

Summary
Structural genomics (SG) programs have developed during the last decade many novel methodologies for faster and more accurate structure determination. These new tools and approaches led to determination of thousands of protein structures. The generation of enormous amounts of experimental data resulted in significant improvements in the understanding of many biological processes at molecular levels. However, the amount of data collected so far is so large that traditional analysis methods are limiting the rate of extraction of biological and biochemical information from 3-D models. This situation has prompted us to review the challenges that remain unmet by structural genomics, as well as the areas in which the potential impact of SG could exceed what has been achieved so far.
Introduction
Structural biology provides invaluable information necessary for understanding the functions of living organisms at a molecular level. However, the number of known protein sequences is growing so rapidly that, despite enormous advances in structure determination protocols, the gap between genomic and structural information is widening. To counteract this trend, a number of structural genomics (SG) programs were created, with the aim of providing ways to limit such disparities. The original task for many public SG centers in the United States was to maximize the structural coverage of protein sequence space through careful target selection and bioinformatics tools, such as homology modeling. Unfortunately, even the most sophisticated bioinformatics tools are not able to classify many gene products, and approximately 30–40% of them are classified as “hypothetical proteins” [1].
SG centers are using (or have used) different approaches to structurally characterize the protein world as completely as possible. For example, the National Institutes of Health (NIH) Protein Structure Initiative (PSI and PSI2) centers focus on structural studies of the representative members of the largest proteins families [2], proteins from human parasites [3], and Mycobacterium tuberculosis [4]. The human proteome was chosen as the target for the Structural Genomics Consortium (SGC) and RIKEN [5], while two National Institute of Allergy and Infectious Diseases (NIAID) funded centers (the Center for Structural Genomics of Infectious Diseases (CSGID) [6] and the Seattle Structural Genomics Center for Infectious Disease (SSGCID) [7]) are determining the structures of proteins from major human pathogens. SPINE2, the successor of Structural Proteomics In Europe – SPINE [8], concentrates on structures of complexes from signaling pathways linking immunology, neurobiology and cancer (http://www.spine2.eu/SPINE2/). Additionally, most SG centers have dedicated a significant part of their efforts to the development of high-throughput (and preferably high-output) methodology, which may now also be used for fast and more accurate determination of structures by both X-ray crystallography and NMR techniques in laboratories not involved in SG efforts. Work on thousands of target proteins has led to the development of efficient protocols for each of the steps of the structure determination process. New experimental protocols that were developed through SG efforts have shifted over time the so-called ‘bottlenecks’ in the pipeline, and it seems that, at present, the analysis of 3-D structures in the context of all biological (functional) and bioinformatics information is the slowest step of the whole process. Similarly, many problems dealing with protein production, crystallization, and other steps on the road from sequence to function can be identified or even overcome by analysis of all available biochemical and bioinformatics data.
During the last decade, SG efforts have shown that it is possible to create pipelines able to generate roughly 200 novel structures per year in a single center. Contrary to expectations, SG pipelines were able to solve many structures that were very difficult. For example, the Midwest Center for Structural Genomics (MCSG) structure [PDB code 3N99] has the best quality parameters among the structures solved to a similar resolution (2.4 Å), despite having a molecular mass close to 1 megadalton in the asymmetric unit.
In this review we would like to concentrate mainly on the application of X-ray diffraction in SG [9], as this methodology is currently used for the determination of ~80% of SG structures. We will address the challenges of SG programs and/or approaches that were not met or areas where the potential impact of SG could surpass that which has been achieved so far. The unmet challenges should not be treated as failures but rather as opportunities to open avenues to new, exciting research.
Protein production and crystallization
A significant part of the SG effort is concentrated on production of soluble and pure proteins. Genes encoding such proteins are overexpressed in a variety of cell types (bacterial, yeast, insect, and mammalian), as well as in cell-free expression systems [10,11]. The latter are currently considered to provide the most important alternative to conventional in vivo expression [12], which has become especially useful in the production of isotopically labeled proteins for structure determination by NMR. Currently, a vast majority (83%) of over 280 protein structures deposited in the Protein Data Bank (PDB) and produced with the utilization of cell-free systems were determined by NMR. Most such proteins are of human origin (Fig. 1) and a majority of their structures were determined at RIKEN. Efforts to optimize expression have resulted in many new vectors, expression systems, and experimental protocols. Despite all these developments, the process from cloning a gene to producing milligram quantities of soluble protein requires substantial effort and resources, even for apparently “easy” bacterial targets. A similar situation is also present in the case of crystallization, and it is not surprising that attrition at these two stages causes a reduction of the overall yield of SG pipelines [13]. A silver bullet for crystallization remains undiscovered, although technologies developed and/or used on a large scale by SG efforts have improved the process. These include surface entropy reduction [14], large-scale reductive methylation of lysine residues [15], in situ proteolysis [16,17], nanoliter volume crystallization [18–21], and a significant degree of automation of the whole process.
Figure 1.

Source organisms of protein structures deposited in the PDB. (A) Structures of proteins produced in cell-free systems. (B) Structures produced by SG centers utilizing E. coli expression systems. Structures determined by NMR and X-ray diffraction methods are separated. “Other” indicates proteins originating mainly from bacterial genomes.
A search of the literature and analysis of PDB deposits [22] shows that the advantage of the unusual stability of many target proteins originating from extremophilic bacteria has not been well exploited to date. Although temperature was shown to be an important factor affecting crystallization, there has been no reported systematic effort to use temperatures above ambient for preparation of proteins from thermophilic organisms such as Thermotoga maritima [22,23]. This is rather surprising, since, based on analysis by the Joint Center for Structural Genomics (JCSG) of the crystallization conditions for several hundred proteins from T. maritima, it has been shown that for the two temperatures of crystallization tested (4 and 20 °C), the higher one gave 25% more hits [24]. Moreover, except for membrane proteins, there is no information about optimization by SG groups of crystallization screens toward particular groups or families of proteins. In contrast, multiple non-SG groups have used crystallization information in the Biological Macromolecule Crystallization Database (BMCD) to identify the best crystallization conditions for particular protein families [25,26]. It seems that the first generation of automation inhibited the flexibility necessary to treat various groups of target proteins differently. For example, application of so-called crystallization chaperones has also been very limited, most likely due to their relatively high cost and to the need for time-demanding protocols [27,28].
