

Abstract
Background
R gene-mediated resistance is one of the most effective mechanisms of immunity against pathogens in plants. To date some components that regulate the primary steps of plant immunity have been isolated, however, the molecular dissection of defense signaling downstream of the R proteins remains to be completed. In addition, R genes are known to be highly variable, however, the molecular mechanisms responsible for this variability remain obscure.
Results
To identify novel factors required for R gene-mediated resistance in rice, we used rice insertional mutant lines, induced by the endogenous retrotransposon Tos17, in a genetic screening involving the rice blast fungus Magnaporthe oryzae. We inoculated 41,119 mutant lines with the fungus using a high throughput procedure, and identified 86 mutant lines with diminished resistance. A genome analysis revealed that 72 of the 86 lines contained mutations in a gene encoding a nucleotide binding site (NBS) and leucine rich repeat (LRR) domain-containing (NBS-LRR) protein. A genetic complementation analysis and a pathogenesis assay demonstrated that this NBS-LRR gene encodes Pish, which confers resistance against races of M. oryzae containing avrPish. The other 14 lines have intact copies of the Pish gene, suggesting that they may contain mutations in the signaling components downstream of Pish. The genome analysis indicated that Pish and its neighboring three NBS-LRR genes are high similar to one another and are tandemly located. An in silico analysis of a Tos17 flanking sequence database revealed that this region is a "hot spot" for insertion. Intriguingly, the insertion sites are not distributed evenly among these four NBS-LRR genes, despite their similarity at the sequence and expression levels.
Conclusions
In this work we isolated the R gene Pish, and identified several other mutants involved in the signal transduction required for Pish-mediated resistance. These results indicate that our genetic approach is efficient and useful for unveiling novel aspects of defense signaling in rice. Furthermore, our data provide experimental evidence that R gene clusters have the potential to be highly preferred targets for transposable element insertions in plant genomes. Based on this finding, a possible mechanism underlying the high variability of R genes is discussed.
Background
Plants have evolved elaborate defense mechanisms to protect themselves from many kinds of pathogens, including fungi, bacteria, viruses, and insects. Defense responses governed by the gene-for-gene hypothesis are triggered in plants when the product of a plant resistance (R) gene directly or indirectly recognizes a specific pathogen effector molecule, which is often the product of a pathogen avirulence (avr) gene [1]. The absence or inactivation of either member of this gene pair results in susceptibility of the host to the pathogen. To date more than 40 R genes have been isolated from several plant species, and most of them exhibit highly conserved structures, despite differences between the types of pathogens that are recognized. Pathogen recognition by any R protein initiates a common set of defense responses, including the production of reactive oxygen species (ROS), expression of pathogen-related (PR) genes, and localized programmed cell death at the site of pathogen challenge, which is known as the hypersensitive response (HR) [2]. This suggests that common downstream components may be shared by R proteins within and among plant species.
The most prevalent class of plant R proteins contains a nucleotide binding site (NBS) and a leucine-rich repeat (LRR) region, and are thus called NBS-and-LRR containing (NBS-LRR) proteins. The NBS domain is a functional ATPase and probably regulates the activity of the R proteins [3]. The LRR domain is required for specific recognition of the pathogen containing corresponding avirulence gene, and changing even a single amino acid in this region alters its recognition specificity, resulting in disease [4]. In addition, there are reports that the N-terminal region of the Toll protein and interleukin-1 receptor (TIR) domain and the putative coiled-coil (CC) domain of some NBS-LRR proteins are involved in specific pathogen recognition [5,6]. Genome analyses have revealed that a large number of NBS-LRR genes exist in plant genomes. The Arabidopsis and rice genomes contain up to 150 and 600 NBS-LRR genes, respectively, and many of them are tightly clustered [1,7].
Rice blast, caused by Magnaporthe oryzae, is one of the most serious diseases of rice world-wide. Many R genes against the fungus have been genetically mapped and some of them have been utilized for breeding in rice [8]. To date, 12 blast R genes have been isolated by map-based cloning, and 11 of these encode NBS-LRR resistance proteins [4,9-18]. The Japonica rice cultivar Nipponbare (NB) has been selected as a standard cultivar for the international rice genome sequencing project. Therefore, NB is a strong tool for the genetic analysis of defense signaling in rice. NB carries R gene-mediated resistance against M. oryzae containing avrPish [19]. Although the Pish locus has been mapped on chromosome 1 by an analysis of quantitative trait loci (QTL), the gene itself has been difficult to isolate because of it confers only moderate resistance [20,21]. The isolation of R genes from NB is important for the molecular dissection of defense mechanisms mediated by R proteins in rice.
The functional and structural diversification of R genes is crucial for the survival of plants, which must fight off rapidly evolving pathogens. The molecular evolution of R genes is thought to be affected by several mechanisms, including mutations caused by transposable elements (TEs). Many types of TEs have been identified in R gene clusters [22,23]. TE-mediated genome reorganization and TE-associated methylation may play roles in R-gene evolution, in addition to the actual insertion of TEs. Therefore, target-site selection by TEs is of great interest in the study of R gene evolution. Many TEs target specific chromosomal sites [24]. For some elements, the target sites are determined by specific DNA sequences, whereas for others, including several retrotransposons and retroviruses, chromatin structure impacts the choice of target site. The large-scale analysis of sequences flanking the rice retrotransposon Tos17 indicated that it prefers gene-dense regions over centromeric heterochromatin regions [25].
Here we report on our screening approach to identifying signaling components required for R gene-mediated disease resistance, using a collection of NB-background mutant lines generated by activation of the rice endogenous retrotransposon Tos17 [26]. We identified many mutant lines with reduced resistance to M. oryzae containing avrPish. While most of the mutants contained Tos17 insertions or deletions in the Pish gene locus, several contained intact copies of the Pish gene. A genome analysis revealed that Pish belongs to the NBS-LRR class of R genes, and three highly conserved NBS-LRR genes lie near it. Interestingly, this gene cluster is one of the hot spots of Tos17 insertion, and the target sites are concentrated within the Pish locus despite its similarity to the other genes at the sequence and expression levels. This suggests that the preference for Tos17-insertion in the Pish locus is regulated at the chromatin level.
Results
Large scale genetic screening
To efficiently screen for loss-of-function mutations in genes encoding components of disease resistance signal transduction, we used insertion mutant lines of NB in which the endogenous retrotransposon Tos17 had been activated by tissue culture [26,27]. While NB carries the Pish gene [19], cv. Kinmaze (KM) does not and is susceptible to the same race of the fungus (Figure 1A, B). To perform high throughput screening, we inoculated 3 week-old seedlings with the fungal isolate containing avrPish. In the first screen, 20 R1 segregating plants per line from 41,119 mutant lines were inoculated. Mutant candidates with susceptible phenotypes (561 lines) were selected for further screening. In the second screen, about 40 plants per selected line were inoculated with the same race of the fungus. We finally identified 86 mutant lines with susceptible phenotypes. These mutant lines were subjected to DNA gel blot analysis to examine co-segregation of Tos17 with the mutant phenotype. Genomic DNA sequences flanking the co-segregating Tos17 insertions were isolated by thermal asymmetric interlaced PCR (TAIL-PCR) [28], or by identifying sequence-specific amplification polymorphisms [29]. Additional flanking sequences were identified by searching the flanking sequence tag (FST) database [30]. In cases where no co-segregating signal was obtained, the causative genes will be isolated by a map-based cloning strategy (Figure 1C).
Figure 1.

High throughput screening of rice mutant lines. Representative phenotypes of NB (A) and KM (B) leaves inoculated with the rice blast fungus Magnaporthe oryzae, isolate Kyu77-07A (avrPish; Race 102.0). Photographs were taken 7 days post inoculation. (C) Experimental scheme for high throughput screening of Tos17 insertional mutant lines.
