

Abstract
Background
FAE1 (fatty acid elongase1) is the key gene in the control of erucic acid synthesis in seeds of Brassica species. Due to oil with low erucic acid (LEA) content is essential for human health and not enough LEA resource could be available, thus new LEA genetic resources are being sought for Brassica breeding. EcoTILLING, a powerful genotyping method, can readily be used to identify polymorphisms in Brassica.
Results
Seven B. rapa, nine B. oleracea and 101 B. napus accessions were collected for identification of FAE1 polymorphisms. Three polymorphisms were detected in the two FAE1 paralogues of B. napus using EcoTILLING and were found to be strongly associated with differences in the erucic acid contents of seeds. In genomic FAE1 sequences obtained from seven B. rapa accessions, one SNP in the coding region was deduced to cause loss of gene function. Molecular evolution analysis of FAE1 homologues showed that the relationship between the Brassica A and C genomes is closer than that between the A/C genomes and Arabidopsis genome. Alignment of the coding sequences of these FAE1 homologues indicated that 18 SNPs differed between the A and C genomes and could be used as genome-specific markers in Brassica.
Conclusion
This study showed the applicability of EcoTILLING for detecting gene polymorphisms in Brassica. The association between B. napus FAE1 polymorphisms and the erucic acid contents of seeds may provide useful guidance for LEA breeding. The discovery of the LEA resource in B. rapa can be exploited in Brasscia cultivation.
Background
Brassica, which comprises six species, three diploid (Brassica rapa, B. oleracea and B. nigra) and three tetraploid (B. napus, B. juncea and B. carianta), is important in global agriculture and food production. Each of the three diploid species contains one of the original genomes, A, B or C; spontaneous hybridization between two of these three original genomes produced the three tetraploid species [1]. According to recent comparative genomic studies [2-4] the three original genomes arose from the same ancestry, so all six species have complex genomes, especially the three tetraploid plants. In general, three paralogous regions were predicted for the diploids and six for the tetraploids. These Brassica species produce an oil that contains essential human dietary components, and some components of the oil are used in industry [5]. During the past 40 years, major traits of Brassica have been significantly improved: yield, plant architecture, seed quality and so on. The erucic acid content of the seeds is also a major trait to be considered in improving Brassica oilseed. A low erucic acid content is necessary for human health, while a high content is necessary for industrial use [5,6]. Therefore, there is a pressing need to collect and identify different kinds of genetic resources, including high and low seed erucic acid contents, for breeding new Brassica cultivars.
FAE1 (fatty acid elongase1) is the key gene in controlling erucic acid synthesis in Brassica seeds. It was originally cloned in Arabidopsis by directed transposon tagging with the maize element Activator (Ac); no intron was found in this gene [7]. The product of FAE1 is a condensing enzyme that extends fatty acid chain lengths from C18 to C20 and C22 [8,9]. Many recent reports have addressed the structure of FAE1 and its relationship to Brassica genomes and seed erucic acid contents. A full cDNA for this gene, as well as the genomic DNA and predicted promoter sequences, are readily available from GenBank. Using this information, full length genomic FAE1 sequences were cloned by screening the B. napus BAC library with one fragment probe of this gene, and two paralogous FAE1 in the genome were found [10]. Two major QTLs were detected using a reference doubled haploid mapping population (TNDH) originating from a Chinese high erucic acid (HEA) cultivar, B. napus cv. Ningyou7, and a European low erucic acid (LEA) cultivar, B. napus cv. Tapidor. These two QTLs, one located on linkage group/chromosome A8 (A genome) and the other on C3 (C genome), accounted for ~71% of the genetic variation [11]. With developed polymorphic markers and the TNDH mapping population, the two paralogues were regarded as genes under the two QTLs for seed erucic acid contents [10]. Similar findings were reported by Rahman et al. [12]. In 2002 and 2004, one SNP was found in FAE1 between a high and a low erucic acid content B. napus cultivar [13,14], and this was also reported by Nath et al. [15]. In addition, a four-base nucleotide deletion within the FAE1 coding region in an LEA compared to an HEA cultivar was reported. Expression these HEA and LEA FAE1 genes in yeast indicated that the SNP or indel causes the differences in erucic acid content [13,14,16]. The B. rapa (A) and B. oleracea (C) genomes also have paralogous FAE1 s [17].