Due to the high degree of automation and to the application of databases, the results of SG crystallization experiments have been relatively well analyzed and both the successful and failed experiments could be considered in order to develop new ones or to modify existing experimental protocols [15,16,29–31]. In addition, SG deposits tend to contain more information on crystallization conditions than deposits coming from traditional structural biology laboratories. In the case of SG, 98% of PDB deposits contain information on crystallization, whereas such information is available for only 86% of non-SG deposits (93% of deposits after October 1, 2000). Such improvement in the completeness of PDB deposits improves the reliability of data mining approaches, especially those which are based on successful experiments. However, the lack of access to all failed experiments limits our ability to extract the entirety of the statistically useful information.
Quality of data and of the resulting structures
Different SG centers not only choose different strategies for target selection, but also use different methodologies for structure determination. As has been noted, the average quality of X-ray structures solved by SG is the same or even better than the average quality of structures determined by traditional structural biology [32]. However, there are quite significant differences in the quality of experimental 3-D structural models elucidated by various consortia and the reasons for such discrepancies merit analysis. Surprisingly, despite almost 5000 structures that have been solved by PSI centers, there is still no precise definition of the terms that could be universally used to assess the quality of macromolecular structures. In an ideal world the resolution should be used as the main parameter describing the accuracy of the model that is coming from a diffraction experiment. However, the resolution limit of diffraction data is not always determined consistently, or, for that matter, even correctly. In most cases, the reported resolution limit depends mainly on the experience of the experimenter, who uses data completeness, I/σI, and Rmerge values to decide which data to use in structure determination and refinement [33]. As a result, two structures reporting the same “nominal” resolution may in fact have significantly different “real” resolution limits, due to measurable differences in the mean I/σI in the highest resolution shell [34]. Structures that were determined using data cutoff at a resolution lower than the true crystal diffraction limits should be of higher quality than other structures determined at the same resolution, if no mistakes that would cancel the benefits of artificially lowered resolution have been made.
Several SG centers (CSGID, JCSG, and SSGCID) make diffraction images publicly available. Availability of such “raw” experimental data not only provides an opportunity to fully examine data quality, but also gives an opportunity to use data for the development of new crystallographic protocols and tools, as well as a unique opportunity to use such data for training purposes. Traditionally, it has been argued that prohibitive cost of storage makes such data availability impractical. In fact, the cost of the hardware to store the raw data from all PDB structures is now much lower than the cost of determining a single structure in the most efficient SG center [9]. The availability of raw diffraction data may further improve validation of macromolecular models.
Some SG centers have developed their own validation protocols, which in many cases use popular validation programs available to the structural biology community since the 1990s. MOLPROBITY [35] is probably the only recently developed validation program that has made a significant impact on the quality of structures. Utilization of this software has led to improvement of the overall quality of structures deposited in the PDB. However, there are still no generally accepted, well-defined criteria describing the quality of PDB deposits. It may be a missed opportunity that no quality standard was created before more than 5000 structures were deposited to the PDB by a single SG initiative (the PSI).
Mandatory deposition to the PDB of not only the coordinates, but also the experimental structure factors allows re-refinement of the published structures and independent evaluation of their quality. Unfortunately, the SG did not develop uniform procedures for interpretation of electron density maps, which limits the usefulness of the PDB for a wider biomedical audience. Uniformity of interpretation is especially important for the map regions of relatively poor quality. For example, in situations where the electron density for an amino acid side chain is missing, three different model-building approaches can be utilized. In the first approach, the side chain is placed in a chemically correct, but arbitrary orientation and its mobility is represented by high B-factor values. The second approach is similar to the first one; however, the occupancy of the missing part of the residue is set to zero. In the third approach, the side chain of the amino acid model is removed outright. Despite various valid arguments, none of these approaches is significantly better than the others, although removal of the missing fragments is most probably the best way to “transform” electron densities into an atomic model. This is especially true when one takes into account not only the mobility of side chains, but also the overall damage to the sample due to the radiation used for diffraction experiments [36].
Such differences in interpretation of the electron density may have a deleterious effect on subsequent analysis of the deposited structures. In fact, one must remember that every improvement of the protocols and software used for analysis of diffraction images, structure refinement, and validation allows for creation of better-fitting models. It was shown recently that a majority of the coordinates in the PDB with available structure factors can be refined further [37,38]. One might conclude that comparisons of the structure quality at the moment of the deposition should be performed not against all available structures, but rather only against the recently deposited structures. Even in the cases where data collection was performed optimally, the resolution of the data is high and the R-factor values of the model are low, there is no guarantee that the derived model is error-free [39].
Currently, re-refinement is the best method for validation of a structure deposited in the PDB (although not always the easiest or the fastest). One of the problems that may be encountered by a user of experimental data deposited in the PDB is the lack of a strict standard determining which and how data should be deposited. For example, a file containing reflection data may contain either experimental structure factors, intensities, or in some cases both. Even in a relatively easy case, such as data for a structure solved by SAD and refined using the same data set, it is not clear whether Bijvoet-pair-merged or -unmerged data should be deposited. Moreover, in many cases structures solved with SAD by the incorporation of Se-Met not only include methionine residues in the deposited model instead of the selenomethionine residues, but also improper residues are used during entire refinement procedure. A similar situation is observed in the case of proteins containing polyhistidine affinity tags (His-tags). The reporting and treatment of His-tags is very inconsistent, and, in extreme cases, the His-tag is reported in the sequence but the electron density clearly corresponding to it is not modeled, or the His-tag is omitted from the sequence although there is clear electron density for it. Such inconsistencies affect data mining attempts, e.g. aimed at analysis of the impact of the presence of a purification tag on crystallization. The lack of consistency is even more pronounced in more complicated situations, especially since the current form of the PDB header information does not allow for detailed description of the diffraction experiment. These inconsistencies cause situations where the originally reported R and Rfree values cannot be reproduced, for example due to missing information about the status of reflections [40]. However, thanks in part to the policy of the PSI SG initiative requiring deposition of structure factors along with structures to the PDB, protein crystallography rates above other methods in terms of having “raw” experimental data available. The recent push for deposition of cryoEM maps to the EMDB (http://www.petitiononline.com/cryoEM/petition.html) is at least 10 years behind the implementation of a universally adopted standard for submission of X-ray diffraction data to the PDB.