Isolation of pish mutants
We analyzed in detail two mutant lines, NC7869 and NC8589, and observed the segregation of individuals exhibiting complete susceptibility to the rice blast isolate containing avrPish in the R1 population of these lines (Figure 2A). The uninfected mutants showed no obvious morphological phenotypes compared with their wild-type siblings (data not shown). Segregants of these lines were screened for the presence of Tos17 insertions. Sixteen and 11 newly transposed copies of Tos17 were detected in the NC7869 and NC8589 lines, respectively, in addition to the two original copies (Figure 2B, left panel). One of the transposed copies (indicated by arrowheads in Figure 2B) was shown to co-segregate with the susceptible mutant phenotype in each line. Genomic sequences flanking the co-segregating Tos17 copies were amplified by TAIL-PCR and used as probes on the same membrane. One of the probes hybridized with both the 4.2-kb band in NC7869 and the 3.9-kb band in NC8589 (Figure 2B, right panel). Because the size of the XbaI fragment detected by this probe was 1.3 kb if Tos17 was not inserted, the wild-type band was not detected in this analysis. Therefore, we carried out a PCR analysis to examine whether the inheritance pattern of the Tos17 insertion was correlated with the susceptibility phenotype. In all of the susceptible plants, homozygous insertions were detected, as indicated by the amplification of only a smaller fragment assumed to be the product of the Tos17 specific primer and one of the flanking sequence primers (Figure 2C). On the other hand, all of the wild-type siblings were heterozygous or did not contain the insertion. These results strongly suggested that the susceptibility phenotype of the NC7869 and NC8589 lines was caused by the Tos17 insertion, and that the insertional mutation was inherited recessively. A BLAST search of the GenBank nucleotide database and the Rice Annotation Project (RAP) database [31] with the flanking sequence revealed that Tos17 was inserted into the 2nd exon of Os01g0782100 in both lines (Figure 3A). This gene contains two introns of 3947 and 109 bp. It encodes a predicted polypeptide of 1290 amino acids with a molecular weight of 147 kDa, which contains conserved NBS and LRR domain motifs (Figure 3B). The NBS domain (169-479 a.a.) contains three sequences, GGAGKS, LLVLDDV, and GSRVLVTSRR, which correspond to the conserved kinase 1a (P-loop), kinase 2, and kinase 3a domains, respectively [32]. The C-terminal region of the protein is composed of 29 irregular LRRs. Further analysis using the Paircoil2 program [33,34] revealed a potential CC domain with a threshold of 0.1 near the N-terminus, indicating the possibility that the encoded protein belongs to the CC-NBS-LRR subclass of R proteins (Figure 3B). Os01g0782100 is located on the long arm of chromosome 1, where Pish has been mapped between the markers RM212 (7.2 cM) and OSR3 (15.2 cM) by QTL analysis [20,21]. Taken together, the results suggest that Os01g0782100 is Pish, and we designated it Pish(t).
Figure 2.

Co-segregation analysis of mutant lines NC7869 and NC8589. (A) Representative phenotypes of the susceptible mutants that segregated in these lines. Numbers on the right of the photographs represent individual seedlings. (B) DNA gel blot analysis of XbaI-digested genomic DNA was carried out to investigate co-segregation between disease phenotypes and Tos17 insertions in individual plants of the NC7869 and NC8589 lines. Disease phenotypes are indicated at the top as R (resistant) and S (susceptible) to the blast fungus containing avrPish. Numbers at the top of each lane represent individual seedlings. NB served as a control for wild-type (resistant) seedlings. The blot was probed first with a 32P labeled Tos17 fragment (left) and then with a labeled fragment flanking the co-segregating Tos17 copy (right). The positions of size markers (λ DNA digested with HindIII) are shown on the left. Co-segregating bands are indicated by arrowheads. (C) Genotyping PCR analysis of genomic DNA from NC7869 and NC8589 plants exhibiting resistance (R) or susceptibility (S). The black triangle indicates the approximately 3.6 kb fragment that was amplified from the wild-type genome using primers at positions indicated by a and b on the diagram below the PCR gel. The white triangles indicate the fragments amplified using primers at positions b and c, designed to amplify the flanking sequence of Tos17. The genotype of each seedling is given at the top of each lane.
Figure 3.

The structure of the Pish(t) gene and its deduced amino acid sequence. (A) Positions of Tos17 insertions within the Pish(t) gene in mutant lines NC7869 and NC8589. Exons are indicated by black boxes. (B) Deduced amino acid sequence of the Pish(t) gene product. Amino acids 52-98 shown in underlined italics, constitute a predicted CC motif. The three conserved motifs in the NBS region are also underlined.
Isolation of allelic mutants and complementation analysis
To confirm whether the null mutation of Pish(t) caused disruption of the resistance mediated by Pish, we screened for other allelic mutants among the 86 selected mutants from our screening. As expected, most of the mutants (72 of the 86 lines) had mutations caused by Tos17 insertions, deletions, or an unknown insertion in this locus. Because these mutant lines were produced by tissue culture and are derived from a relatively small number of induced calli, some of these mutations are shared in multiple independently regenerated plants [27]. By considering which calli the mutants were derived from and by examining each mutation pattern (i.e., the positions of Tos17 insertions or the deletion sizes in those mutants), we determined that there are 56 independent mutant alleles at the Pish(t) locus among the 72 mutant lines (Table 1). Of these, 46 alleles were caused by Tos17 insertion. The direction of Tos17 was not always the same and the insertion sites were dispersed evenly at this locus, suggesting that the insertion site within the locus was random rather than depending on specific DNA sequences. Nine independent deletion mutations were detected among thirteen lines. The deletion sizes were diverse, ranging from 24 bp to over 50 kb. One allele in the ND2032/ND2105/ND2452/ND2562 lines contained an unidentified insert of about 2.5-kb. In these mutants, the transcription of Pish(t) was barely detectable or not detected at all (typical examples are shown in Figure 4A, B).
Table 1.
Allele of pish(t) obtained in this screening
| Line Name | Mutation | Directiona) | Positionb) | Originc) | |
|---|---|---|---|---|---|
| 1 | NC7869 | Tos17 insertion | ↑ | +2.4 | N0 |
| 2 | NC8589 | Tos17 insertion | ↑ | +2.2 | N0 |
| 3 | ND0085 | Tos17 insertion | ↓ | +1.1 | N1 |
| 4 | ND2294 | Tos17 insertion | ↓ | +2.8 | N3 |
| 5 | ND2607 | Tos17 insertion | ↓ | +1.7 | N3 |
| 6 | ND2794 | Tos17 insertion | ↓ | +3.6 | N3 |
| 7 | ND3377 | Tos17 insertion | ↑ | +2.4 | N4 |
| 8 | ND3760 | Tos17 insertion | ↑ | +1.5 | N4 |
| 9 | ND5163 | Tos17 insertion | ↑ | +1.1 | N7 |
| 10 | ND5216/ND5490/ND5546 | Tos17 insertion | ↑ | +1.3 | N7 |
| 11 | ND5907 | Tos17 insertion | ↑ | +1.0 | N7 |
| 12 | ND6103 | Tos17 insertion | ↓ | +2.7 | N8 |