EcoTILLING is a new approach developed from TILLING (Targeting Induced Local Lesions In Genome) and is mainly used to detect SNPs in natural populations. The first successful application of EcoTILLING was to detect variations in several genes in a natural Arabidopsis population in 2004 [18]. Many SNPs in a dozen genes were also identified in wild populations of Populus trichocarpa [19]. Several reports have indicated that EcoTILLING is a highly efficient approach to detecting genetic variations associated with target traits for crop improvement. Nieto et al. detected one haplotype comprising several grouped SNPs controlling virus susceptibility in a natural melon population. Key SNPs in the resistance gene were also identified and these may be exploited for resistance breeding [20]. However, despite several reports on EcoTILLING, it is relatively difficult to apply the technique to crop improvement, especially for polyploid plants with complex genomes.
Here we report the application of EcoTILLING to the identification of LEA cultivars among natural populations of three Brassica species. About 100 accessions of modern cultivars of B. napus, seven of B. rapa and nine of B. oleracea were collected. Variations in the two paralogous, B. napus FAE1s, among these cultivars were detected using EcoTILLING. In total, three SNPs/indels were found to be associated with differences in seed erucic acid contents. A new LEA genetic resource was found in the natural B. rapa population using sequence alignment. Analysis of the evolutionary relationships and sequence similarities among these three Brassica species showed a closer pedigree relationship between the Brassica A and C genomes than between the A/C genomes and Arabidopsis genome; 18 SNPs were found in the coding region of FAE1 and may be used as genome-specific markers to differentiate the A and C genomes.
Results
1. Phenotyping for erucic acid contents of seeds of Brassica accessions
One hundred and one accessions of B. napus cultivars were collected, mostly modern cultivars. Many of them perform excellently in terms of key traits such as oil content, fatty acid composition, yield and so on. These accessions were derived from different geographic origins: 71 from China, 9 from Sweden, 6 from Germany, 5 from France, 4 from Australia, 1 from Canada, 1 from Denmark, 1 from Poland, and 3 from unknown regions [Additional file 1]. To learn more about the LEA genetic resource, the original LEA ancestors of B. napus, 'Liho' and 'Oro', were also collected. Seven accessions of B. rapa (5 from China, 1 from Australia and 1 from Finland) (Table 1) and nine of B. oleracea (all from China) were also collected (Table 1).
Table 1.
Accession collection of B. rapa and B. oleracea accessions for determining erucic acid contents of seeds and their FAE1 polymorphisms at coding region
| Cultivar No. | Cultivar name | Erucic acid content of seeds (%) | Origin | Positions of nucleotides and amino acid polymorphisms | |||||
|---|---|---|---|---|---|---|---|---|---|
| B. rapa | 591a | 735 | 898 | 968 | 1020 | 1265 | |||
| P197=b | A245= | R300= | T323I | K340= | F422= | ||||
| 1 | Kunshan maquedan | 48.58 | China | G | C | C | C | G | T |
| 2 | Sanyue huang | 49.76 | China | G | C | C | C | G | T |
| 3 | Hanzhong aiyoucai | 45.70 | China | G | C | C | C | G | T |
| 4 | Xinxian huangyoucai | 52.02 | China | G | C | C | C | G | T |
| 5 | 7801_1 | 51.26 | China | G | C | C | C | G | T |
| 6 | Jumbuck | 1.93 | Australia | A | T | C | T | A | T |