Homology modeling
Homology modeling is currently the most accurate computational technique able to generate three-dimensional models of proteins in cases when a de novo model derived from NMR or X-ray experiments is not available [41]. When sequence identity between the target and a template structure is low, determination of an accurate homology model becomes very difficult. In order to provide the largest possible number of templates for homology modeling, the PSI centers have concentrated their efforts on proteins with no known homologs in the PDB, defined as having less than 30% sequence identity to any protein with a deposited structure. Such a systematic effort to provide experimental data for structural coverage of genomes [1,2] resulted in a significant number of new templates for modeling [42,43]. Millions of models are now easily available through PSI webpage (www.proteinmodelportal.org/), as well as through other large databases of protein models [44,45]. Due to their lack of sequence similarity to previously deposited structures, SG targets are ideal cases for CASP (Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction competition [46]). In fact, in recent editions of CASP, SG groups have provided the vast majority (121 out of 128 in CASP8) of experimental structures used to assess the quality of homology modeling [47].
Even if well-defined templates for domains are available, modeling of larger complexes may be problematic as the determination of the relative orientation of different domains may be very difficult. In many such cases the use of small-angle X-ray scattering and/or cryo-electron microscopy techniques has been shown to be beneficial [48].
The PSI centers have been an invaluable source of structural data for tests of structure prediction algorithms. For example, 90% of targets in the CASP7 and CASP8 (Critical Assessment of Techniques for Proteins Structure Prediction) competitions originated from SG groups [47]. The structures of many new proteins have been found to be similar to already known structures despite the lack of detectable sequence similarity, and thus selection of these structures for SG efforts might be treated either as a failure of target selection for SG centers or as a success in identification of new physicochemical ways of accommodating existing folds. However, it is possible that protein molecules could be described using some new concepts, such as, for example, the protein meta-structure [49].
Small molecule ligands
Almost 70% of all crystal structures deposited to the PDB include one or more ligand(s) [34]. In many cases, the ligands could be as simple as metal cations (such as magnesium or calcium), or anions (such as sulfate, phosphate, etc.), but quite often the ligands are large and complicated molecules. The presence of ligands may create serious problems during structure validation, as the standard validation tools are not able to assess the “correctness” of many small molecule compounds. Taking into account that the functions of 26% of SG structures are unknown, it is also not surprising that SG structures contain unknown ligands more often than do those structures solved by traditional means (the fraction of structures with unknown ligands is 3.0% vs. 0.2%, respectively). In many cases, annotations are simply wrong, which may be worse for biologists than no annotation at all. Careful analysis of the electron density and of the details of ligand binding sites have led in some cases to the identification of the proper ligands and structure redeposition (e.g. 2NYD and 3LNL; 1PB6 and 3LOC). When performed correctly, such analysis, albeit time consuming, can provide considerable insights into the function of the protein in question [50].
One example of often overlooked method for identification of a bound ligand involves x-ray fluorescence measurements during data collection. Such experiments can provide unambiguous identification of metal ions, nevertheless do not answer the question of whether they are bound to the protein, or is simply present in solution. Rapid structure determination performed while the crystal is still mounted on the goniometer head allows for subsequent measurement(s) of anomalous differences, which can be used to generate a difference map that can precisely show the location of metal ions [51]. Unfortunately, these more complex experiments are performed only infrequently, despite the fact that 11% of SG structures contain transition metals (Cd, Co, Cu, Fe, Mn, Ni, and Zn). Our experience shows that fast structure determination is not only desirable, but also readily achievable in practice, and critical for determining whether sufficient data have been collected. The presence of ordered metals bound to a protein can also facilitate structure elucidation, if they are used for phasing. It should be readily apparent that proper identification of metal ions is a prerequisite for correct refinement and validation. The most common errors in metal ion refinement are caused by their misidentification during the refinement process. Most of these errors can be relatively easily spotted by analysis of distances between an ion and its coordinating atoms [52]. It is most likely that the lack of a proper validation tool capable of checking the geometrical likelihood of a metal-binding environment leads to a situation that the chemistry of metal coordination seems to be resolution-dependent. For example, the mean values for metal-oxygen distances calculated using high- or medium- resolution structures are typically different [52]. Such a phenomenon should not be observed if the refinement was carried out correctly. Furthermore, if proper metal-oxygen distance restraints are used, the standard deviations of distributions at different resolutions should be similar.
It is impossible to overestimate the importance of structure quality. Sometimes even a single incorrect structure may destroy data mining research. For example, analysis of the distances between Na+ ions and their coordinating oxygen atoms for PDB structures determined at the resolution of 1.2 Å or better shows a bimodal distribution, in contrast to the unimodal distribution observed for equivalent distances in small molecule compounds (Fig. 2). A more detailed analysis shows that the second maximum is caused by a single PDB deposit, 3FJ0 (Fig. 3). Re-refinement of this structure indicates that many water molecules were wrongly assigned as sodium cations, and after reinterpretation of the electron density and correction of the model, this “unusual” chemistry is no longer observed. The re-refined structure exhibit dramatically better R-factors, as well as more idealized geometrical parameters as analyzed by MOLPROBITY (Table 1). This is a telling example that shows why the structures used in bioinformatics analyses should be extremely carefully reviewed, taking into account not only the resolution or scope of the set, but also other parameters describing the correctness of the models.
Figure 2.

(A) Distribution of Na+-O distances in the PDB structures determined at resolution 1.2 Å or better (blue bars), and in the Cambridge Structural Database (CSD) (red bars). (B) The same distribution after re-refinement of a single structure (PDB code 3FJ0), which was solved by a traditional (i.e., non-SG) structural biology laboratory.
Figure 3.

Crystal structure of β-glucosidase (PDB code 3FJ0) [76]. (A) Overall structure shown in ribbon representation, with a reaction intermediate shown in stick representation. (B) The structure has an unusually large number of Na+ ions (purple spheres). Water molecules are marked as red spheres. The inset shows the binding site of the reaction intermediate in greater detail. The automatic procedures described in [37,38] do improve the R factors, but do not correct the misidentification of waters as sodium ions: after automatic rerefinement, the resulting structure contains the same erroneous number of Na+ atoms (252).
Table 1.
