally complete liquid diet (Shake & Pour Bioserv, Inc., Frenchtown, NJ, USA) for a 1 week acclimation period. The use of a liquid diet is based on the scenario that ethanol self-administration resulted in less nutritional deficiencies and less stress to the animals in comparison to forced-feeding regimens, intravenous administration, or aerosolized inhalation [16]. Upon completion of the acclimation period, half of the C57 and LID mice were maintained on the regular liquid diet (without ethanol), and the remaining half began a 15 week period of isocaloric 4% (vol/vol) ethanol diet feeding. An isocaloric pair-feeding regimen was employed to eliminate the possibility of nutritional deficits. Control mice were offered the same quantity of diet ethanol-consuming mice drank the previous day [11].
Measurement of blood ethanol level
On the last day of diet feeding, mice were killed under anaesthesia (ketamine/xylazine: 3:1, 1.32 mg/kg, i.p.). Blood was collected and stored in sealed vials. A volume of 100 μl plasma from each sample was put into an autosampler vial. Six microlitres of n-propanol and 194 μl H2O were then added to the vial. Following a 20 min. incubation at 50°C, 50 μl aliquot of headspace gas was removed and transferred to an Agilent 6890 Gas Chromatograph (Agilent Technologies, Inc, Wilmington, DE, USA) equipped with a flame ionization detector. Ethanol, n-propanol and other components such as acetaldehyde were separated on a 60 m VOCOL capillary column (Supelco, Inc., Bellefonte, PA, USA) with film of 1.8 μm in thickness and an inner diameter of 320 μm. The carrier gas was helium at a flow rate of 18.0 ml/min. Quantitation was achieved by calibrating peak areas against those from headspace samples of known ethanol standards [12].
Murine cardiomyocyte isolation and in vitro drug treatment
Mouse hearts were removed under anaesthesia (ketamine/xylazine at 3:1, 1.32 mg/kg) and were perfused with Krebs-Henseleit bicarbonate buffer containing (in mM): 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 4-(2-hydroxyerhyl)piperazine-1-erhanesulfonic acid (HEPES), 11.1 glucose and 10 butanedione with 5% CO2–95% O2. Hearts were subsequently digested with 0.1 mg/ml Liberase Blendzymes (Roche Diagnostics, Indianapolis, IN, USA) for 10 min. at 37°C. After perfusion, left ventricles were removed and minced. Extracellular Ca2+ was added back to 1.25 mM. Functional studies were conducted within 6 hrs of isolation and myocytes with obvious sarcolemmal blebs or spontaneous contractions were not used. The percentage of rod-shaped (viable) cardiomyocytes was approximately 70% which was not affected by either IGF-1 deficiency or chronic alcohol feeding [12]. To examine the effect of IGF-1 replenishment on ethanol-induced cardiac contractile response, cardiomyocytes from LID mice were treated with ethanol (240 mg/dl) at 37°C for 4 hrs with or without exogenous recombinant IGF-1 (5 nM) supplementation [5] prior to the mechanical assessment. To directly assess the role of AMPK activation on ethanol-induced cardiomyocyte contractile response, cardiomyocytes were treated with ethanol (240 mg/dl) at 37°C for 4 hrs in the absence or presence of the AMPK agonist 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR, 500 μM) [17] before mechanical function was assessed.
Cell shortening/relengthening
Mechanical properties of cardiomyocytes were assessed using a SoftEdge MyoCam® system (IonOptix Corporation, Milton, MA, USA) [12]. In brief, cardiomyocytes were placed in a chamber mounted on the stage of an inverted microscope (Olympus Incorporation, Model IX-70, Tokyo, Japan) and superfused at 25°C with a buffer containing (in mM): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, at pH 7.4. The cells were field stimulated with suprathreshold voltage at a frequency of 0.5 Hz (unless otherwise stated), 3 msec. duration, using a pair of platinum wires placed on opposite sides of the chamber connected to a FHC stimulator (Brunswick, NE, USA). The myocyte being studied was displayed on a computer monitor using an IonOptix MyoCam camera. An IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed including peak shortening (PS), time-to-PS (TPS), time-to-90% relengthening (TR90) and maximal velocities of shortening/relengthening (±dL/dt). In case of altering stimulus frequency, the steady-state contraction of myocytes was achieved (usually after the first six beats) before PS was recorded [12].
Intracellular Ca2+ transient measurement
Intracellular Ca2+ was measured using a dual-excitation, single-emission photomultiplier system (IonOptix) in myocytes loaded with fura 2-AM (0.5 μM). Myocytes were placed on an inverted microscope and imaged through an Olympus (IX-70) Fluor ×40 oil objective. Myocytes were exposed to light emitted by a 75 W halogen lamp through either a 360 or 380 nm filter while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm by a photomultiplier tube after initial illumination at 360 nm for 0.5 sec. and then at 380 nm for the duration of the recording protocol. The 360 nm excitation reading was repeated at the end of the protocol. Qualitative evaluation of intracellular Ca2+ was inferred from fura fluorescence intensity (FFI) changes (ΔFFI). A Chebyshev equation was used to evaluate the intracellular Ca2+ decay constant [18].