| 7 | Hja 96337 | 0.90 | Finland | A | T | A | T | G | C |
| B. oleracea | 489c | 542 | 1079 | 1422 | 1458 | ||||
| Q163= | T181K | F360S | S474= | Y486= | |||||
| 1 | ChunFeng | 40.75 | China | A | A | T | A | C | |
| 2 | Zhonggan11 | 45.90 | China | A | A | T | G | C | |
| 3 | Xiaguang Oleracea | 43.27 | China | G | C | C | G | T | |
| 4 | Zhengchun Oleracea | 47.63 | China | A | A | T | A | C | |
| 5 | Jingfeng2 | 48.20 | China | A | C | T | G | Y | |
| 6 | Xinfeng | 41.40 | China | A | A | Y | G | Y | |
| 7 | Zaofeng | 51.52 | China | A | A | Y | A | Y | |
| 8 | Improved | ||||||||
| Niuxin Oleracea | 42.07 | China | A | A | T | A | C | ||
| 9 | Hanchun3 | 41.39 | China | G | A | T | G | C | |
a591 indicates the position 63 bp from the base 'A' in the start codon of B. rapa FAE1. P197=b indicates amino acid diversity due to this nucleotide polymorphism. c489 indicates the position 63 bp from the base 'A' in the start codon of B. oleracea FAE1. Three haplotypes of FAE1 can be inferred among these seven B. rapa accessions. Accessions Nos. 1-5 are B. rapa haplotype 1; accession No. 6 is B. rapa haplotype 2 and accession No. 7 is B. rapa haplotype 3. Seven haplotypes of FAE1 can be inferred among these nine B. oleracea accessions. Accessions Nos. 1, 4 and 8 are B. oleracea haplotype 1; Accessions No. 2, 3, 5, 6, 7 and 9 are B. oleracea haplotypes 2 to 7, respectively.
The erucic acid contents of the seeds of the 101 B. napus accessions ranged from approximately zero to more than 57% [Additional file 1]. In 60 accessions, the erucic acid contents were between zero and 10%; in one, between 10% and 20%; in eight, between 20% and 30%; in five, between 30% and 40%; and in 27, more than 40%. Thus, the collection comprised B. napus pools with low, medium and high erucic acid contents in seeds. In the seven B. rapa accessions, two were LEA with almost zero erucic acid contents (Table 1). We found no LEA accession among the nine cultivars of B. oleracea (Table 1).
2. FAE1 polymorphisms in the three Brassica species
Two paralogous FAE1 s were cloned by Wang et al. [10] through screening the B. napus BAC library. They were designated as Bn.FAE1-A8 and Bn.FAE1-C3. Open Reading Frame (ORF) analysis indicated that the coding regions of the two FAE1 paralogues were 1521 bp long with no intron and the similarity between them in the HEA B. napus cv. Ningyou7 genome was 98.6%. Using primer pairs designed according to these sequences (Table 2, Figure 1) and EcoTILLING, polymorphisms were detected in the 101 B. napus accessions. Overall, one position in Bn.FAE1-A8 showed polymorphism and two positions in Bn.FAE1-C3 showed polymorphisms in the coding regions. Using EcoTILLING, seven accessions were identified individually as heterozygous on Bn.FAE1-A8 or Bn.FAE1-C3. Figure 2 illustrates the polymorphisms in the second half region of Bn.FAE1-C3 with EcoTILLING screening.
Table 2.
Primer sequences used for PCR amplification
| Primer name | Forward primer sequence (5'→3') | Reverse primer sequence (5'→3') |
|---|---|---|
| FAE1-A8 | GGCACCTTTCATCGGACTAC | GATAGAACTCGGGGTTTTAGTTG |
| FAE1-C3 | GGCACCTTTCATCGGACTAC | TTAACAGAAGATCCTTAACCCC |
| ECOT1 | CGGACCACAAAAGAGGATCC | GTCTCCTTGTTGCACGCAACG |
| ECOT2 | CGTATGCTCTTGTGGTGAGC | GAGAAACATCGTAGCCATC |
| FAE1-rapa | ATGACGTCCGTTAACGTTAAGC | AAAAGAAACGAAAGAGAGCA |
| FAE1-oleracea | CTCCGACACACACACTGAGCA | GGGTTTTAGGGTTAAAGATGGTC |
| M13 | CACGACGTTGTAAAACGAC | GGATAACAATTTCACACAGG |
Figure 1.