Refinement statistics for the PDB deposit 3FJ0 before and after correction.
| PDB code | 3FJ0 | Re-refined 3FJ0 |
|---|---|---|
| R (%) | 21.0 | 12.1 |
| Rfree (%) | 21.6 | 14.7 |
| Rmsd bond length (Å) | -* | 0.016 |
| Rmsd bond angles (°) | 1.2 | 1.6 |
| Number of non-H atoms | 4121 | 4159 |
| Number of water molecules | 234 | 550 |
| Clashscore | 8.12 | 4.50 |
| Clashscore percentile | 48 | 80 |
| Rotamer outliers (%) | 1.66 | 0 |
| MolProbity score | 1.67 | 1.23 |
| Molprobity score percentile | 54 | 91 |
| Ramachandran plot favored (%) | 97.71 | 98.63 |
Not reported in the deposit
SG structures and drug discovery
One of the frequently mentioned potential benefits of protein crystallography is its use in the process of drug discovery and development. However, the discovery of novel drugs is still extremely challenging and it is well appreciated that the success rate of such projects can be very disappointing [53]. The lack of optimism becomes even more profound when it is considered that despite growing research and development expenditures, the number of newly approved drugs is decreasing [54]. SG projects have not yet significantly influenced this field of research [34], although some preliminary results seem to be promising [4,55–58]. Moreover, it is possible that technologies developed by SG are not yet fully implemented in the traditional and commercial laboratories. As shown in Fig. 4, SG delivers structures at a much faster rate than traditional structural biology. In the future, the fast delivery of structures, and, more importantly, structures of small-molecule complexes of human proteins and/or proteins derived from different human pathogens may vastly benefit drug discovery programs [59,60].
Figure 4.
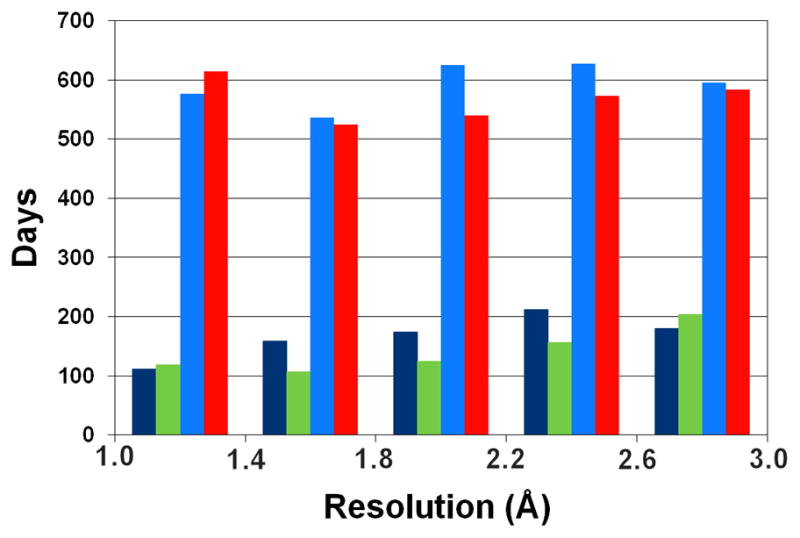
Average time (in days) between data collection and deposition for SG and non-SG structures. Dark blue and green bars represent SG structures, whereas light blue and red bars represent non-SG structures deposited in 2000–2004 and 2005–2009, respectively. Structures were binned by reported resolution limit (0.4 Å bin width).
SG had a definitely positive influence on homology modeling, but the same is not true for molecular docking. This is quite intriguing, since structures deposited by PSI centers could be very easily leveraged to create high quality test sets for docking studies. Even more importantly, such structures could be used for validation of more challenging modeling studies, such as molecular docking to homology models [61–63]. In addition to high diversity, the representative structures selected for such studies should have the highest possible quality, a criterion which is almost never examined when test sets of structures are selected from the PDB (Fig. 5).
Figure 5.


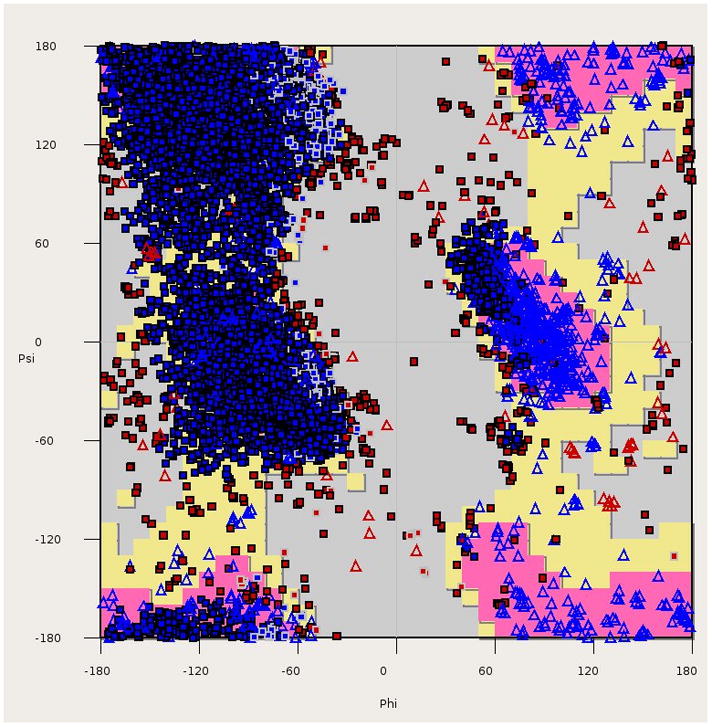
Quality indicators for protein structures. (A) Clashscore (calculated with MOLPROBITY) as a function of resolution for all crystal structures in the PDB (box plots) vs. the structures of protein targets used in evaluation of templates in molecular docking [77] (red circles), high-resolution docking (blue circle) [78] and modeling (green circles) [79]. The box plots are labeled as follows: red lines mark the clashscore median for a particular resolution range, the boxes include structures with clashscores between the 25th and 75th percentile, and the dashed lines include structures with clashscores between 25% − 1.5IQR (the interquartile range) and 75% + 1.5IQR. (B) R-factors as a function of resolution for crystal structures of protein targets used in evaluation of templates in molecular docking [77], high-resolution docking [78], and modeling [79]. Dark blue diamonds represent models with structure factors deposited, while light blue diamonds mark structures without structure factors. The blue line shows the linear regression of R-factor as a function of resolution for all PDB structures, while the green, purple, yellow and red lines are the analogous linear regression fits for structures deposited by SG in general, MCSG, JCSG and CSGID respectively. (C) A Ramachandran plot for all structures of protein targets used in evaluation of molecular docking [77]. This panel was created with COOT [80].