| 13 | ND6886 | Tos17 insertion | ↑ | +1.0 | N8 |
| 14 | ND7244/ND7372 | Tos17 insertion | ↑ | +0.4 | N9 |
| 15 | ND7350/ND7813 | Tos17 insertion | ↓ | -0.4 | N9 |
| 16 | ND7933 | Tos17 insertion | ↓ | +3.7 | N9 |
| 17 | ND9375 | Tos17 insertion | ↓ | +2.9 | N12 |
| 18 | ND9468 | Tos17 insertion | ↓ | +1.3 | N12 |
| 19 | NE0979 | Tos17 insertion | ↓ | +0.7 | N13 |
| 20 | NE1033 | Tos17 insertion | ↑ | +3.5 | N14 |
| 21 | NE1136 | Tos17 insertion | ↓ | +0.1 | N14 |
| 22 | NE1348 | Tos17 insertion | ↓ | +2.4 | N14 |
| 23 | NE1445 | Tos17 insertion | ↑ | +3.3 | N14 |
| 24 | NE1544/NE1588/NE1602/NE1617 | Tos17 insertion | ↓ | +2.1 | N15 |
| 25 | NE1662 | Tos17 insertion | ↓ | +1.0 | N15 |
| 26 | NE1710/NE1864 | Tos17 insertion | ↓ | +2.4 | N15 |
| 27 | NE2127 | Tos17 insertion | ↓ | +3.4 | N16 |
| 28 | NE3788 | Tos17 insertion | ↑ | -0.9 | N18 |
| 29 | NE3814 | Tos17 insertion | ↑ | +2.3 | N18 |
| 30 | NE9963 | Tos17 insertion | ↓ | +3.4 | N19 |
| 31 | NF1237 | Tos17 insertion | ↑ | +3.4 | N28 |
| 32 | NF1437/NF1457 | Tos17 insertion | ↓ | +0.6 | N28 |
| 33 | NF2942 | Tos17 insertion | ↑ | +3.6 | N39 |
| 34 | NF6474 | Tos17 insertion | ↑ | +2.4 | N33 |
| 35 | NF6868 | Tos17 insertion | ↓ | +2.0 | N44 |
| 36 | NF7813 | Tos17 insertion | ↑ | +1.4 | N42 |
| 37 | NF7959 | Tos17 insertion | ↓ | +2.2 | N42 |
| 38 | NG0430 | Tos17 insertion | ↑ | +1.9 | N47-2 |
| 39 | NG2085 | Tos17 insertion | ↑ | +1.7 | N51 |
| 40 | NG3372 | Tos17 insertion | ↑ | +2.8 | N54 |
| 41 | NG3664 | Tos17 insertion | ↓ | +2.8 | N55 |
| 42 | NG5334 | Tos17 insertion | ↑ | -2.4 | N61 |
| 43 | NG6314 | Tos17 insertion | ↓ | +3.9 | N65 |
| 44 | NG7885 | Tos17 insertion | ↑ | +2.6 | N68 |
| 45 | NG8190 | Tos17 insertion | ↓ | +3.3 | N69 |
| 46 | NG8193 | Tos17 insertion | ↓ | +2.3 | N69 |
| 47 | NC7524/NC8928 | Large deletion | N0 | ||
| 48 | NC7925 | Large deletion | N0 | ||
| 49 | ND2043 | Large deletion | N3 | ||
| 50 | ND2602/ND2810 | 330bp deletion | N3 | ||
| 51 | ND6227 | Large deletion | N8 | ||
| 52 | ND8614 | Large deletion | N11 | ||
| 53 | NE2386 | 24bp deletion | N16 | ||
| 54 | NF4922/NF4946/NF4950 | 56bp deletion | N41 | ||
| 55 | NG2263 | Large deletion | N52 | ||
| 56 | ND2032/ND2105/ND2452/ND2562 | Unknown insertion | N3 |
a) Downward arrows indicate insertions with Tos17 oriented in the same direction as Pish and the Npi37-1 to Npi37-3 genes. Upward arrows indicate insertions in the opposite orientation.
b) Positions (kb) of Tos17 insertions relative to the Pish ATG initiation codon.
c) Name of the flasks in which their origin calli were cultured.
Figure 4.
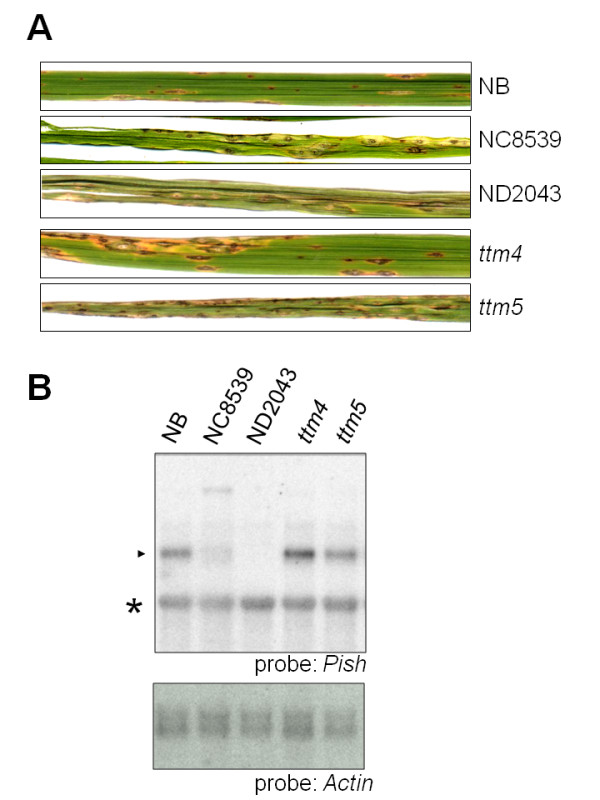
The susceptible phenotype of selected mutant lines. (A) Three-week-old NB, pish mutant lines, NC8589 andND2043, and ttm mutant lines were inoculated with M. oryzae containing avrPish. The photographs were taken 7 days post inoculation. (B) Expression of the Pish gene. Total RNA was extracted from leaf tissues of NB and each mutant. Samples (1 μg) of poly(A)+ RNA were separated by gel electrophoresis, blotted, and hybridized with radiolabeled probes as indicated. The Actin probe was used as a loading control. The closed arrowhead indicates the size of the Pish mRNA. The asterisk indicates a non-specific signal detected by the Pish probe.
On the other hand, 14 of the 86 lines with diminished Pish-mediated resistance contained neither mutations, insertions, nor deletion in this locus (data not shown; typical examples are shown in Figure 4A). In these mutants, the expression of Pish(t) was no different from in the wild type plants (typical examples are shown in Figure 4B). Therefore, we concluded that they are not pish(t) mutants and designated them ttm (tissue-culture triggered mutation). Although we cannot exclude the possibility that some of the ttm mutants have mutations in unknown R genes that correspond to the blast isolate carrying additional avr genes other than avrPish, others are likely to have mutations in components required for activation of Pish-mediated disease resistance.
To examine whether Pish(t) confer race-specific resistance, we transformed KM plants with an empty vector control and a construct containing the Pish(t) cDNA under the control of the cauliflower mosaic virus 35 S promoter (Figure 5). We obtained more than five independent transgenic lines for each construct, and used the T1 and T2 generations for the following analyses. Transgenic plants containing the Pish(t) construct were as healthy as KM plants transformed with the empty vector (data not shown). When infected with a rice blast isolate containing avrPish, three independent transgenic lines expressing Pish(t) exhibited a resistance phenotype, whereas the lines containing the empty vector were susceptible (Figure 5A and 5B). Thus, expression of the Pish(t) cDNA conferred Pish-mediated resistance on KM. To determine the resistance spectrum of Pish(t), the transgenic lines were inoculated with seven additional rice blast isolates (Table 2). As expected, the transgenic KM plants containing Pish(t) exhibited the same pattern of resistance specificity as the donor cultivar NB. Thus, we concluded that Pish(t) is the Pish gene.
Figure 5.

Complementation tests of transgenic plants. (A) A cDNAs of Pish(t) under the control of the 35 S promoter was introduced into KM. Control plants were transformed with the empty vector (Vec). Three-week-old plants of the T1 and T2 generations were challenged with a rice blast isolate containing avrPish by spray inoculation. The photographs show typical phenotypes of leaf blades 7 days post inoculation. Results were reproduced in at least three independent infection experiments using five or more plants each of three independent transgenic lines. (B) RT-PCR analysis was performed on total RNA isolated from leaves of T1 plants transformed with the empty vector or Pish(t). An Action gene was used as a control for RNA template amounts.
Table 2.
Disease reactions of transgenic KM and NB plants to eight M. oryzae isolates
| Transgenic lines | |||
|---|---|---|---|
| Isolate | NB (Pish) | Vectora) | Pisha) |
| Kyu77-07A | Rb | Sb | R |
| 84-81A | R | S | R |
| H07-1-1 | S | S | S |
| Kyu89-246 | S | S | S |
| Ina86-137 | S | S | S |
| TH68-126 | S | S | S |
| 24-22-1-1 | S | S | S |
| Kyu9439013 | S | S | S |
a) The constructs were transformed into the susceptible cultivar KM. Three independent transgenic lines for each construct were assayed.
b) R: resistance, S: susceptible
Expression analysis of Pish
Quantitative real-time RT-PCR analysis was carried out to investigate the expression pattern of Pish after infection with various M. oryzae races. The analysis revealed that there were no distinguishable alterations in Pish expression levels at different time points after inoculation with either incompatible or compatible races of the fungus, or after mock treatment (Figure 6). This result is consistent with previous reports that other Pi genes were constitutively expressed and not induced by pathogen challenge [10,11,13-15]. It is likely that most Pi genes are expressed before pathogen invasion and are post-transcriptionally regulated for activation of the signal transduction pathways leading to resistance responses.