Regions of the B. napus FAE1 genomic sequences amplified by the primer pairs. Lines represent 5' and 3' genomic sequences of FAE1 (~1 kb 5' and 3' for both Bn.FAE1-A8 and Bn.FAE1-C3) and the rectangles represent the coding region (1-521 bp). FAE1-A8 (line with triangles at the end) indicates the specific primer for Bn.FAE1-A8, and FAE1-C3 (line with diamonds at the end) indicates the specific primer for Bn.FAE1-C3. ECOT-1 and ECOT-2 (lines with circles at the end) both amplify the Bn.FAE1-A8 and Bn.FAE1-C3 coding regions (with a short part of the 5' or 3' regions). Sequences alignment for the region of specific primers (reverse primers of FAE1-A8 and FAE1-C3) were showed in corresponding areas.
Figure 2.

Detection of polymorphisms for part of Bn.FAE1-C3 using EcoTILLING. DNA from each accession mixed 1:1 with B. napus cv. Huayou5 and DNA from each accession alone were used for EcoTILLING, and these two samples were loaded on adjacent lanes on the gel. Lanes with triangle indicate the mixed samples for testing. New bands appeared in both lanes, indicating that the polymorphism was heterozygous. The sizes of DNA ladders and digested PCR products are labelled with corresponding numbers and the unit is bp.
To confirm these EcoTILLING results, we randomly sequenced 5-10 samples of each polymorphism and confirmed them at all the positions tested. In total, 19 sequences were obtained; ten of these were selected to confirm the polymorphism on Bn.FAE1-A8 (GenBank No.: HM362915 to HM362924); nine were selected to confirm the two polymorphisms on Bn.FAE1-C3 (GenBank No.: HM362925 to HM362933, HM362925 and HM362926 contained two polymorphisms). The sequencing results showed that the polymorphic locus of Bn.FAE1-A8 was at 845 bp. (We designated the nucleotide 'A' in the start codon of the coding region as position number '1' and the 5' flanking sequence as plus positions; all positions were numbered on the FAE1 sequence of the reference sample B. napus cv. Huayou5.) In some accessions the position was C; in others it was T [Additional file 1]. Two polymorphic loci of Bn.FAE1-C 3 were located at 1368-1371 bp (an indel, AGGC deletion) and 1422-1423 bp (an indel, AA deletion). Additional file 1 shows the positions of all the polymorphisms within the HEA FAE1 coding region.
Two primer pairs, FAE1-rapa and FAE1-olearacea, were selected to amplify the genomic FAE1 sequences of B. rapa and B. oleracea, respectively (Table 2). Sequencing of the PCR products from each sample of B. rapa revealed six polymorphisms in the FAE1 coding regions of the seven B. rapa accessions (Table 1). Different geographical origins may contribute to sequence differences because five accessions from China had identical sequences and the other two, one from Australia and one from Finland, had different sequences. Five polymorphisms in the coding regions of FAE1 were detected from Chinese accessions of B. oleracea which showed much greater divergence (Table 1, Additional file 1).
3. Association of FAE1 polymorphisms with differences in seed erucic acid contents among the three Brassica species
According to the EcoTILLING results that identified FAE1 polymorphisms in the 101 B. napus accessions, two haplotypes of Bn.FAE1-A8 and four of Bn.FAE1-C3 were inferred. The sequences with nucleotide C and T in the 845 bp position on Bn.FAE1-A8 were designated haplotype A8-H0 and A8-H1, respectively. No deletion of Bn.FAE1-C3 was designated haplotype C3-H0. A four-base (AGGC) deletion in Bn.FAE1-C3 1368-1371 bp and a two-base (AA) deletion in Bn.FAE1-C3 1422-1423 bp were designated C3-H1 and C3-H2, respectively. A haplotype with deletions in both positions (1368-1371 bp and 1422-1423 bp) was named C3-H3 (Table 3). The reference accession B. napus cv. Huayou5 contained A8-H0 and C3-H0 in its genome and was an HEA cultivar. Furthermore, we found that when the nucleotide changed from C (A8-H0) to T (A8-H1) in FAE1, the predicted translated amino acid changed from serine to phenylalanine. Both deletions in Bn.FAE1-C3 led to premature stop codon in the predicted ORF.
Table 3.