It is clear that the reliability of in silico screening of macromolecular ligands is a serious bottleneck in drug discovery research [34,64,65]. It is hard to estimate how the outcomes of in silico docking experiments are affected by the use of poor quality structures in analysis. Poor quality structures may negatively affect computational methods, both when they are used in docking studies and when they are used to test the algorithms implementing docking protocols. It is possible that some of the errors in the experimental models are eliminated during optimization of the models prior to ligand docking, especially when the ligand-binding site is known and its chemistry could be analyzed and corrected. These are areas that clearly merit further examination as SG efforts continue to expand and mature. Unfortunately, the use of poor quality structures cannot be avoided in some cases, as they represent the only available experimental models. In many cases, structures of very important drug targets were determined many years ago, using tools much less advanced than those currently available and used by SG. Many of such structures cannot be re-refined, since the structure factors were not deposited (Fig. 5).
From structure to function?
As many as 26% of all SG structures deposited to PDB are described as proteins of unknown function, or quite often their function is referred to as putative. The putative functions are most often assigned based on sequence similarity. High sequence identity usually allows for annotation transfer from a protein with a known function to the one that was newly directly investigated. Such transfer of an annotation is connected with some probability level that this annotation is true, yet the information about the probability that ‘transferred’ annotations are true is never shown. Experimenters must be especially careful checking gene or structure annotations which were done completely automatically. In many cases, curation of the data is necessary to avoid serious errors, caused, for example, by the use of the same names to describe different proteins across species [66]. Even a high degree of structural conservation does not guarantee that the function is also conserved. Some proteins having completely different functions may still have the same overall fold, and, if they are enzymes, very similar active sites.
Automation of the annotation process may not only fail at the sequence level, but may also be unsuccessful after experimental confirmation and publication of the protein’s function. This effect is mainly caused by the fact that the function of a protein is annotated differently in different databases, and annotation of a protein in a single database may also be tied to homologous proteins as well. Automated correction of one database or of one record in a database may not be propagated to other databases or records, respectively. Despite the fact that information about a protein’s function is explicitly stated in the title of the relevant publication, the function is sometimes marked as unknown elsewhere. Similarly, there are many PDB deposits that list unknown function in the deposit title despite of the fact that their authors published papers in which they established a function of the protein. Some efforts have used a collaborative method to expand and correct annotation information on sites such as Proteopedia [67] or TOPSAN (www.topsan.org). Another approach is to utilize the ISee concept, which uses an intuitive and interactive approach to disseminate structural information to the larger biomedical community [68].
Structural data provided by the SG community should be linked to a particular function and biological process. It turns out that, in many cases, the availability of a structure alone does not necessarily lead to properly assigned function. This situation prompted development of different bioinformatics approaches which should help in a search for functional clues [69]. The tools used to predict function from structure were developed not only by SG groups [70], but also by many scientists not involved directly in structural genomics [71]. It is not currently possible to evaluate the number of cases in which prediction of the function of SG targets from structure and/or sequence analysis was successful and subsequently verified by other experimental data [69].
On the other hand, the availability of a large number of purified proteins of unknown function also resulted in the development of experimental approaches directed to function assignment. For that purpose, some SG centers created panels of enzymatic assays [72,73] or tests for ligand binding [60,74].
Conclusions
During the last ten years SG programs have generated enormous amounts of experimental data. It seems that a major bottleneck of the whole program is the ability to analyze data and immediately leverage the derived information for optimization of experimental pipelines. For example, large attrition rates on the path from gene to soluble protein and crystals could be treated, at least partially, as a failure of the target selection process. Many of the target proteins should not have been selected for high-throughput programs in the first place, e.g. due to their intrinsic properties. Of course, for many targets the high probability of failure could not have been predicted at the time of their selection, and only later it was possible to learn from experimental techniques, such as light scattering or NMR, that such proteins might be difficult to crystallize.
Most probably, the major unmet challenge of SG is the insufficient rate of conversion of experimental data into biomedical information. In fact, this might be a result of the success of some SG programs that generated such vast amount of data that the currently available tools are not able to transform them into biologically useful information. Hopefully, creation of more sophisticated databases, like the PSI Structural Genomics Knowledgebase [75], will improve extraction of information. The existence of such a database may also prompt creation of stricter and more precise standards, for example for determining deposition of structural data. It seems that such a step may be not only beneficial for traditional structural biology, but also necessary for the success of all large-scale analysis attempts.
Acknowledgments
The authors would like to thank Matthew Zimmerman, Ian Wilson, Jack Johnson, Tom Terwilliger, Steve Almo, Samar Hasnain, Wayne Anderson, Andrzej Joachimiak, Zbyszek Dauter, and Heping Zheng for valuable comments on the manuscript. We especially thank Dr. Almo for turning our attention to structure libraries used for modeling studies. This work was supported by grants GM74942, GM53163, with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200700058C, and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Maksymilian Chruszcz, Email: maks@iwonka.med.virginia.edu.
Marcin Domagalski, Email: marcin_d@iwonka.med.virginia.edu.
Tomasz Osinski, Email: tomek_o@iwonka.med.virginia.edu.