Figure 6.
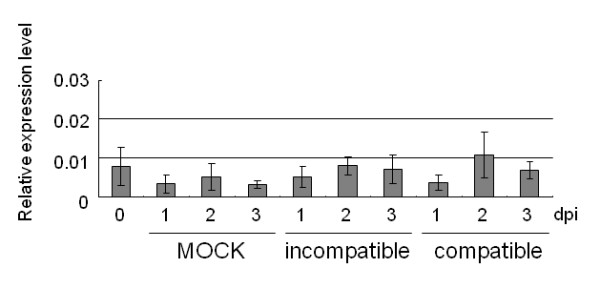
Quantitative real-time RT-PCR analysis of Pish expression. Total RNA was isolated from NB leaves at 0, 1, 2, and 3 days after inoculation with incompatible or compatible races of M. oryzae, or after mock inoculations. The samples were quantified using Actin mRNA as a reference. Error bars indicate standard deviations.
The genomic organization in the Pish region
Pish (Os01g0782100) is present on the long-arm of chromosome 1. The RAP database revealed that three other NBS-LRR genes (Os01g0781100, Os01g0781200 and Os01g0781700) are arranged as tandem repeats near Pish within a 55-kb interval (Figure 7A). An allele of Os01g0781700 in the rice cv. St. No.1 encodes the R protein Pi37 [10]. Therefore, we named the genes Npi37-1, Npi37-2 and Npi37-3. Their deduced amino acid sequences show 67, 89, and 98% similarity, respectively, to Pish (Table 3). Npi37-3 shows the highest similarity to Pish, with differences at only 19 residues in the LRR domain (Figure 7B).
Figure 7.
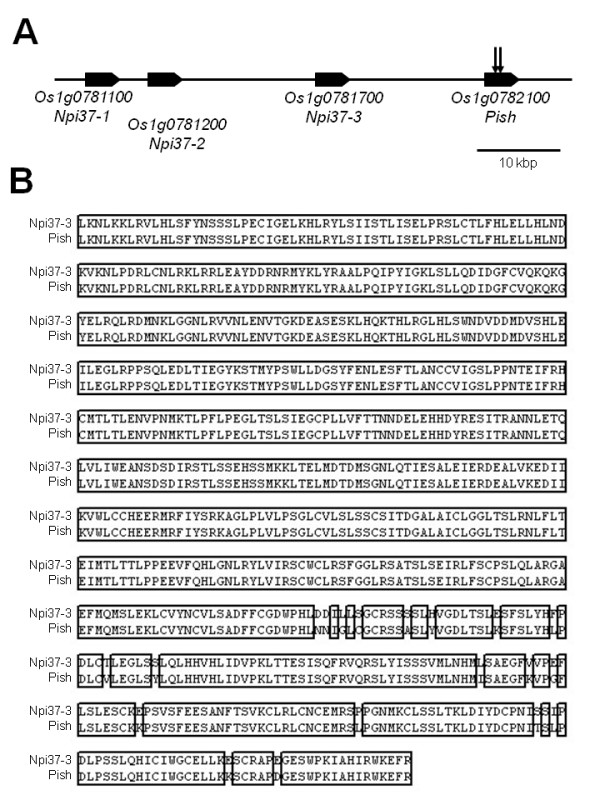
Physical map of the region containing the Pish locus on chromosome 1. (A) The NLR gene cluster in the Pish region. Arrows indicate the positions of the Tos17 insertions in the NC7869 and NC8589 lines. (B) Alignment of the LRR domains of Npi37-3 and Pish. Identical amino acid residues are enclosed by open boxes.
Table 3.
Amino acid identity among the Npi37-1, Npi37-2, Npi37-3 and Pish proteins
| Npi37-1 | Npi37-2 | Npi37-3 | Pish | |
|---|---|---|---|---|
| Npi37-1 | 67 | 67 | 67 | |
| Npi37-2 | 91 | 89 | ||
| Npi37-3 | 98 |
The amino acid sequences of the four NBS-LRR proteins were compared with each other using ClustalW http://www2.ebi.ac.uk/clustalw/.
The Pish gene cluster is a hot spot of Tos17 insertions
In this screening, we obtained 46 independent mutant alleles of Pish that were caused by Tos17 insertions. This suggested the possibility that the Pish locus is a hot spot of Tos17 insertions. It has been reported that Tos17 insertions are not distributed randomly in the rice genome, and several hot spots have been identified in NB, including genes encoding protein kinases and disease/defense-related genes [25]. An in silico analysis of the FST database revealed that 1,295 sequences flanking Tos17 insertion sites were present within the 55-kb genomic region including the four NBS-LRR genes (Table 4). These flanking sequences were particularly concentrated (1,294/1,295) within a region of about 47-kb that includes Npi37-2, Npi37-3, and Pish, but not the Npi37-1 locus. Based on the original callus lines and the sites of insertion, we determined that 193 of these insertions were independent. We also examined the 50-kb regions on either side of the 55-kb region. Even though each of these regions contains six predicted genes, the Tos17 insertions were highly concentrated in three of the four NBS-LRR genes (Table 4), indicating that these genes constitute a hot spot for Tos17 insertion. The four genes Npi37-1, Npi37-2, Npi37-3, and Pish contain highly similar sequences, and the sequences derived from FST are very short. Therefore, it was difficult to identify the exact site of each Tos17 insertion based on the FST sequences. To explore this further we randomly selected 50 independent insertions from the database and identified the insertion sites for 44 of them by PCR using specific primers. Unexpectedly, most of them were concentrated in either Npi37-3 or Pish. More than half of the insertions (27 of 44; 61%) were detected in the Pish gene, and 15 of 44 (34%) were in Npi37-3 (Figure 8A, Additional file 1). On the other hand, only two insertions were found in Npi37-2 and none were found in the Npi37-1 locus. This is surprising given that the Npi37-2 DNA sequence is 93% and 92% identical to those of Npi37-3 and Pish, respectively. Tos17 is activated by tissue culture, and we therefore speculated that the frequency of the insertion in a particular gene may depend on its expression level in cultured cells. To test this possibility, we analyzed the expression levels of each of the NBS-LRR genes in cultured cells. RT-PCR analysis demonstrated that the transcript levels of these four genes were not significantly different from one another (Figure 8B). These results suggest that neither gene structure nor relative expression levels are sufficient to explain the location of this hot spot.
Table 4.
The numbers and positions of Tos17 insertions in a region of 155 kb including the Pish locus
| Region | Independenta/FST databaseb | Length (bp) | Gene number |
|---|---|---|---|
| upstream region | 0/0 | 50,000 | 6 |
| Npi37-1 | 1/1 | 4,740 | 1 |
| IG1c | 0/0 | 1,711 | 0 |
| Npi37-2 | 4-96/294* | 6,778 | 1 |
| IG2c | 0/0 | 11,317 | 0 |
| Npi37-3 | 0-168/500* | 8,958 | 1 |
| IG3c | 0/0 | 11,721 | 0 |
| Pish | 20-180/500* | 8,695 | 1 |
| downstream region | 39/76 | 50,000 | 6 |
| Total | 232 (193d)/1,371 (1,295d) | 153,920 | 16 |
a) Number of non-redundant flanking sequences.
b) Number of flanking sequences described in the FST database including redundant sequences.
c) Intergenic regions between Npi37-1 and Npi37-2 (IG1), between Npi37-2 and Npi37-3 (IG2) and between Npi37-3 and Pish (IG3).
d) Number of flanking sequences in the 55 kb genomic region including Npi37-1, Npi37-2, Npi37-3 and Pish.
* Data including sequences whose exact positions were not determined.
Figure 8.