Association of differences in seed erucic acid contents with the different FAE1 haplotypes of B. napus
| C3-H0 | C3-H1 | C3-H2 | C3-H3 | LSD test for A8 haplotypes | |
|---|---|---|---|---|---|
| A8-H0 | 46.91 ± 4.82a (n = 29b) | No plants | 30.39 ± 8.53 (n = 3) | No plants | 45.36 A |
| A8-H1 | 23.71 ± 1.42 (n = 3) | 0.67 ± 0.72 (n = 9) | 1.02 ± 1.19 (n = 47) | 0.55 ± 0.47 (n = 3) | 2.04 B |
| LSDc test for C3 haplotypes | 44.73 Ad | 2.78 B | 0.673 C | 0.55 D |
aErucic acid content of seeds (%)
bNo. of plants with the two kinds of haplotype in combination
cLeast significant difference multiple comparisons
dMeans with different letters following are significantly different (P < 0.01)
The erucic acid content of A8-H0 seeds (mean = 45.36%) was significantly higher than that of A8-H1 (mean = 2.04%) according to ANOVA (GLM model) and an LSD (least significant difference) All-Pairwise Comparison Test (P < 0.01), indicating that C in the 845 bp position of Bn.FAE1-A8 is essential for gene function and T may result in loss of function. We found that C3-H0, C3-H1, C3-H2, C3-H3, were also significantly associated with differences in seed erucic acid contents (P < 0.01). The erucic acid content related to C3-H0 (mean = 44.73%) was far higher than that in the other three haplotypes. The erucic acid content of C3-H2 seeds (mean = 0.67%) was significantly lower than that of C3-H1 (mean = 2.78%), showing that the two-base (AA) deletion at 1422-1423 bp in Bn.FAE1-C3 inhibited erucic acid synthesis more efficiently that the four-base (AGGC) deletion at 1368-1371 bp. The erucic acid content of haplotype C3-H3 (mean = 0.55%), which harboured both deletions in Bn.FAE1-C3, was significantly lower than those of C3-H1 and C3-H2, which harboured one but not both of the deletions. In conclusion, nucleotide C at the 845 bp position of Bn.FAE1-A8 is essential for gene function and T might lead to loss of function; the two premature stop codons resulting from the two deletions on Bn.FAE1-C3 might also cause loss of function.
Too few B. rapa and B. oleracea samples were collected for ANOVA to be applied to their data. Alignment of the seven FAE1 coding sequences of B. rapa showed that three FAE1 haplotypes can be inferred. Accession Nos. 1-5 were B. rapa haplotype 1, accession No. 6 was haplotype 2 and accession No. 7 haplotype 3 (Table 1). Three of the six polymorphisms were found at positions 591 bp (G/A), 735 bp (C/T) and 968 bp (C/T), and these may be associated with differences in the erucic acid contents of seeds. Plants with nucleotide G at 591 bp and C at 735 bp and 968 bp in B. rapa FAE1 had high seed erucic acid contents; those with A at 591 bp and T at 735 bp and 968 bp had low contents. The nucleotide changes at 591 bp and 735 bp did not alter the amino acids predicted from the ORF, but when the C changed to T at 968 bp, the corresponding amino acid changed from threonine to isoleucine. Thus, the difference at position 968 bp in FAE1 may have caused the phenotypic difference. Comparing these findings with the nucleotide change in position 845 bp in the A genome of B. napus, it may be concluded that the LEA genetic resources in the A genomes in our collection differed between B. rapa and B. napus.
Alignment of the nine B. oleracea FAE1 coding sequences indicated seven haplotypes. Accessions Nos. 1, 4 and 8 were B. oleracea haplotype 1; accessions Nos. 2, 3, 5, 6, 7 and 9 were B. oleracea 2 to 7, respectively (Table 1). Five polymorphisms were found in these nine FAE1 coding regions (Table 1). Since no LEA accession was collected for B. oleracea in this study, no rule could be found for the genotypes of the HEA accessions of this species.