Alexander Wlodawer, Email: wlodawer@nih.gov.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1••.Jaroszewski L, Li Z, Krishna SS, Bakolitsa C, Wooley J, Deacon AM, Wilson IA, Godzik A. Exploration of uncharted regions of the protein universe. PLoS Biol. 2009;7:e1000205. doi: 10.1371/journal.pbio.1000205. An analysis of the NIH PSI effort to determine representative structures of novel protein families. It arrives at the conclusion that the majority of these novel families represent highly divergent homologs of previously characterized protein families. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dessailly BH, Nair R, Jaroszewski L, Fajardo JE, Kouranov A, Lee D, Fiser A, Godzik A, Rost B, Orengo C. PSI-2: structural genomics to cover protein domain family space. Structure. 2009;17:869–881. doi: 10.1016/j.str.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan E, Baker D, Fields S, Gelb MH, Buckner FS, Van Voorhis WC, Phizicky E, Dumont M, Mehlin C, Grayhack E, et al. Structural genomics of pathogenic protozoa: an overview. Methods Mol Biol. 2008;426:497–513. doi: 10.1007/978-1-60327-058-8_33. [DOI] [PubMed] [Google Scholar]
- 4•.Ioerger TR, Sacchettini JC. Structural genomics approach to drug discovery for Mycobacterium tuberculosis. Curr Opin Microbiol. 2009;12:318–325. doi: 10.1016/j.mib.2009.04.006. A review of the methodology used by the Tuberculosis Structural Genomics Consortium. This review also addresses the impact of the Consortium on the development of treatments for drug-resistant tuberculosis. [DOI] [PubMed] [Google Scholar]
- 5•.Edwards A. Large-scale structural biology of the human proteome. Annu Rev Biochem. 2009;78:541–568. doi: 10.1146/annurev.biochem.78.070907.103305. A review that analyzed the impact of structural genomics on the determination of structures of human proteins. It identifies the most important protein families that are highly relevant for improvement of human health. [DOI] [PubMed] [Google Scholar]
- 6•.Anderson WF. Structural genomics and drug discovery for infectious diseases. Infect Disord Drug Targets. 2009;9:507–517. doi: 10.2174/187152609789105713. A description of the methods and approaches used by the Center for Structural Genomics of Infectious Diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Myler PJ, Stacy R, Stewart L, Staker BL, Van Voorhis WC, Varani G, Buchko GW. The Seattle Structural Genomics Center for Infectious Disease (SSGCID) Infect Disord Drug Targets. 2009;9:493–506. doi: 10.2174/187152609789105687. A description of the methods and approaches used by the Seattle Structural Genomics Center for Infectious Diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albeck S, Alzari P, Andreini C, Banci L, Berry IM, Bertini I, Cambillau C, Canard B, Carter L, Cohen SX, et al. SPINE bioinformatics and data-management aspects of high-throughput structural biology. Acta Crystallogr D Biol Crystallogr. 2006;62:1184–1195. doi: 10.1107/S090744490602991X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9••.Joachimiak A. High-throughput crystallography for structural genomics. Curr Opin Struct Biol. 2009;19:573–584. doi: 10.1016/j.sbi.2009.08.002. This review of high-throughput crystallography discusses the methodology and the resulting trends of this approach. It summarizes the application of synchrotron radiation, new phasing techniques, and automation in structure determination pipelines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makino S, Goren MA, Fox BG, Markley JL. Cell-free protein synthesis technology in NMR high-throughput structure determination. Methods Mol Biol. 2010;607:127–147. doi: 10.1007/978-1-60327-331-2_12. [DOI] [PubMed] [Google Scholar]
- 11.Tyler RC, Aceti DJ, Bingman CA, Cornilescu CC, Fox BG, Frederick RO, Jeon WB, Lee MS, Newman CS, Peterson FC, et al. Comparison of cell-based and cell-free protocols for producing target proteins from the Arabidopsis thaliana genome for structural studies. Proteins. 2005;59:633–643. doi: 10.1002/prot.20436. [DOI] [PubMed] [Google Scholar]
- 12.Torizawa T, Shimizu M, Taoka M, Miyano H, Kainosho M. Efficient production of isotopically labeled proteins by cell-free synthesis: a practical protocol. J Biomol NMR. 2004;30:311–325. doi: 10.1007/s10858-004-3534-2. [DOI] [PubMed] [Google Scholar]
- 13.Payne DJ, Gwynn MN, Holmes DJ, Rosenberg M. Genomic approaches to antibacterial discovery. Methods Mol Biol. 2004;266:231–259. doi: 10.1385/1-59259-763-7:231. [DOI] [PubMed] [Google Scholar]
- 14••.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallogr D Biol Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. A description of different approaches for engineering protein surfaces in order to improve the success rate of crystallization experiments. [DOI] [PubMed] [Google Scholar]
- 15•.Kim Y, Quartey P, Li H, Volkart L, Hatzos C, Chang C, Nocek B, Cuff M, Osipiuk J, Tan K, et al. Large-scale evaluation of protein reductive methylation for improving protein crystallization. Nat Methods. 2008;5:853–854. doi: 10.1038/nmeth1008-853. The most extensive study analyzing the effects of reductive lysine methylation on protein crystallization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Dong A, Xu X, Edwards AM, Chang C, Chruszcz M, Cuff M, Cymborowski M, Di Leo R, Egorova O, Evdokimova E, et al. In situ proteolysis for protein crystallization and structure determination. Nat Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. A description of a large scale application of the in situ proteolysis approach. This methodology was successfully utilized in a number of projects for which crystals could not be obtained using traditional methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Wernimont A, Edwards A. In situ proteolysis to generate crystals for structure determination: an update. PLoS One. 2009;4:e5094. doi: 10.1371/journal.pone.0005094. An update of recent developments and improvements of the in situ proteolysis method. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerdts CJ, Elliott M, Lovell S, Mixon MB, Napuli AJ, Staker BL, Nollert P, Stewart L. The plug-based nanovolume Microcapillary Protein Crystallization System (MPCS) Acta Crystallogr D Biol Crystallogr. 2008;64:1116–1122. doi: 10.1107/S0907444908028060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hazes B, Price L. A nanovolume crystallization robot that creates its crystallization screens on-the-fly. Acta Crystallogr D Biol Crystallogr. 2005;61:1165–1171. doi: 10.1107/S0907444905017336. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Mustafi D, Fu Q, Tereshko V, Chen DL, Tice JD, Ismagilov RF. Nanoliter microfluidic hybrid method for simultaneous screening and optimization validated with crystallization of membrane proteins. Proc Natl Acad Sci U S A. 2006;103:19243–19248. doi: 10.1073/pnas.0607502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng B, Gerdts CJ, Ismagilov RF. Using nanoliter plugs in microfluidics to facilitate and understand protein crystallization. Curr Opin Struct Biol. 2005;15:548–555. doi: 10.1016/j.sbi.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koclega KD, Chruszcz M, Zimmerman MD, Bujacz G, Minor W. ‘Hot’ macromolecular crystals. Cryst Growth Des. 2009;10:580. doi: 10.1021/cg900971h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Thiele I, Weekes D, Li Z, Jaroszewski L, Ginalski K, Deacon AM, Wooley J, Lesley SA, Wilson IA, et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325:1544–1549. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Page R, Grzechnik SK, Canaves JM, Spraggon G, Kreusch A, Kuhn P, Stevens RC, Lesley SA. Shotgun crystallization strategy for structural genomics: an optimized two-tiered crystallization screen against the Thermotoga maritima proteome. Acta Crystallogr D Biol Crystallogr. 2003;59:1028–1037. doi: 10.1107/s0907444903007790. [DOI] [PubMed] [Google Scholar]