Histological examination
Mice were anesthetized with ketamine/xylazine (3:1, 1.32 mg/kg, i.p.). Hearts were arrested in diastole with saturated KCl. Transverse slices of the heart were cut at mid-level and fixed in neutral-buffered formalin at room temperature for 24 hrs. The specimen was then embedded in paraffin, cut in 5 μm sections and stained with haematoxylin and eosin. Cardiomyocyte cross-sectional areas from three to five randomly selected high power fields from three to five mice per group were calculated on a digital microscope using the NIH ImageJ software (Version 1.43r). Mean cardiomyocyte cross-sectional areas were averaged from approximately 200 cells per group [19].
Caspase-3 assay
The caspase-3 activity was determined according to the published method [20]. Briefly, 1 ml phosphate-buffered solution was added to a flask containing left ventricular tissue homogenates prior to centrifugation at 10,000 × g at 4°C for 10 min. The supernatant was discarded and homogenates were lysed in 100 μl of ice-cold cell lysis buffer [50 mM HEPES, pH 7.4, 0.1% 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), 1 mM dithiothreitol (DTT), 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% NP40]. The assay was carried out in a 96-well plate with each well containing 30 μl cell lysate, 70 μl of assay buffer (50 mM HEPES, 0.1% CHAPS, 100 mM NaCl, 10 mM DTT and 1 mM EDTA) and 20 μl of caspase-3 colorimetric substrate Ac-DEVD-pNA (Sigma-Aldrich, St. Louis, MO, USA). The 96-well plate was incubated at 37°C for 1 hr, during which time the caspase in the sample was allowed to cleave the chromophore p-NA from the substrate molecule. Absorbency was detected at 405 nm with caspase-3 activity being proportional to colour reaction. Protein content was determined using the Bradford method [21]. The caspase-3 activity was expressed as picomoles of pNA released per microgram of protein per minute.
Western blot analysis
Heart tissue from C57 and LID mice with or without chronic ethanol treatment were homogenized and sonicated in a lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM ethylenediaminetetraacetic acid (EGTA), 1% Triton, 0.1% SDS and protease inhibitor cocktail. Protein levels of cleaved caspase-3, Bcl-2, c-Jun NH2-terminal kinase (JNK), ALDH2, AMPK and pAMPK were examined by standard Western immunoblotting [12]. Membranes were probed with the anti-caspase-3, anti-Bcl-2, anti-JNK, anti-ALDH2, anti-AMPK, anti-phosphor-AMPK (pAMPK, Thr172) and anti-α-tubulin (internal loading control) antibodies followed by incubation with horseradish peroxidase-coupled secondary antibody (Cell Signaling Technology, Inc., Beverly, MA, USA). After immunoblotting, the film was scanned and detected with a Bio-Rad Calibrated Densitometer and the intensity of immunoblot bands was normalized to the loading control α-tubulin.
Statistical analysis
Data are presented as mean ± S.E.M. Statistical significance (P < 0.05) was determined by anova followed by a Tukey’s post hoc analysis.
Results
General features of C57 and LID mice fed with alcohol chronically
The LID mice weighed less compared with the age-matched C57 mice. The 15 week chronic alcohol feeding regime did not significantly affect body, liver and kidney weights (and their respective organ size) although the heart was significantly enlarged (both weight and size) in C57 mice compared with the non-ethanol-treated mice. LID did not significantly affect organ weight or size although it significantly attenuated alcohol-induced cardiac hypertrophy (both absolute weight and size). Blood alcohol levels were significantly elevated equally in alcohol consuming C57 and LID mice (Table 1).
Table 1.
General features of C57 and LID mice with or without chronic alcohol treatment
| C57-con | C57-ethanol | LID-con | LID-ethanol | |
|---|---|---|---|---|
| Body weight (g) | 28.2 ± 1.4 | 27.4 ± 0.8 | 25.4 ± 0.9* | 24.9 ± 0.6* |
| Heart weight (mg) | 153 ± 6 | 174 ± 7* | 149 ± 5 | 148 ± 4 |
| HW/BW (mg/g) | 5.46 ± 0.13 | 6.38 ± 0.24* | 5.95 ± 0.28 | 5.95 ± 0.13 |
| Liver weight (g) | 1.34 ± 0.04 | 1.35 ± 0.04 | 1.31 ± 0.08 | 1.31 ± 0.02 |
| LW/BW (mg/g) | 48.2 ± 1.6 | 49.3 ± 0.7 | 51.7 ± 2.8 | 52.6 ± 1.4 |
| Kidney weight (g) | 0.34 ± 0.02 | 0.34 ± 0.01 | 0.33 ± 0.02 | 0.32 ± 0.02 |
| KW/BW (mg/g) | 12.1 ± 0.3 | 12.4 ± 0.4 | 12.9 ± 0.6 | 12.9 ± 0.5 |
| Blood alcohol (mM) | 0 ± 0 | 5.13 ± 0.63* | 0 ± 0 | 5.49 ± 0.60* |
HW: heart weight; LW: liver weight; KW: kidney weight; Mean ± S.E.M.