4. Evolutionary relationship of FAE1 among the three Brassica species
In order to investigate the pedigree relationship among the different Brassica species and explore the genetic basis of the LEA genetic resource, a phylogenetic tree was constructed using 17 FAE1 sequences: six B. napus FAE1 haplotypes, seven B. oleracea haplotypes, three B. rapa haplotyes, and one Arabidopsis sequence (Figure 3). This phyogenetic tree indicated that the divergence between the A and C genome FAE1 s occurred later than that between Arabidopsis and Brassica species, which is consistent with other reports [4,21,22]. The tree also showed that the FAE1 sequences of the A genome formed two groups, viz. the sequences from B. napus and B. rapa. However, the C genome revealed more complex groups, making it difficult to draw clear conclusions about the pedigree relationship. In addition, 18 SNPs differed between the A and C genomes, as shown by multiple alignments of 16 FAE1 sequences from the three Brassica species (Table 4). Analysis of these SNPs showed that many restriction enzymes could distinguish between the A and C genomes. Changes in the nucleotides of 15 of these 18 SNPs led to no changes in the amino acid encoded.
Figure 3.
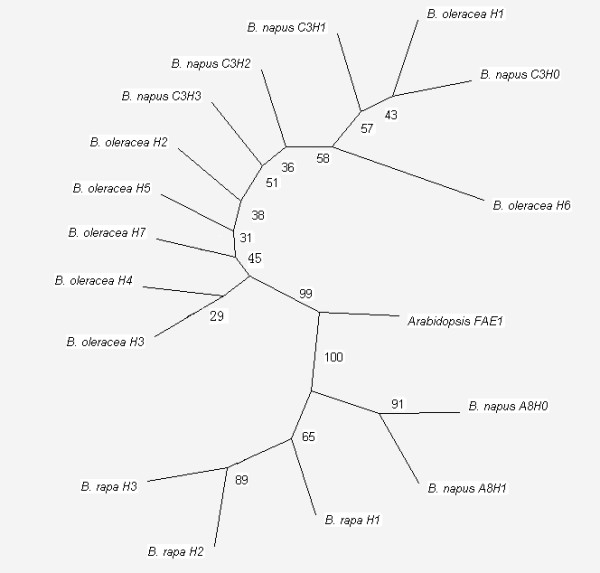
Phylogenetic tree constructed using Phylip 3.68 software with neighbour-joining (N-J) algorithm. A total of 17 Brassica sequences - four haplotypes of Bn.FAE1-C3 (B. napus C3H0 to C3H3), two of Bn.FAE1-A8 (B. napus A8H0 and A8H1), three of B. rapa (B. rapa H1 to H3), seven of B. oleracea (B. oleracea H1 to H7), and Arabidopsis FAE1- were used for this analysis. These 16 sequences have been submitted to GenBank and their accessions are from GU325717 to GU325732.
Table 4.
FAE1 polymorphisms between A and C genomes
| No. | Positions | Nucleotide Differences Between A and C Genomes | Amino acid difference | Restriction Enzymes |
|---|---|---|---|---|
| 1 | 63a | C Tb | C21=c | |
| 2 | 72 | G A | P24= | AccI, HindII, HpaI, MjaIV, TspGWI |
| 3 | 168 | C T | I56= | BsrDI, BccI, BtgZI |
| 4 | 312 | T C | Y104= | |
| 5 | 363 | G C | S121= | EcoRII, ScrFI, Hpy99I |
| 6 | 417 | T C | T139= | DsaI, SecI |
| 7 | 531 | C T | N177= | AclI, MaeII |
| 8 | 591 | A T | P197= | BccI, BtgZI |
| 9 | 856 | G A | G286R | Eco57MI, EcoRII, GsuI, ScrFI |
| 10 | 1036 | T C | L346= | BseYI, AclI, MaeII |
| 11 | 1113 | T C | D371= | |
| 12 | 1128 | C T | Y376= | |
| 13 | 1140 | T C | D380= | |
| 14 | 1146 | A G | K382= | AluI, Cac8I, CviJI, HindIII |
| 15 | 1155 | T C | I385= | TaqI |
| 16 | 1184 | G A | R395K | |
| 17 | 1217 | C G | A406G | AvrII, HaeIII, SecI, StuI, StyI |
| 18 | 1464 | G C | V488= | MseI, Hin4I |
a63 indicates the position 63 bp from the base 'A' in the start codon.