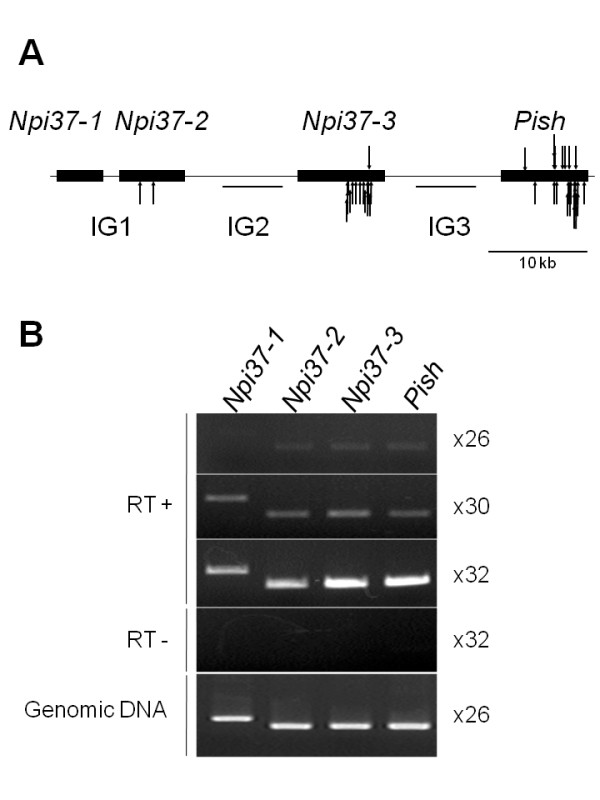
Tos17 insertions and expression of genes near the Pish locus. (A) Tos17 insertion sites in the 55-kb genomic region containing the Pish locus are indicated by arrows. Downward arrows indicate insertions with Tos17 oriented in the same direction as Pish and the Npi37-1 to Npi37-3 genes. Upward arrows indicate insertions in the opposite orientation. The black boxes indicate genes. (B) RT-PCR analysis of the four NLR genes. One microgram of total RNA isolated from NB callus was used. A series of 26, 30, and 32 cycles of amplification was used for the PCR. The experiment was repeated three times, and typical results are shown. Genomic DNA isolated from NB callus was used as a control for PCR efficiency by each primer pair.
The FST database also demonstrated that no Tos17 insertions occurred in the intergenic regions within this 55-kb genomic region (Table 4). The intergenic regions between Npi37-2 and Npi37-3 (IG2) and between Npi37-3 and Pish (IG3) are each more than 10 kb in length and show high levels of sequence similarity because of their duplication. An investigation of the intergenic regions using the RepeatMasker [35] program demonstrated that many ancient retrotransposon and other TE sequences are present in IG2 and IG3 (Additional file 2). In addition, a search of the rice MPSS database [36] indicated that the target sites of many small RNAs (sRNA) are present in IG2 and IG3 (data not shown). It is possible that these TE sequences constitute micro-heterochromatic regions due to the accumulation of these sRNA target sites. Because Tos17 insertions occur more frequently in gene-dense regions than in heterochromatin at the chromosomal level, this may explain why we found no Tos17 insertion in these intergenic regions.
Discussion
Large-scale screening using the retrotransposon Tos17 in rice
We screened 41,119 mutant lines induced by the rice retrotransposon Tos17 with the goal of obtaining signal components required for disease resistance in rice. This mutant population is estimated to carry over 400,000 insertions, based on our previous reports that the average number of insertions per line is about 10 [25,26]. We estimated that 400,000 insertions are required to provide a 99% chance of finding a mutant of any one gene [25,26]. Furthermore, since the lines were derived from cultured cells, they were expected to contain many deletions and substitutions (Table 1, [37]). Thus the number of mutant lines we employed here should have been enough for the identification of signaling mutants by genetic screening.
Among the mutations isolated by this screen, loss-of-function alleles of Pish were the most abundant. These mutants were easy to distinguish from wild-type plants because they exhibited complete susceptibility to a M. oryzae isolate containing avrPish. On the other hand, since the strength of resistance mediated by Pish is moderate, it was difficult to identify other mutations that caused reductions in resistance rather than a complete collapse in resistance. Another reason for the abundance of pish mutations in our screen is that the Pish locus is one of the hot spots for Tos17 insertions (Figure 8A). Therefore, it might have been inevitable that we would obtain a lot of pish mutants. The finding that most of the mutations isolated were loss-of-function alleles of an R gene is consistent with previous reports of screens for disease resistance mutations in Arabidopsis [38-40], tomato [41], and barley [42,43]. In Arabidopsis, Tornero and colleagues recovered 110 mutants that exhibited no HR after avrRpm1 induction, and 95 of them were loss-of-function mutations in the R gene Rpm1, which confers resistance to the bacterial pathogen Pseudomonas syringae pv. tomato expressing the avrRpm1 gene [40]. Jørgensen and colleagues performed a mutant screening of barley and isolated over 20 loss-of-function alleles of the Mla12 gene, which confers resistance to powdery mildew, and only three additional mutants in two other loci (RAR1 and RAR2) [43,44]. Thus, our ratio of 72 pish mutant lines to 14 other mutations was not surprising. The other 14 mutant lines have not yet been characterized, however, we expect that the analysis and isolation of the causative genes may provide novel insights into disease resistance signaling in rice.
An NBS-LRR gene cluster in the Pish region
Genome sequencing revealed that there are up to 600 NBS-LRR genes in the rice genome and they account for about 1% of all of predicted ORFs [7]. The majority of R genes in the NBS-LRR class are physically clustered with closely related genes [45,46]. We found three additional NBS-LRR genes near Pish, arranged as tandem repeats (Figure 7A). Although one of these (Npi37-3) exhibits high similarity to Pish, with differences at only 19 residues in the LRR domain, it does not confer resistance to M. oryzae containing avrPish (Additional file 3). It is likely that some of these 19 residues are important for maintaining the target recognition site of Pish. This is consistent with reports that the LRR domain is required for the specific recognition of avirulence factors directly or indirectly, and that mutations in this domain often change gene-for-gene specificities [4,14]. On the other hand, the NBS domain may also be involved in the specific recognition of the target factors. Npi37-3 is an allele of another R gene, Pi37 [10]. The only difference between them is in two amino acid residues (V239A and I247M) in the NBS domain (Additional file 4). As discussed previously [10], these substitutions in the NBS domain are presumably involved in avrPi37 recognition.
Pi35(t) was identified in a QTL analysis of a population derived from the Japonica rice cultivar Hokkai 188 and the Indica rice cultivar Danghang-Shali. The gene was mapped on the long arm of chromosome 1 between the SSR markers RM1216 and RM1003, and is closely linked with Pish [47]. Interestingly, the resistance conferred by Pi35(t) to M. oryzae is classified as partial resistance (quantitative) rather than true resistance (qualitative). Although the race specificity and defense strength of Pi35(t) is different from that of Pish, it is possible that Pi35(t) is allelic to either Pish or Pi37. If so, the region containing the Pish locus is one of the clusters of R genes in rice. This is consistent with the idea that the evolution of R genes is driven by selection of allelic variations created by mutations or recombination within these clusters. This phenomenon has also been observed in the case of other R gene clusters such as the one containing Pi9, Pi2 and Piz-t on chromosome 6 [13,14]. The identification and characterization of Pi35(t) will allow us to understand the difference between partial and true resistance at the molecular level, and will also shed light on the evolution of these R genes during rice domestication.
Evolution of the Pish gene cluster
Classical genetics and genome analysis have demonstrated that R genes tend to be clustered. The 55-kb region containing the Pish locus contains three other NBS-LRR genes, all oriented in the same direction (Figure 7A). Among the proteins encoded by these genes, Npi37-1 exhibits little similarity to the other three, while Npi37-3 shows high similarity to Npi37-2 (91%) and Pish (98%) (Table 3). Intriguingly, the N-terminal half of Npi37-3 is identical to that of Pish and the other half is identical to that of Npi37-2 (data not shown). These data suggest that at first the ancestral NBS-LRR gene was duplicated to produce Npi37-1 and Npi37-2-pre. This would be followed by a second duplication event in which Npi37-2-pre was duplicated to produce Npi37-2 and Pish. More recently, a crossover and/or duplication presumably occurred between Npi37-2 and Pish, resulting in Npi37-2, Npi37-3, and Pish. Npi37-1 and Pish are identical between the cultivars NB and St. No.1 [10], suggesting that the two paralogs, Npi37-2 and Npi37-3, probably mutated independently in NB and/or St. No.1 after the duplication events described above.