P < 0.05 versus C57-Control (CON) group, n = 9–10 mice per group.
Cardiomyocyte contractile and intracellular Ca2+ property
Chronic alcohol intake did not significantly affect the resting cell length in C57 or LID mice. Chronic alcohol intake significantly reduced PS and ± dL/dt as well as prolonged TR90 without affecting TPS in cardiomyocytes from C57 mice. Importantly, LID significantly attenuated or abolished chronic alcohol intake-induced cardiomyocyte mechanical abnormalities (Fig. 1). In addition, cardiomyocytes from alcohol-fed C57 mice displayed significantly depressed resting and peak intracellular Ca2+ levels, dampened rise in intracellular Ca2+ in response to electrical stimulus (ΔFFI) and prolonged intracellular Ca2+ decay time. LID attenuated or negated alcohol-induced changes in resting, peak intracellular Ca2+, ΔFFI and intracellular Ca2+ decay (Fig. 2A–D).
Fig 1.
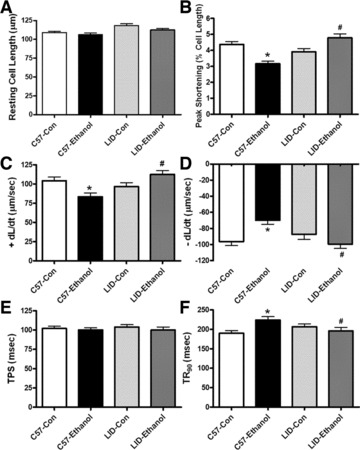
Effect of LID on chronic ethanol intake-induced cardiomyocyte contractile defects. (A) Resting cell length; (B) PS (normalized to resting cell length); (C) Maximal velocity of shortening (+dL/dt); (D) Maximal velocity of relengthening (–dL/dt); (E) TPS and (F) TR90. Mean ± S.E.M., n = 144–147 cells from five mice per group, *P < 0.05 versus C57 control (C57-Con) group, #P < 0.05 versus C57-Ethanol group.
Fig 2.
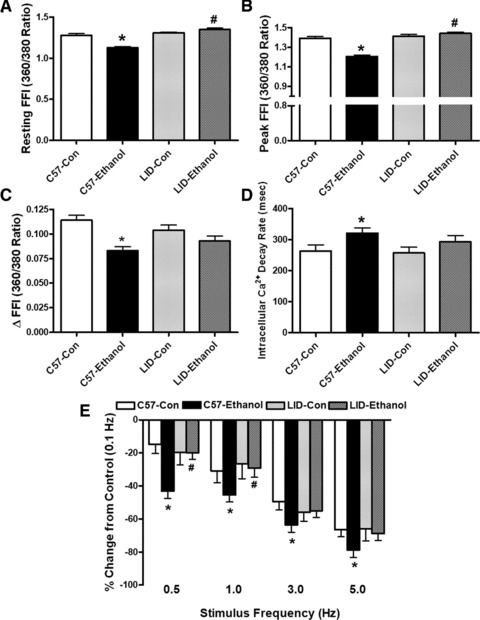
Effect of LID on chronic ethanol intake-induced intracellular Ca2+ homeostasis and SR Ca2+ store evaluated by frequency (0.1–5.0 Hz) dependent shortening response in murine cardiomyocytes. (A) Resting FFI; (B) Peak FFI; (C) ΔFFI in response to electrical stimuli; (D) Intracellular Ca2+ transient decay rate; and (E) Frequency (0.1–5.0 Hz) response. Change in PS at each stimulus frequency was made in reference to that of 0.1 Hz from the same cell. Mean ± S.E.M., n = 91–95 cells (A–D) or 22 cells (E) from five mice per group, *P < 0.05 versus C57 control (C57-Con) group, #P < 0.05 versus C57-Ethanol group.
Increased stimulus frequency-induced cardiomyocyte contractile response
Mouse hearts beat at high frequencies (>400/min. at 37°C) with sarcoplasmic reticulum (SR) Ca2+ store being the primary determinant of the frequency dependent response. We initially stimulated cardiomyocytes to contract at 0.5 Hz for 5 min. to ensure steady-state before stepwise altering the frequency from 0.1 to 5 Hz (300 beat/min.). All recordings were normalized to the PS obtained at 0.1 Hz of the same cell. Cardiomyocytes from alcohol-fed C57 mice exhibited significantly exaggerated depression in PS at 0.5 Hz and higher. LID itself did not alter the pattern of PS response at all frequencies although it significantly alleviated or ablated chronic alcohol-induced loss in PS amplitude between 0.5 and 5 Hz (Fig. 2E).