bThe first base is in the A genome and the second is in the C genome.
cC21 = indicates no change in the amino acid (synonymous variation)
Discussion
EcoTILLING, a method developed from TILLING (Targeting Induced Local Lesions in Genome), was successfully applied to B. napus, which is considered one of the most complex polyploid plants. We may infer that EcoTILLING is a very powerful method for identifying polymorphisms and for association mapping and developing functional markers for crops. However, gene-specific primer design is the key step for successful application of EcoTILLING to crop improvement. From the first application of TILLING to Arabidopsis [23], many studies have detailed the difficulty of developing primers for TILLING and EcoTILLING for crops, because most crops harbour very complex genomes and genomic sequence information is insufficient [10,20,24,25]. Of course, it is relatively easy to design gene-specific primers for plants that have been completely sequenced, such as Arabidopsis and rice [18,26]. Two procedures for developing primers for TILLING and EcoTILLING have been reported for crops with complex genomes. First, gene-specific primers can be designed according to sequence differences among multi-copy genes if the relevant genomic information is obtained. Most TILLING and EcoTILLING studies have been carried out using this procedure [18,19,24,25]. Secondly, co-amplified primers can be used for genes that are present in multiple copies and show minor sequence differences among paralogues. Two studies have successfully applied this procedure in TILLING and EcoTILLING [10,27]. In the present study, genomic sequence information was obtained first, and then gene-specific primers were designed for the two paralogues of FAE1.
It has been reported that erucic acid is an antinutritional component of seed oil [6]. Therefore, a major objective for rapeseed breeding is to achieve and apply genetic resources with low seed erucic acid contents. During the 1960s, the first LEA B. napus germplasm was found in an animal feed rape 'Liho', and then the first B. napus LEA cultivar 'Oro' was developed by introducing an LEA genetic resource from 'Liho' [28]. As far as we know, no other LEA genetic resource for breeders has been reported. In this research, we investigated the LEA resources in a collection of 101 modern B. napus accessions. The polymorphisms found for Bn.FAE1-A8 and Bn.FAE1-C3 readily revealed the SNPs/indels associated with differences in seed erucic acid contents. Thus, our results are consistent with other studies showing that SNPs/indels in FAE1 corresponded to loss of function of this gene in yeast [13,14,16]. However, comparing these SNPs/indels to the first LEA germplams 'Liho' and 'Oro', we could find only one polymorphisim (1368-1371 bp of Bn. FAE1-C3) which may be a new LEA resource. But this deletion was also on the background of the original LEA germplams 'Liho' and 'Oro'. This indicated that no more other LEA resource came out during the past 40 years. Therefore, the LEA genetic resource for B. napus is not sufficient and more are required to avoid genetic erosion.
New LEA mutants with a high erucic acid content genetic background were obtained by screening a large B. napus EMS mutant population with TILLING [10]. This would be a very efficient method for augmenting LEA genetic resources. In this study, one new LEA genetic resource in B. rapa was also found, which differed from that in B. napus. There have been many successful reports about the exchange of beneficial traits among six Brassica species in the U'Triangle [29-32]. Thus, the LEA genetic resource from B. rapa could be introduced to B. napus to improve this crop.
The All-Pairwise Comparison Test revealed differences in seed erucic acid contents, related to the four haplotypes of Bn.FAE1-C3 in our B. napus collection. Both the two-base deletion at 1422-1423 bp and the four-base deletion at 1367-1371 bp affected Bn.FAE1-C3 function. The two-base deletion reduced the seed erucic acid content more efficiently than the four-base deletion, and the combination of the two deletions knocked out gene function (Table 3) more effectively than either deletion alone. Thus, to take advantage of these LEA resources, it would be better to introduce both deletions into the target plants so as to breed cultivars with less erucic acid in the seeds.