How did these frequent gene duplications in the Pish region occur? Although R gene loci in general tend to be duplicated, the mechanism for this duplication remains obscure. Gene duplication is sometimes caused by the misrepair of chromosomal double-strand breaks (DSBs), which arise spontaneously during the life of a cell. One possible inducer of DSBs is the endonuclease activity encoded by TEs. In Drosphila melanogaster, it has been reported that DSBs are important triggers of segmental duplication (SD), and the distribution of SDs correlates positively with that of TEs [48]. Here we showed that the Pish locus is one of the hot spots for Tos17 insertions (Figure 8). Therefore, the Pish locus was presumably attacked frequently by the Tos17 endonuclease, resulted in DSBs, which might have caused gene duplication. In addition to this effect of endonuclease activity, the insertion of TEs is thought to be important for the molecular evolution of R genes. Actually, many types of TEs have been identified in R gene clusters [22,23]. Recently, Hayashi and Yoshida [22,23] reported that the insertion of a retrotransposon Renovator in the promoter region of the blast R gene Pit promoted its expression and reactivation, demonstrating that the insertion of TEs has contributed to R gene evolution.
Our results indicating that the Pish locus is a hot spot for Tos17 insertions is consistent with a previous report that disease resistance genes are among the preferred targets for Tos17 insertion [25]. However, the disease resistance genes annotated in the report were predicted from their DNA sequence similarity with sequences encoding NBS and/or LRR domains, therefore there was no direct evidence indicating they truly function as R genes or components of defense signaling. Here, we demonstrated that the functional R gene Pish is actually a hot spot of Tos17 insertion in the NB genome, and is the preferred target site among four highly conserved and closely linked NBS-LRR genes, even though the genes are highly similar at both the nucleotide sequence and expression levels. These results suggest that Tos17 inserts most frequently in functional genes within hot spot regions. A search of the FST database indicates that not all NBS-LRR gene loci are hot spots for Tos17 insertion. It is possible that the NBS-LRR gene loci that are hot spots for TE insertions are functional R genes that have not yet been identified. Thus, it may be possible to predict novel functional R genes in the FST database by looking for regions that are hot spots for TE insertions. This possibility should be assessed in the future.
The molecular mechanisms that determine TE integration site specificity in plants are still unknown. Studies of the Ty retrotransposons of yeast have revealed that interactions with bound chromosomal proteins can tether the Ty integration machinery to chromosomes and thereby direct integration to nearby sites [49]. The human immunodeficiency virus (HIV) integrates preferentially into actively transcribed genes at sites with transcription-associated histone modifications. Therefore, it is possible that the insertion of Tos17 is regulated by chromatin structure or through interaction with chromatin binding proteins, rather than being controlled directly by the structures or expression levels of the targeted sequences.
Conclusions
The genetic screening described here was a high-throughput system that allowed us to identify several mutants involved in R gene-mediated signal transduction and also to isolate the R gene Pish. We are confident that these mutants will be useful tools for the genetic analysis of defense signaling mechanism downstream of an R gene. Furthermore, our data provided experimental evidence that an R gene cluster is a highly preferred target for TE insertions. Our observations raise the hypothesis that recurrent gene duplication during the evolution of the Pish locus depended on this unique feature of being a preferred target site of Tos17 insertion. Further studies of the Pish locus will provide new insights into the functional and structural diversification of R genes, which is crucial for the survival of plants in their fight against rapidly evolving pathogens.
Methods
Plant and pathogen materials
The rice (Oryza sativa) cultivars Nipponbare (NB) and Kinmaze (KM) were used in this study. The mutant lines described in this article will be made available at the Rice Genome Resource Center of the National Institute of Agrobiological Sciences, Japan [50]. The M. oryzae isolate Kyu77-07A was used as an incompatible race against NB for our screening [19]. Other isolates, 84-81A (race 102.0), H07-1-1 (race 003.0), Kyu89-246 (race 003.0), Ina86-137 (race 007.0), TH68-126 (race 033.1), 24-22-1-1 (race 037.1), and Kyu9439013 (race 047.0), of which the country of origin is Japan, were used in complementation tests. M. oryzae was grown on oatmeal agar medium (30 g/l oatmeal, 5 g/l sucrose, and 16 g/l agar) at 22°C. Seedlings were inoculated at the 4-6 leaf stage by spraying to runoff with an aqueous spore suspension containing 1.5 × 105 spores per ml. Inoculated seedlings were kept in a dark chamber with a moisture-saturated atmosphere at 24°C for 20 h, and then maintained at 27°C and 70-80% relative humidity in a greenhouse. Disease development was monitored one week after inoculation.
Co-segregation analysis and isolation of Tos17 flanking sequences
Linkage between the susceptible phenotype and transposed Tos17 fragments was analyzed by DNA gel blot hybridization or PCR. One microgram of rice genomic DNA was digested with XbaI, and the fragments were separated by electrophoresis in a 0.8% (w/v) agarose gel, then transferred to Amersham Hybond-N+ membranes (GE Healthcare). A 376 bp DNA probe specific to Tos17 was generated by digestion of a Tos17 genomic sequence with EcoRI and BamHI. A DNA probe specific to Pish(t) was generated by PCR using the primers AOL45 and AOL48 (Additional file 5). Sequences flanking Tos17 insertions were amplified by thermal asymmetric interlaced PCR (TAIL-PCR) as described previously [51]. Genotyping was performed by PCR using the Pish-specific primers AOL52 and AOL54 in combination with the Tos17-specific primer Tos17-448R (Additional file 5).
Rice transformation
For complementation analyses, cDNA fragments corresponding to Npi37-3 and Pish were amplified by PCR using the primer sets AOL51-AOL53 and AOL51-AOL52 (Additional file 5). The coding sequences were cloned into the Ti-based vector pPZP2Ha3(+) downstream of the cauliflower mosaic virus 35 S promoter, and Agrobacterium tumefaciens-mediated transformation of rice callus was performed according to a published protocol [52,53]. Control calli were transformed with the empty vector. Plants regenerated from hygromycin-resistant calli were grown in an isolated greenhouse. More than five independent transgenic lines were produced for each construct. Expression of the transgenes and Actin (as a control) was confirmed by RT-PCR using the specific primer sets: AOL64-AOL53 for Npi37-3, AOL64-AOL52 for Pish, and OsAct1U- OsAct1L for Actin (Genbank accession number:AK100267).
RNA Analysis
Poly(A)+ mRNA was purified using an Oligotex™-dT30 <Super> mRNA Purification kit (Takara) according to the manufacturers' instructions. As the probe, the DNA fragment corresponding to Pish was amplified by PCR from genomic DNA using the specific primer pairs AOL64-AOL71 (Additional file 5). The rice gene for actin was used as the loading control. Quantitative real-time RT-PCR was conducted using the iQ SYBR Green Supermix (BioRad, Hercules, CA, USA) and an iCycler (BioRad) according to the manufacturers' instructions. At least three independent biological samples were used with specific primers for each gene. The primers pairs were AOL351-AOL353 for Pish and OsAct1U- OsAct1L for an actin (Additional file 5). The data were normalized using actin gene expression levels. Gene expression in cultured cells was analyzed using specific primers for Npi37-1 (AOL212-AOL213), Npi37-2 (AOL234-AOL369), Npi37-3 (AOL62-AOL234), and Pish (AOL62-AOL233).
Identification of Tos17 insertion sites
Genomic DNA was extracted from mutant lines selected from the flanking sequence tag (FST) database. Tos17 insertion sites were identified by PCR using specific primers for Tos17 (TAIL3 or T17-242R) in combination with the primers AOL71, AOL72, AOL356, or AOL357, which are specific for Pish, Npi37-1, Npi37-2, and/or Npi37-3 (Additional file 5).