Effects of chronic ethanol treatment on myocardial histology
Chronic alcohol intake leads to cardiac hypertrophy [19]. To examine if IGF-1 deficiency affects chronic alcohol ingestion-induced change in cardiomyocyte size, if any, cardiomyocyte cross-sectional area was measured in myocardium from C57 and LID mice following chronic ethanol intake. Consistent with the enhanced ventricular weight and heart-to-body weight ratio, chronic ethanol intake led to a significant increase in cardiomyocyte cross-sectional area in the ethanol-treated C57 mice.
Interestingly, IGF-1 deficiency ablated chronic ethanol intake-induced increase in cardiomyocyte cross-sectional area (Fig. 3), in agreement with its beneficial effect against ventricular hypertrophy, cardiomyocyte mechanical and intracellular Ca2+ anomalies following chronic alcohol intake.
Fig 3.
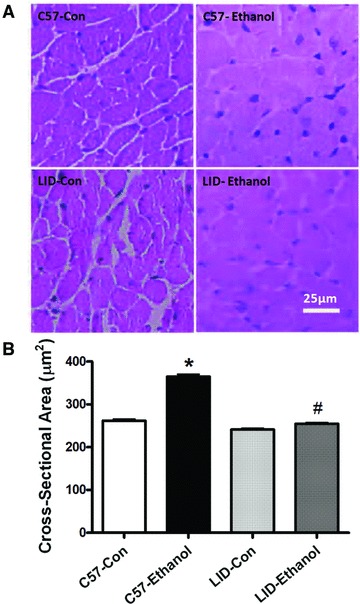
Histological analyses of hearts from C57 and LID mice with or without chronic ethanol treatment. (A) Representative haematoxylin and eosin stained micrographs showing transverse sections of left ventricular myocardium (×400) from C57 and LID mice with or without ethanol treatment; (B) Quantitative analysis of cardiomyocyte cross-sectional (transverse) area from approximately 200 cells from three to five mice per group. Mean ± S.E.M., *P < 0.05 versus C57 control (C57-Con) group, #P < 0.05 versus C57-Ethanol group.
Effect of IGF-1 replenishment on ethanol-induced cardiomyocyte contractile response
To consolidate the role of IGF-1 deficiency in ethanol-induced cardiac contractile response, recombinant IGF-1 (5 nM) [5] was replenished to cardiomyocytes isolated from LID mice before the ethanol (240 mg/dl) induced cardiomyocyte contractile response was evaluated. Neither IGF-1 supplementation nor acute ethanol exposure significantly affected resting cell length in LID mice. Consistent with data obtained from the chronic setting of ethanol treatment shown in Figure 1, acute ethanol exposure failed to elicit notable effect on cardiomyocyte mechanical properties in LID mice. Intriguingly, exogenous recombinant IGF-1 replenishment unmasked an ethanol exposure-induced cardiomyocyte contractile depression including decreased PS and ± dL/dt without affecting TPS and TR90 in cardiomyocytes from LID mice (Fig. 4). This observation favours a likelihood protective role of IGF-1 deficiency against ethanol-induced cardiac mechanical anomalies.
Fig 4.
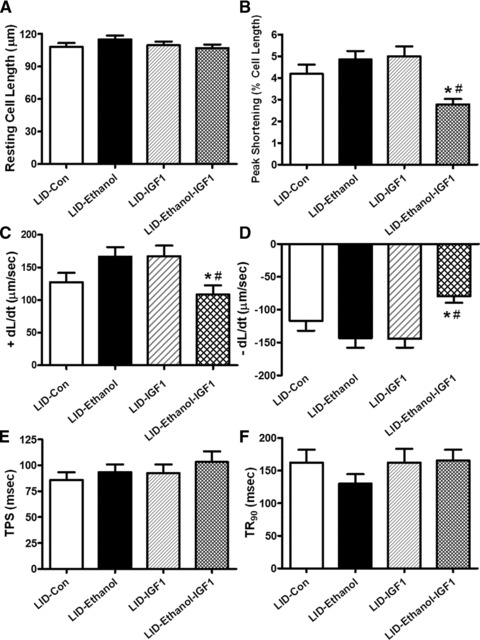
Effect of IGF-1 replenishment on acute ethanol exposure-induced cardiac contractile response in myocytes from LID mice. Freshly isolated cardiomyocytes from LID mice were incubated with ethanol (240 mg/dl) in the presence or absence of IGF-1 (5 nM) for 4 hrs. (A) Resting cell length; (B) PS (normalized to resting cell length); (C) Maximal velocity of shortening (+dL/dt); (D) Maximal velocity of relengthening (–dL/dt); (E) TPS and (F) TR90. Mean ± S.E.M., n = 34–39 cells from three mice, *P < 0.05 versus LID control (LID-Con) group, #P < 0.05 versus LID-Ethanol group.