- 25.Farr RG, Perryman AL, Samudzi CT. Re-clustering the database for crystallization of macromolecules. Journal of Crystal Growth. 1998;183:653–668. [Google Scholar]
- 26.Hennessy D, Buchanan B, Subramanian D, Wilkosz PA, Rosenberg JM. Statistical methods for the objective design of screening procedures for macromolecular crystallization. Acta Crystallogr D Biol Crystallogr. 2000;56:817–827. doi: 10.1107/s0907444900004261. [DOI] [PubMed] [Google Scholar]
- 27.Koide S. Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol. 2009;19:449–457. doi: 10.1016/j.sbi.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kossiakoff AA, Koide S. Understanding mechanisms governing protein-protein interactions from synthetic binding interfaces. Curr Opin Struct Biol. 2008;18:499–506. doi: 10.1016/j.sbi.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Price WN, 2nd, Chen Y, Handelman SK, Neely H, Manor P, Karlin R, Nair R, Liu J, Baran M, Everett J, et al. Understanding the physical properties that control protein crystallization by analysis of large-scale experimental data. Nat Biotechnol. 2009;27:51–57. doi: 10.1038/nbt.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik A. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics. 2007;23:3403–3405. doi: 10.1093/bioinformatics/btm477. [DOI] [PubMed] [Google Scholar]
- 31•.Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, et al. Protein production and purification. Nat Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. A broad review of the methods used by several different structural genomics projects for protein production and purification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. A detailed analysis of 3D macromolecular models determined by structural genomics consortia. The quality of SG models is compared with the quality of the structures elucidated by traditional structural biology projects. [DOI] [PubMed] [Google Scholar]
- 33•.Chruszcz M, Borek D, Domagalski M, Otwinowski Z, Minor W. Structural Genomics, Part C. Elsevier Academic Press Inc; 2009. X-ray diffraction experiment - the last experiment in the structure elucidation process; pp. 23–40. Advances in Protein Chemistry and Structural Biology, vol 77. A description of experimental protocols used in collecting macromolecular X-ray diffraction data. This paper describes current experimental trends and provides statistical analysis of diffraction data reported to the PDB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34••.Grabowski M, Chruszcz M, Zimmerman MD, Kirillova O, Minor W. Benefits of structural genomics for drug discovery research. Infect Disord Drug Targets. 2009;9:459–474. doi: 10.2174/187152609789105704. A review of the possible impact of SG programs on drug discovery research. This review provides insights into the strengths and weaknesses of SG programs and the role of such programs in drug development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Borek D, Cymborowski M, Machius M, Minor W, Otwinowski Z. Diffraction data analysis in the presence of radiation damage. Acta Crystallogr D Biol Crystallogr. 2010;66:426–436. doi: 10.1107/S0907444909040177. A discussion of radiation damage and its influence on diffraction data collection, processing and phasing. The paper analyzes the effects of radiation damage on the crystal sample and also shows how sample decay might influence the proper choice of experimental strategy during the diffraction experiment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joosten RP, Vriend G. PDB improvement starts with data deposition. Science. 2007;317:195–196. doi: 10.1126/science.317.5835.195. [DOI] [PubMed] [Google Scholar]
- 38.Joosten RP, Womack T, Vriend G, Bricogne G. Re-refinement from deposited X-ray data can deliver improved models for most PDB entries. Acta Crystallogr D Biol Crystallogr. 2009;65:176–185. doi: 10.1107/S0907444908037591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39••.Wlodawer A, Minor W, Dauter Z, Jaskolski M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. Febs J. 2008;275:1–21. doi: 10.1111/j.1742-4658.2007.06178.x. A review discussing in detail all aspects of structure quality and the limitations of X-ray crystallography. A “must read” for scientists who are entering the field of structural biology, or need to use structural biology results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joosten RP, Salzemann J, Bloch V, Stockinger H, Berglund AC, Blanchet C, Bongcam-Rudloff E, Combet C, Da Costa AL, Deleage G, et al. PDB_REDO: automated re-refinement of X-ray structure models in the PDB. Journal of Applied Crystallography. 2009;42:376–384. doi: 10.1107/S0021889809008784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J. Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry. 2010;49:2987–2998. doi: 10.1021/bi902153g. Practical application of computational methods for modeling protein structures with the Rosetta suite. This paper presents several of the most popular computational methods utilized in that suite of programs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moult J. Comparative modeling in structural genomics. Structure. 2008;16:14–16. doi: 10.1016/j.str.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Montelione GT, Rost B. Novel leverage of structural genomics. Nat Biotechnol. 2007;25:849–851. doi: 10.1038/nbt0807-849. [DOI] [PubMed] [Google Scholar]
- 44.Kanou K, Hirata T, Iwadate M, Terashi G, Umeyama H, Takeda-Shitaka M. HUMAN FAMSD-BASE: high quality protein structure model database for the human genome using the FAMSD homology modeling method. Chem Pharm Bull (Tokyo) 2010;58:66–75. doi: 10.1248/cpb.58.66. [DOI] [PubMed] [Google Scholar]
- 45.Yura K, Yamaguchi A, Go M. Coverage of whole proteome by structural genomics observed through protein homology modeling database. J Struct Funct Genomics. 2006;7:65–76. doi: 10.1007/s10969-006-9010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Critical assessment of methods of protein structure prediction - Round VIII. Proteins. 2009;77 (Suppl 9):1–228. doi: 10.1002/prot.22589. [DOI] [PubMed] [Google Scholar]