Authors' contributions
AT carried out all the genetic analyses, pathological tests, and molecular biology work, and participated in writing the first manuscript draft, and its revision. NH participated in the experimental design and provided expertise and all isolates of M. oryzae. AM provided all of the mutant seeds. HH conceived of the study, participated in the experimental design, participated in the coordination of the work, helped to draft the manuscript and contributed to its revision. All authors have read and approved the final manuscript.
Supplementary Material
Insertion sites of Tos17 in the Pish locus. The table lists the positions of the Tos17 insertion sites and their directions in the mutant lines.
Investigation of the intergenic regions of the Pish locus. The table lists ancient retrotransposon and transposon sequences presented within the 55-kb genomic region including the Pish locus. SW score: Smith-Waterman score of the match, usually complexity adjusted. perc div.: % substitutions in matching region compared with the consensus. perc del.: % of bases opposite a gap in the query sequence (deleted bp). perc ins.: % of bases opposite a gap in the repeat consensus (inserted bp). position in query: starting or ending position of match in query sequence. direction: (C) = match is with the Complement of the consensus sequence in the database. matching repeat: name of the matching interspersed repeat. repeat class/family: the class of the repeat
Complementation tests of Npi37-3 transgenic plants. A cDNA of Npi37-3 under the control of the 35 S promoter was introduced into KM. The empty vector was used as a control. The photographs show leaf blades 7 days after the plants were inoculated as described for the experiment shown in Figure 5. The expression of the transgene was confirmed by RT-PCR analysis. An Actin gene was used as a control for RNA template amounts.
Alignment of the predicted amino acid sequences of Pish, Npi37-3, and Pi37. Identical amino acid residues are enclosed by open boxes. Stars indicate two amino acid residues that differ between Npi37-3 and Pi37.
Primer pairs utilized in this work. The table lists the primers sequence with their direction and target genes.
Contributor Information
Akira Takahashi, Email: atakaha@affrc.go.jp.
Nagao Hayashi, Email: nhayash@affrc.go.jp.
Akio Miyao, Email: miyao@affrc.go.jp.
Hirohiko Hirochika, Email: hirohiko@nias.affrc.go.jp.
Acknowledgements
This work was supported by a grant from the Ministry of Agriculture, Forestry, and Fisheries of Japan (Green Technology Project, IP-4002 and Genomics for Agricultural Innovation, PMI0005).
References
- Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- Greenberg JT, Yao N. The role and regulation of programmed cell death in plant-pathogen interactions. Cell Microbiol. 2004;6:201–211. doi: 10.1111/j.1462-5822.2004.00361.x. [DOI] [PubMed] [Google Scholar]
- van Ooijen G, Mayr G, Kasiem MM, Albrecht M, Cornelissen BJ, Takken FL. Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J Exp Bot. 2008;59:1383–1397. doi: 10.1093/jxb/ern045. [DOI] [PubMed] [Google Scholar]
- Bryan GT, Wu KS, Farrall L, Jia Y, Hershey HP, McAdams SA, Faulk KN, Donaldson GK, Tarchini R, Valent B. A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell. 2000;12:2033–2046. doi: 10.1105/tpc.12.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck JE, Lawrence GJ, Dodds PN, Shepherd KW, Ellis JG. Regions outside of the leucine-rich repeats of flax rust resistance proteins play a role in specificity determination. Plant Cell. 2000;12:1367–1377. doi: 10.1105/tpc.12.8.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rairdan GJ, Collier SM, Sacco MA, Baldwin TT, Boettrich T, Moffett P. The coiled-coil and nucleotide binding domains of the Potato Rx disease resistance protein function in pathogen recognition and signaling. Plant Cell. 2008;20:739–751. doi: 10.1105/tpc.107.056036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Wang Y, Chen JQ, Araki H, Jing Z, Jiang K, Shen J, Tian D. Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NBS-LRR genes. Mol Genet Genomics. 2004;271:402–415. doi: 10.1007/s00438-004-0990-z. [DOI] [PubMed] [Google Scholar]
- Ballini E, Morel JB, Droc G, Price A, Courtois B, Notteghem JL, Tharreau D. A genome-wide meta-analysis of rice blast resistance genes and quantitative trait loci provides new insights into partial and complete resistance. Mol Plant Microbe Interact. 2008;21:859–868. doi: 10.1094/MPMI-21-7-0859. [DOI] [PubMed] [Google Scholar]
- Shang J, Tao Y, Chen X, Zou Y, Lei C, Wang J, Li X, Zhao X, Zhang M, Lu Z. et al. Identification of a new rice blast resistance gene, Pid3, by genomewide comparison of paired nucleotide-binding site--leucine-rich repeat genes and their pseudogene alleles between the two sequenced rice genomes. Genetics. 2009;182:1303–1311. doi: 10.1534/genetics.109.102871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Chen S, Que Z, Wang L, Liu X, Pan Q. The blast resistance gene Pi37 encodes a nucleotide binding site leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome 1. Genetics. 2007;177:1871–1880. doi: 10.1534/genetics.107.080648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shang J, Chen D, Lei C, Zou Y, Zhai W, Liu G, Xu J, Ling Z, Cao G. et al. A B-lectin receptor kinase gene conferring rice blast resistance. Plant J. 2006;46:794–804. doi: 10.1111/j.1365-313X.2006.02739.x. [DOI] [PubMed] [Google Scholar]
- Wang ZX, Yano M, Yamanouchi U, Iwamoto M, Monna L, Hayasaka H, Katayose Y, Sasaki T. The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J. 1999;19:55–64. doi: 10.1046/j.1365-313X.1999.00498.x. [DOI] [PubMed] [Google Scholar]
- Qu S, Liu G, Zhou B, Bellizzi M, Zeng L, Dai L, Han B, Wang GL. The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics. 2006;172:1901–1914. doi: 10.1534/genetics.105.044891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Qu S, Liu G, Dolan M, Sakai H, Lu G, Bellizzi M, Wang GL. The eight amino-acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol Plant Microbe Interact. 2006;19:1216–1228. doi: 10.1094/MPMI-19-1216. [DOI] [PubMed] [Google Scholar]
- Liu X, Lin F, Wang L, Pan Q. The in silico map-based cloning of Pi36, a rice coiled-coil nucleotide-binding site leucine-rich repeat gene that confers race-specific resistance to the blast fungus. Genetics. 2007;176:2541–2549. doi: 10.1534/genetics.107.075465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yoshida H. Refunctionalization of the ancient rice blast disease resistance gene Pit by the recruitment of a retrotransposon as a promoter. Plant J. 2009;57:413–425. doi: 10.1111/j.1365-313X.2008.03694.x. [DOI] [PubMed] [Google Scholar]
- Ashikawa I, Hayashi N, Yamane H, Kanamori H, Wu J, Matsumoto T, Ono K, Yano M. Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer pikm-specific rice blast resistance. Genetics. 2008;180:2267–2276. doi: 10.1534/genetics.108.095034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Song MY, Seo YS, Kim HK, Ko S, Cao PJ, Suh JP, Yi G, Roh JH, Lee S. et al. Rice Pi5-mediated resistance to Magnaporthe oryzae requires the presence of two coiled-coil-nucleotide-binding-leucine-rich repeat genes. Genetics. 2009;181:1627–1638. doi: 10.1534/genetics.108.099226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbe T, Matsumoto S. Inheritance of resistance of rice varieties to the blast fungus strains virulent to the variety "Reiho". Japan J Breed. 1985;35:332–339. [Google Scholar]
- Fukuta Y, Yanoria MJ, Mercado-Escueta D, Ebron LA, Fujita Y, Araki E, Khush GS. Rice Blast: Interaction with Rice and Control, Proceeding of the 3rd International Rice Blast Conference: 2004. Kluwer Accdemic Publishers; 2004. Quantitative trait loci (QTL) reaction to rice blast isolates from Japan and the Philippines; pp. 113–121. [Google Scholar]