Impact of IGF-1 deficiency on caspase-3 activity, expression of caspase 3, Bcl-2, JNK, ALDH2 and AMPK
To determine the potential mechanism(s) involved in the LID-elicited antagonism against chronic alcoholism-induced cardiomyocyte mechanical defect, we evaluated apoptotic signals such as caspase-3, Bcl-2 and JNK, energy regulator AMPK and the key ethanol metabolizing enzyme ALDH2 in myocardium using caspase-3 assay and Western blot analysis. Our data suggested that chronic alcohol feeding significantly enhanced caspase-3 activity, up-regulated cleaved caspase-3 expression and down-regulated Bcl-2 expression without effecting JNK expression in C57 mice. LID attenuated alcoholism-induced increase in caspase-3 activity and protein expression but failed to alter ethanol-induced response in Bcl-2 and JNK expression. LID itself did not affect the activity or protein expression of caspase-3, Bcl-2 and JNK in the absence of chronic alcohol treatment (Fig. 5). Our study further revealed that chronic alcohol feeding down-regulated the acetaldehyde metabolizing enzyme ALDH2 and suppressed phosphorylation of the cellular energy regulator AMPK without affecting AMPK expression. IGF-1 deficiency itself did not affect ALDH2 and AMPK expression and/or activation although LID rescued decreased ALDH2 expression and AMPK phosphorylation (Fig. 6).
Fig 5.
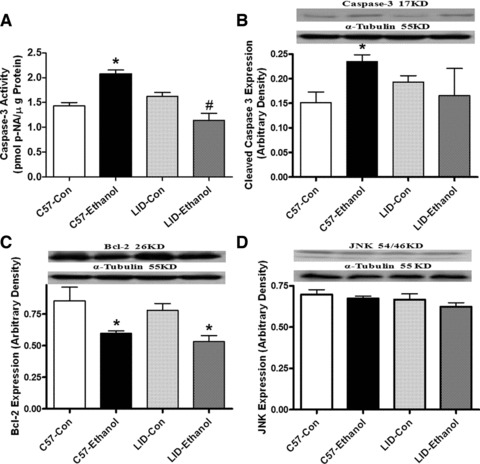
Caspase-3 activity (A) and protein expression of cleaved caspase-3 (B), Bcl-2 (C) and JNK (D) in myocardium from C57 and LID mice with or without chronic ethanol treatment. Insets: Representative gel blots of caspase-3, Bcl-2, JNK and α-tubulin (loading control) using specific antibodies against caspase-3, Bcl-2, JNK and α-tubulin. Mean ± S.E.M., n = 4–6 per group, *P < 0.05 versus C57 control (C57-Con) group.
Fig 6.
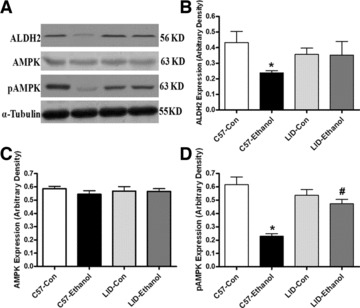
Protein expression of ALDH2 (B), AMPK (C) and pAMPK (D) in myocardium from C57 and LID mice with or without chronic ethanol treatment. (A) Representative gel blots of ALDH2, AMPK, pAMPK and α-tubulin (loading control) using specific antibodies against ALDH2, AMPK, pAMPK and α-tubulin. Mean ± S.E.M., n = 5–6 per group, *P < 0.05 versus C57 control (C57-Con) group, #P < 0.05 versus C57-Ethanol group.
Effect of AMPK activation on ethanol-induced changes in cell shortening
To further examine the role of AMPK activation in ethanol-induced cardiac contractile defects, freshly isolated cardiomyocytes from wild-type C57 mice were treated with ethanol (240 mg/dl) for 4 hrs in the absence or presence of the AMPK agonist AICAR (500 μM) [17]. Our data shown in Figure 7 indicated that the short-term treatment of ethanol significantly decreased PS and ± dL/dt as well as prolonged TR90 without affecting resting cell length and TPS in cardiomyocytes. Interestingly, AICAR significantly alleviated the short-term ethanol incubation-induced mechanical defects without eliciting any effects on cardiomyocyte mechanics by itself. These data provided direct evidence for a role of AMPK activation in the ethanol exposure-induced cardiac contractile dysfunction.
Fig 7.