- 47.Tress ML, Ezkurdia I, Richardson JS. Target domain definition and classification in CASP8. Proteins. 2009;77 (Suppl 9):10–17. doi: 10.1002/prot.22497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konrat R. The protein meta-structure: a novel concept for chemical and molecular biology. Cell Mol Life Sci. 2009;66:3625–3639. doi: 10.1007/s00018-009-0117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Binkowski TA, Joachimiak A. Protein functional surfaces: global shape matching and local spatial alignments of ligand binding sites. BMC Struct Biol. 2008;8:45. doi: 10.1186/1472-6807-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ascone I, Strange R. Biological X-ray absorption spectroscopy and metalloproteomics. J Synchrotron Radiat. 2009;16:413–421. doi: 10.1107/S0909049509010425. [DOI] [PubMed] [Google Scholar]
- 52.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. Data mining of metal ion environments present in protein structures. J Inorg Biochem. 2008;102:1765–1776. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53••.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. An excellent review of the difficulties in finding new antibacterial drugs. It presents the major challenges of drug discovery programs. [DOI] [PubMed] [Google Scholar]
- 54.Shah S, Federoff HJ. Drug discovery dilemma and Cura quartet collaboration. Drug Discov Today. 2009;14:1006–1010. doi: 10.1016/j.drudis.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Arcus VL, Lott JS, Johnston JM, Baker EN. The potential impact of structural genomics on tuberculosis drug discovery. Drug Discov Today. 2006;11:28–34. doi: 10.1016/S1359-6446(05)03667-6. [DOI] [PubMed] [Google Scholar]
- 56.Weigelt J, McBroom-Cerajewski LD, Schapira M, Zhao Y, Arrowsmith CH. Structural genomics and drug discovery: all in the family. Curr Opin Chem Biol. 2008;12:32–39. doi: 10.1016/j.cbpa.2008.01.045. [DOI] [PubMed] [Google Scholar]
- 57.Artz JD, Dunford JE, Arrowood MJ, Dong A, Chruszcz M, Kavanagh KL, Minor W, Russell RG, Ebetino FH, Oppermann U, et al. Targeting a uniquely nonspecific prenyl synthase with bisphosphonates to combat cryptosporidiosis. Chem Biol. 2008;15:1296–1306. doi: 10.1016/j.chembiol.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 58••.Weigelt J. Structural genomics-Impact on biomedicine and drug discovery. Exp Cell Res. 2010 doi: 10.1016/j.yexcr.2010.02.041. An outline of the impact of SG programs on drug discovery. The impact of structural genomics is analyzed in several different areas, such as technology, structures and methodology development. [DOI] [PubMed] [Google Scholar]
- 59.Edwards AM, Bountra C, Kerr DJ, Willson TM. Open access chemical and clinical probes to support drug discovery. Nat Chem Biol. 2009;5:436–440. doi: 10.1038/nchembio0709-436. [DOI] [PubMed] [Google Scholar]
- 60.Van Voorhis WC, Hol WG, Myler PJ, Stewart LJ. The role of medical structural genomics in discovering new drugs for infectious diseases. PLoS Comput Biol. 2009;5:e1000530. doi: 10.1371/journal.pcbi.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 62••.Kalyanaraman C, Imker HJ, Fedorov AA, Fedorov EV, Glasner ME, Babbitt PC, Almo SC, Gerlt JA, Jacobson MP. Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening. Structure. 2008;16:1668–1677. doi: 10.1016/j.str.2008.08.015. A description of a computational approach which was successfully used in the determination of enzymatic function. This approach combines homology modeling and ligand docking methods, the results of which could be validated by structural biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song L, Kalyanaraman C, Fedorov AA, Fedorov EV, Glasner ME, Brown S, Imker HJ, Babbitt PC, Almo SC, Jacobson MP, et al. Prediction and assignment of function for a divergent N-succinyl amino acid racemase. Nat Chem Biol. 2007;3:486–491. doi: 10.1038/nchembio.2007.11. [DOI] [PubMed] [Google Scholar]
- 64.Antonyuk S, Strange RW, Hasnain SS. Structural discovery of small molecule binding sites in Cu-Zn human superoxide dismutase familial amyotrophic lateral sclerosis mutants provides insights for lead optimization. J Med Chem. 2010;53:1402–1406. doi: 10.1021/jm9017948. [DOI] [PubMed] [Google Scholar]
- 65.Nowak RJ, Cuny GD, Choi S, Lansbury PT, Ray SS. Improving binding specificity of pharmacological chaperones that target mutant superoxide dismutase-1 linked to familial amyotrophic lateral sclerosis using computational methods. J Med Chem. 2010;53:2709–2718. doi: 10.1021/jm901062p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaudet P, Lane L, Fey P, Bridge A, Poux S, Auchincloss A, Axelsen K, Braconi Quintaje S, Boutet E, Brown P, et al. Collaborative annotation of genes and proteins between UniProtKB/Swiss-Prot and dictyBase. Database (Oxford) 2009;2009:bap016. doi: 10.1093/database/bap016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hodis E, Prilusky J, Martz E, Silman I, Moult J, Sussman JL. Proteopedia - a scientific ‘wiki’ bridging the rift between three-dimensional structure and function of biomacromolecules. Genome Biol. 2008;9:R121. doi: 10.1186/gb-2008-9-8-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raush E, Totrov M, Marsden BD, Abagyan R. A new method for publishing three-dimensional content. PLoS One. 2009;4:e7394. doi: 10.1371/journal.pone.0007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:W89–93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gherardini PF, Helmer-Citterich M. Structure-based function prediction: approaches and applications. Brief Funct Genomic Proteomic. 2008;7:291–302. doi: 10.1093/bfgp/eln030. [DOI] [PubMed] [Google Scholar]
- 72.Proudfoot M, Kuznetsova E, Sanders SA, Gonzalez CF, Brown G, Edwards AM, Arrowsmith CH, Yakunin AF. High throughput screening of purified proteins for enzymatic activity. Methods Mol Biol. 2008;426:331–341. doi: 10.1007/978-1-60327-058-8_21. [DOI] [PubMed] [Google Scholar]
- 73.Baran R, Reindl W, Northen TR. Mass spectrometry based metabolomics and enzymatic assays for functional genomics. Curr Opin Microbiol. 2009;12:547–552. doi: 10.1016/j.mib.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 74.Nettleship JE, Brown J, Groves MR, Geerlof A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 2008;426:299–318. doi: 10.1007/978-1-60327-058-8_19. [DOI] [PubMed] [Google Scholar]
- 75.Berman HM, Westbrook JD, Gabanyi MJ, Tao W, Shah R, Kouranov A, Schwede T, Arnold K, Kiefer F, Bordoli L, et al. The protein structure initiative structural genomics knowledgebase. Nucleic Acids Res. 2009;37:D365–368. doi: 10.1093/nar/gkn790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nam KH, Sung MW, Hwang KY. Structural insights into the substrate recognition properties of beta-glucosidase. Biochem Biophys Res Commun. 2010;391:1131–1135. doi: 10.1016/j.bbrc.2009.12.038. [DOI] [PubMed] [Google Scholar]
- 77.Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A. Molecular docking screens using comparative models of proteins. J Chem Inf Model. 2009;49:2512–2527. doi: 10.1021/ci9003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Movshovitz-Attias D, London N, Schueler-Furman O. On the use of structural templates for high-resolution docking. Proteins. 2010;78:1939–1949. doi: 10.1002/prot.22710. [DOI] [PubMed] [Google Scholar]
- 79.Kundrotas PJ, Vakser IA. Accuracy of protein-protein binding sites in high-throughput template-based modeling. PLoS Comput Biol. 2010;6:e1000727. doi: 10.1371/journal.pcbi.1000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]