- Araki E, Yanoria MJT, Ebron LA, Mercado-Escueta D, Takai T, Fukuta Y. Mapping of a rice blast resistance gene Pish. Breeding research. 2003;5(Suppl 2):333. [Google Scholar]
- Song WY, Pi LY, Wang GL, Gardner J, Holsten T, Ronald PC. Evolution of the rice Xa21 disease resistance gene family. Plant Cell. 1997;9:1279–1287. doi: 10.1105/tpc.9.8.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Wing RA, Wise RP. Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley. Plant Cell. 2002;14:1903–1917. doi: 10.1105/tpc.002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell. 2003;115:135–138. doi: 10.1016/S0092-8674(03)00760-8. [DOI] [PubMed] [Google Scholar]
- Miyao A, Tanaka K, Murata K, Sawaki H, Takeda S, Abe K, Shinozuka Y, Onosato K, Hirochika H. Target site specificity of the Tos17 retrotransposon shows a preference for insertion within genes and against insertion in retrotransposon-rich regions of the genome. Plant Cell. 2003;15:1771–1780. doi: 10.1105/tpc.012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirochika H, Guiderdoni E, An G, Hsing YI, Eun MY, Han CD, Upadhyaya N, Ramachandran S, Zhang Q, Pereira A. et al. Rice mutant resources for gene discovery. Plant Mol Biol. 2004;54:325–334. doi: 10.1023/B:PLAN.0000036368.74758.66. [DOI] [PubMed] [Google Scholar]
- Hirochika H. Contribution of the Tos17 retrotransposon to rice functional genomics. Curr Opin Plant Biol. 2001;4:118–122. doi: 10.1016/S1369-5266(00)00146-1. [DOI] [PubMed] [Google Scholar]
- Liu YG, Whittier RF. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics. 1995;25:674–681. doi: 10.1016/0888-7543(95)80010-J. [DOI] [PubMed] [Google Scholar]
- Waugh R, McLean K, Flavell AJ, Pearce SR, Kumar A, Thomas BB, Powell W. Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP) Mol Gen Genet. 1997;253:687–694. doi: 10.1007/s004380050372. [DOI] [PubMed] [Google Scholar]
- Rice Tos17 Insertion Mutant Database. http://tos.nias.affrc.go.jp/index.html.en
- Rice Annotation Project database. http://rapdb.dna.affrc.go.jp/
- Traut TW. The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur J Biochem. 1994;222:9–19. doi: 10.1111/j.1432-1033.1994.tb18835.x. [DOI] [PubMed] [Google Scholar]
- Paircoil2. http://groups.csail.mit.edu/cb/paircoil2/
- McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–358. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- RepeatMasker. http://www.repeatmasker.org/
- Rice MPSS database. http://mpss.udel.edu/rice/
- Hirochika H, Miyao A, Yamazaki M, Takahashi A, Agrawal GK, Cheng C, Yamashita Y, Harada M, Nakamura H, Hakata M, Proceedings of the Fifth International Rice Genetics Symposium: 2007. World Scientific Publishing Co.; 2007. Tissue culture-induced mutations and overexpression of full-length cDNAs as a tool for functional analysis of rice genes; pp. 77–84. full_text. [Google Scholar]
- Bisgrove SR, Simonich MT, Smith NM, Sattler A, Innes RW. A disease resistance gene in Arabidopsis with specificity for two different pathogen avirulence genes. Plant Cell. 1994;6:927–933. doi: 10.1105/tpc.6.7.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Century KS, Holub EB, Staskawicz BJ. NDR1, a locus of Arabidopsis thaliana that is required for disease resistance to both a bacterial and a fungal pathogen. Proc Natl Acad Sci USA. 1995;92:6597–6601. doi: 10.1073/pnas.92.14.6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornero P, Chao RA, Luthin WN, Goff SA, Dangl JL. Large-scale structure-function analysis of the Arabidopsis RPM1 disease resistance protein. Plant Cell. 2002;14:435–450. doi: 10.1105/tpc.010393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeron JM, Barker SJ, Carland FM, Mehta AY, Staskawicz BJ. Tomato mutants altered in bacterial disease resistance provide evidence for a new locus controlling pathogen recognition. Plant Cell. 1994;6:511–520. doi: 10.1105/tpc.6.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torp J, Jørgensen JH. Modification of powdery mildew resistance gene Mla12 by induced mutation. Can J Genet Cytol. 1986;28:725–731. [Google Scholar]
- Jørgensen JH. Genetic analysis of barley mutants with modifications of the powdery mildew resistance gene Mla12. Genome. 1988;30:129–132. doi: 10.1139/g88-022. [DOI] [Google Scholar]
- Freialdenhoven A, Scherag B, Hollricher K, Collinge DB, Thordal-Christensen H, Schulze-Lefert P. Nar-1 and Nar-2, Two Loci Required for Mla12-Specified Race-Specific Resistance to Powdery Mildew in Barley. Plant Cell. 1994;6:983–994. doi: 10.1105/tpc.6.7.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelmore RW, Meyers BC. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 1998;8:1113–1130. doi: 10.1101/gr.8.11.1113. [DOI] [PubMed] [Google Scholar]
- Richter TE, Ronald PC. The evolution of disease resistance genes. Plant Mol Biol. 2000;42:195–204. doi: 10.1023/A:1006388223475. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Koizumi S, La TN, Zenbayashi KS, Ashizawa T, Yasuda N, Imazaki I, Miyasaka A. Pi35(t), a new gene conferring partial resistance to leaf blast in the rice cultivar Hokkai 188. Theor Appl Genet. 2006;113:697–704. doi: 10.1007/s00122-006-0337-8. [DOI] [PubMed] [Google Scholar]
- Fiston-Lavier AS, Anxolabehere D, Quesneville H. A model of segmental duplication formation in Drosophila melanogaster. Genome Res. 2007;17:1458–1470. doi: 10.1101/gr.6208307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Devine SE. Yeast retrotransposons: finding a nice quiet neighborhood. Cell. 1998;93:1087–1089. doi: 10.1016/S0092-8674(00)81450-6. [DOI] [PubMed] [Google Scholar]
- Rice Genome Resource Center. http://www.rgrc.dna.affrc.go.jp/index.html
- Yamazaki M, Tsugawa H, Miyao A, Yano M, Wu J, Yamamoto S, Matsumoto T, Sasaki T, Hirochika H. The rice retrotransposon Tos17 prefers low-copy-number sequences as integration targets. molecular genetics and genomics. 2001;265:336–344. doi: 10.1007/s004380000421. [DOI] [PubMed] [Google Scholar]
- Fuse T, Sasaki T, Yano M. Ti-plasmid vectors useful for functional analysis of rice genes. Plant Biotechnology. 2001;18:219–222. [Google Scholar]
- Hiei Y, Ohta S, Komari T, Kumashiro T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994;6:271–282. doi: 10.1046/j.1365-313X.1994.6020271.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Insertion sites of Tos17 in the Pish locus. The table lists the positions of the Tos17 insertion sites and their directions in the mutant lines.
Investigation of the intergenic regions of the Pish locus. The table lists ancient retrotransposon and transposon sequences presented within the 55-kb genomic region including the Pish locus. SW score: Smith-Waterman score of the match, usually complexity adjusted. perc div.: % substitutions in matching region compared with the consensus. perc del.: % of bases opposite a gap in the query sequence (deleted bp). perc ins.: % of bases opposite a gap in the repeat consensus (inserted bp). position in query: starting or ending position of match in query sequence. direction: (C) = match is with the Complement of the consensus sequence in the database. matching repeat: name of the matching interspersed repeat. repeat class/family: the class of the repeat
Complementation tests of Npi37-3 transgenic plants. A cDNA of Npi37-3 under the control of the 35 S promoter was introduced into KM. The empty vector was used as a control. The photographs show leaf blades 7 days after the plants were inoculated as described for the experiment shown in Figure 5. The expression of the transgene was confirmed by RT-PCR analysis. An Actin gene was used as a control for RNA template amounts.
Alignment of the predicted amino acid sequences of Pish, Npi37-3, and Pi37. Identical amino acid residues are enclosed by open boxes. Stars indicate two amino acid residues that differ between Npi37-3 and Pi37.
Primer pairs utilized in this work. The table lists the primers sequence with their direction and target genes.