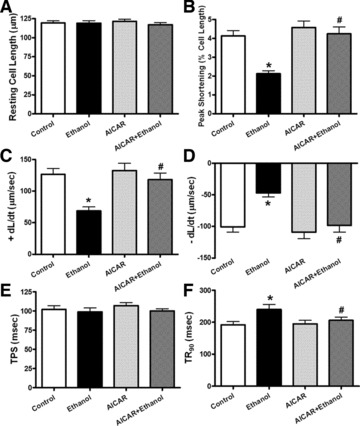
Effect of the AMPK activator AICAR on ethanol-induced cardiomyocyte contractile defects. Freshly isolated cardiomyocytes from normal mice were incubated with ethanol (240 mg/dl) in the presence or absence of AICAR (500 μM) for 4 hrs. (A) Resting cell length; (B) PS (normalized to resting cell length); (C) Maximal velocity of shortening (+dL/dt); (D) Maximal velocity of relengthening (–dL/dt); (E) TPS and (F) TR90. Mean ± S.E.M., n = 64–66 cells from three mice per group, *P < 0.05 versus control group, #P < 0.05 versus ethanol group.
Discussion
Data from our current study revealed that IGF-1 deficiency effectively attenuated chronic alcohol intake-induced cardiac contractile dysfunction, intracellular Ca2+ derangement, enlarged cardiomyocyte cross-sectional area and cardiac hypertrophy, enhanced expression or activity of key apoptotic marker caspase-3, down-regulated ALDH2 expression and dampened AMPK activation. Interestingly, the beneficial effect of IGF-1 deficiency against ethanol-elicited cardiac contractile response was ablated by replenishment of exogenous recombinant IGF-1. Moreover, the AMPK agonist AICAR effectively alleviated acute ethanol exposure-induced cardiomyocyte contractile dysfunction. These results have provided supporting evidence for a beneficial role of reduced IGF-1 levels in cardiac morphological, contractile and intracellular Ca2+ defects associated with chronic alcohol administration where AMPK activation may play a key role.
The hallmarks of alcoholic cardiomyopathy are cardiac hypertrophy and poor myocardial contractility [11, 12], somewhat similar to the characteristic cardiac morphological and functional changes in other forms of myopathic conditions such as diabetes and hypertension [20, 22]. This notion received support from our observation at the cardiomyocyte level of enlarged cross-sectional area and reduced cardiomyocyte shortening capacity (PS and ± dL/dt) in C57 mice following chronic alcohol consumption. Our data further revealed that impaired cardiomyocyte mechanical function following chronic alcohol ingestion may be underscored by intracellular Ca2+ mishandling (depressed basal, peak and rise in FFI). The prolonged intracellular Ca2+ decay time is in line with the prolonged relengthening duration (TR90) in cardiomyocytes from alcohol-fed mice. This is consistent with the earlier notion that impaired intracellular Ca2+ homeostasis such as compromised SR Ca2+ uptake and Ca2+ regulatory function may contribute to cardiac contractile dysfunction following chronic alcohol exposure [11, 12]. Interestingly, our data indicated that exacerbated cardiac depression with increasing stimulus frequencies (0.5 to 5.0 Hz) following chronic alcohol ingestion, suggesting a reduced SR Ca2+ loading capacity following alcohol intake. Although IGF-1 deficiency itself did not elicit any notable effect on cardiomyocyte contractile function and intracellular Ca2+ homeostasis, it attenuated or negated chronic alcoholism-induced cardiac hypertrophy (heart weight/size or cardiomyocyte cross-sectional area), cardiomyocyte contractile and intracellular Ca2+ abnormalities as well as intolerance to the increased stimulus frequency. Although IGF-1 is known to be essential to the maintenance of myocardial structure and contractile function [1], deficiency of the growth factor has been shown to rescue oxidative stress and aging-induced decline in myocardial contractile function [5, 6], supporting the apparently paradoxical role of IGF-1 deficiency in the preservation of cardiomyocyte mechanical function under pathological condition such as chronic alcoholism. It is noteworthy that IGF-1 deficiency appears to dampen cardiac contractile function early on in life [5, 6]. By mid age such as in the current experimental setting, severely IGF-1 deficiency did not exhibit overt cardiac morphological, contractile and intracellular Ca2+ alteration, indicating a possible role of the residue IGF-1 in LID mice in the maintenance of cardiac function over time.
Results from our study further revealed that IGF-1 deficiency-elicited beneficial effects against chronic alcohol intake-induced cardiac morphological, contractile and intracellular Ca2+ changes were correlated with antagonism against alcoholism-elicited increase in caspase-3 as well as reduction in ALDH2 and AMPK phosphorylation. Our data did not favour any major role of Bcl-2 or JNK in the IGF-1 deficiency elicited beneficial effect in alcoholic hearts. A plethora of evidence accumulated over the past decades has indicated a tie between AMPK signalling and cardiac function, which may underscore the regulation of cardiac function by growth factor, nutrient, intrinsic and extrinsic stress signals [23]. Our data revealed that chronic alcohol intake reduced AMPK activation, which is known to regulate apoptosis, cell metabolism, differentiation and DNA repair [23]. Regulation of these signal cascades in alcoholic hearts may play an important role in the development of the eventual alcoholic cardiomyopathy. IGF-1 deficiency significantly alleviated alcoholism-induced loss of AMPK activation and increase in apoptosis (manifested as enhanced expression of cleaved caspase-3 and caspase-3 activity), which is expected to be pivotal in both cell survival and cardiac function. Our earlier study using the very LID model depicted that IGF-1 deficiency is capable of activating AMPK [6]. AMPK has recently emerged as a chief regulator of energy balance, the activation of which plays an essential role in the protection against cardiac ischemia-reperfusion injury [23]. Reduced AMPK activation has been shown to be associated with ethanol challenge in hepatoma cells via inhibition of the AMPK upstream kinase LKB1 [24]. Data from our in vitro study added convincing evidence that AMPK activation itself using AICAR is capable of rescuing ethanol exposure-associated cardiomyocyte contractile dysfunction, consolidating a role of dampened AMPK activation in ethanol-induced cardiac dysfunction. In addition to AMPK, possible interactions between IGF-1 and alcohol at the convergence of Akt or PKC may also contribute to IGF-1 deficiency elicited cardioprotection against alcoholism. Alcohol has been shown to alter IGF-1-stimulated Akt phosphorylation and ultimately induction of critical gene expression through Akt signalling in brain [25]. On the other hand, chronic alcohol alters PKC isozyme expression and activity to suppress IGF-1 signalling in embryonic heart cells [26]. Nonetheless, further study is warranted to elucidate the precise interactions between chronic alcoholism and IGF-1 (or IGF-1 deficiency) on these signalling cascades and how these interactions contribute to the onset and development of alcoholic cardiac injury. Last but not the least, our current study revealed down-regulated ALDH2 in myocardium following alcohol challenge, the effect of which was reconciled by LID. These data favour a possibly role of ALDH2 in the IGF-1 deficiency-offered cardioprotection against alcoholic injury, consistent with the notion of ALDH2 against myocardial injury in response to chronic alcohol exposure and ischemia-reperfusion [19, 27].
In conclusion, our present study has indicated that IGF-1 deficiency abrogates chronic alcohol intake-elicited undesirable cardiac morphological and functional changes. Our findings are consistent with the recent report that IGF-1 deficiency enhances the overall resistance against the oxidative stress and aging-induced cardiomyocyte dysfunction [5, 6]. Our study suggests that AMPK phosphorylation and ALDH2 may contribute to the IGF-1 deficiency induced beneficial effects in cardiomyocytes. This is supported by our in vitro finding that the AMPK activator AICAR alleviates ethanol-induced cardiomyocyte contractile dysfunction. Given that moderate alcohol intake in healthy individuals results in a profound decline in circulating IGF-1 levels [28], it may be postulated that the loss in circulating IGF-1 in response to moderate alcohol intake may contribute to the beneficial effect of light-to-moderate alcohol intake on myocardial function. Nevertheless, little information is available with regards to the application of IGF-1 analogue(s) in clinical management against alcoholic complications. The precise role of the IGF-1 cascade and IGF-1-induced myocardial response following alcohol intake remains a puzzle for alcoholic cardiomyopathy. Our present study using LID mice should shed some lights towards the ultimate elucidation of this question.
Acknowledgments
The authors are grateful to Dr. Derek LeRoith from the Mount Sinai School of Medicine (New York, NY, USA) for providing the LID founder mice. Monoclonal antibody to ALDH2 was a generous gift by Dr. Henry Weiner from Purdue University, West Lafayette, IN, USA. Technical assistance in blood ethanol measurement from Dr. E. Kurt Dolence from University of Wyoming College of Health Sciences is greatly appreciated. This work was supported in part by NIH 1R01 AA013412INBRE and P20 RR016474 (J.R.).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Ren J, Samson WK, Sowers JR. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol. 1999;31:2049–61. doi: 10.1006/jmcc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 2.Gola M, Bonadonna S, Doga M, et al. Cardiovascular risk in aging and obesity: is there a role for GH. J Endocrinol Invest. 2005;28:759–67. doi: 10.1007/BF03347561. [DOI] [PubMed] [Google Scholar]
- 3.Yakar S, Sun H, Zhao H, et al. Metabolic effects of IGF-I deficiency: lessons from mouse models. Pediatr Endocrinol Rev. 2005;3:11–9. [PubMed] [Google Scholar]
- 4.Climent V, Marin F, Pico A, et al. Growth hormone therapy, is it always good for the heart. Eur Heart J. 2004;25:183. doi: 10.1016/j.ehj.2003.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Li Q, Yang X, Sreejayan N, et al. Insulin-like growth factor I deficiency prolongs survival and antagonizes paraquat-induced cardiomyocyte dysfunction: role of oxidative stress. Rejuvenation Res. 2007;10:501–12. doi: 10.1089/rej.2007.0552. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Ceylan-Isik AF, Li J, et al. Deficiency of insulin-like growth factor 1 reduces sensitivity to aging-associated cardiomyocyte dysfunction. Rejuvenation Res. 2008;11:725–33. doi: 10.1089/rej.2008.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corpas E, Harman SM, Blackman MR. Human growth-hormone and human aging. Endocrine Reviews. 1993;14:20–39. doi: 10.1210/edrv-14-1-20. [DOI] [PubMed] [Google Scholar]
- 8.Holzenberger M, Dupont J, Ducos B, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–7. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 9.Pecherskaya A, Rubin E, Solem M. Alterations in insulin-like growth factor-I signaling in cardiomyocytes from chronic alcohol-exposed rats. Alcohol Clin Exp Res. 2002;26:995–1002. doi: 10.1097/01.ALC.0000021149.50494.87. [DOI] [PubMed] [Google Scholar]
- 10.Rojdmark S, Brismar K. Decreased IGF-I bioavailability after ethanol abuse in alcoholics: partial restitution after short-term abstinence. J Endocrinol Invest. 2001;24:476–82. doi: 10.1007/BF03343879. [DOI] [PubMed] [Google Scholar]
- 11.Hintz KK, Relling DP, Saari JT, et al. Cardiac overexpression of alcohol dehydrogenase exacerbates cardiac contractile dysfunction, lipid peroxidation, and protein damage after chronic ethanol ingestion. Alcohol Clin Exp Res. 2003;27:1090–8. doi: 10.1097/01.ALC.0000075823.73536.DD. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Ren J. Cardiac overexpression of metallothionein rescues chronic alcohol intake-induced cardiomyocyte dysfunction: role of Akt, mammalian target of rapamycin and ribosomal p70s6 kinase. Alcohol Alcohol. 2006;41:585–92. doi: 10.1093/alcalc/agl080. [DOI] [PubMed] [Google Scholar]
- 13.Lang CH, Lynch CJ, Vary TC. Alcohol-induced IGF-I resistance is ameliorated in mice deficient for mitochondrial branched-chain aminotransferase. J Nutr. 2010;140:932–8. doi: 10.3945/jn.109.120501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dees WL, Srivastava V, Hiney JK. Actions and interactions of alcohol and insulin-like growth factor-1 on female pubertal development. Alcohol Clin Exp Res. 2009;33:1847–56. doi: 10.1111/j.1530-0277.2009.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yakar S, Liu JL, Stannard B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–9. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keane B, Leonard BE. Rodent models of alcoholism: a review. Alcohol Alcohol. 1989;24:299–309. doi: 10.1093/oxfordjournals.alcalc.a044916. [DOI] [PubMed] [Google Scholar]
- 17.Nyblom HK, Sargsyan E, Bergsten P. AMP-activated protein kinase agonist dose dependently improves function and reduces apoptosis in glucotoxic beta-cells without changing triglyceride levels. J Mol Endocrinol. 2008;41:187–94. doi: 10.1677/JME-08-0006. [DOI] [PubMed] [Google Scholar]
- 18.Yang X, Doser TA, Fang CX, et al. Metallothionein prolongs survival and antagonizes senescence-associated cardiomyocyte diastolic dysfunction: role of oxidative stress. FASEB J. 2006;20:1024–6. doi: 10.1096/fj.05-5288fje. [DOI] [PubMed] [Google Scholar]
- 19.Doser TA, Turdi S, Thomas DP, et al. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119:1941–9. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Fang CX, Dong F, Thomas DP, et al. Hypertrophic cardiomyopathy in high-fat diet-induced obesity: role of suppression of forkhead transcription factor and atrophy gene transcription. Am J Physiol Heart Circ Physiol. 2008;295:H1206–15. doi: 10.1152/ajpheart.00319.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Wold LE, Relling DP, Duan J, et al. Abrogated leptin-induced cardiac contractile response in ventricular myocytes under spontaneous hypertension: role of Jak/STAT pathway. Hypertension. 2002;39:69–74. doi: 10.1161/hy0102.100777. [DOI] [PubMed] [Google Scholar]
- 23.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009:89–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 24.Liangpunsakul S, Wou SE, Zeng Y, et al. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1173–81. doi: 10.1152/ajpgi.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiney JK, Srivastava VK, Les DW. Insulin-like growth factor-1 stimulation of hypothalamic KiSS-1 gene expression is mediated by Akt: effect of alcohol. Neuroscience. 2010;166:625–32. doi: 10.1016/j.neuroscience.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ila R, Solem M. Chronic-alcohol exposure alters IGF1 signaling in H9c2 cells via changes in PKC delta. Alcohol. 2006;39:169–78. doi: 10.1016/j.alcohol.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Chen CH, Budas GR, Churchill EN, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rojdmark S, Rydvald Y, Aquilonius A, et al. Insulin-like growth factor (IGF)-1 and IGF-binding protein-1 concentrations in serum of normal subjects after alcohol ingestion: evidence for decreased IGF-1 bioavailability. Clin Endocrinol. 2000;52:313–8. doi: 10.1046/j.1365-2265.2000.00908.x. [DOI] [PubMed] [Google Scholar]