In the molecular evolutionary analysis of the FAE1 sequences of the three Brassica species and Arabidopsis, we found 18 SNPs that differed between the A and C genomes. Fifteen of these 18 SNPs are silent variations and the original types of Brassica species with a full function of FAE1 which could produce high erucic acid in seeds. From these results, we may deduce that it is difficult for plants with loss of functional FAE1 to survive long in evolution. In other words, plants with higher seed erucic acid contents have a greater capacity to adapt to the environment.
Conclusions
EcoTILLING has been successfully applied to the identification of FAE1 variations in Brassica. Polymorphisms in Bn.FAE1-A8 and Bn.FAE1-C3 were strongly associated with differences in the erucic acid contents of seeds. Different nucleotide deletions in Bn. FAE1-C3 reduced the erucic acid content to different degrees. The discovery of a new LEA resource enlarged the pool of genetic resources and could be used for LEA breeding.
Methods
1. Plant materials
Seeds of most of the B. napus and B. rapa accessions were obtained from the Chinese Crop Germplasms Information System (CGRIS, a germplasm repository for collecting worldwide genetic resource of oilseed crop) and seeds of the two LEA ancestor cultivars, 'Liho' and 'Oro', were obtained from the Australian Temperate Field Crops Collection (ATFCC). Seeds of the nine accessions of B. oleracea were obtained from the Horticulture Department of Huazhong Agricultural University (HAU). The plants were grown in the field and young leaves were picked to extract DNA with a JYZ-3-1-2 isolation kit (Genebase Gene-Tech Co. Ltd, Shanghai, China). Self-pollinated seeds from B. napus and B. oleracea cultivars were harvested for the determination of erucic acid content. Owing to the self-incompatibility of B. rapa, sib-crossings between two plants with uniform genetic backgrounds were done for each accession collected, and seeds were then harvested.
2. Erucic acid contents of seeds
The erucic acid contents of self-pollinated seeds were determined by gas chromatography (GC) as described in [33] with little modification. First, 30-50 oven-dried seeds were crushed and transferred to 50 ml screw-capped centrifuge tubes. Thereafter, 1 ml ether/Sherwood oil reagent (1 ether: 1 Sherwood oil, by volume) and 1 ml methanol reagent (23 g potassium hydroxide in 1000 ml methanol) were added. The mixture was incubated at room temperature (25°C) for at least 40 min then 20-30 ml distilled water was added to the tubes. Finally 0.4-0.8 μl upper layer was loaded on to the GC instrument (HP 6890 series). Erucic acid content was determined by measuring the area of the peak.
3. Screening of B. napus FAE1 polymorphisms by EcoTILLING
Two complete paralogous FAE1 genomic sequences, EU543282 (Bn.FAE1-A8) and EU543283 (Bn.FAE1-C3), were retrieved from NCBI. Because these two paralogues are very similar, it was difficult to design gene-specific primer pairs for the coding region. Therefore, two primer pairs, FAE1-A8 and FAE1-C3, were designed with 5' and 3' flanking sequences of the two paralogous genes (Table 2, Figure 1). The two forward primers corresponding to the 5' flanking sequence were the same (their position in genes is -167 to -147 bp), whereas the reverse primers corresponding to the 3' flanking sequence differed between Bn.FAE1-A8 and Bn.FAE1-C3 (1711 to 1734 bp for Bn.FAE1-A8, 1728 to 1750 bp for Bn.FAE1-C3, Figure 1). PCR products amplified with the two primer pairs were both about 1.9 kb and were unsuitable for EcoTILLING because 1.5 kb is the longest size for the detection instrument [34]. Another two primer pairs, designated ECOT-1 and ECOT-2, (Table 2), were designed according to the two paralogous coding regions, and were used to amplify the first half fragment (-74 to 1025) and the second half fragment (727-1589) of Bn.FAE1-A8 or Bn.FAE1-C3, respectively (Figure 1). Sequence of universal primers M13F was added to 5' ends of ECOT-1F and ECOT-2F as adaptors; and M13R was also added to ECOT-1R and ECOT-2R (Table 2). The universal primers M13F and M13R were labelled at the 5' end with IRD 700 and IRD 800, separately (MWG Biotech, Inc., Ebersberg, Germany). FAE1-A8 and FAE1-C3 were used to amplify the two paralogous genes separately in the first PCR reaction, and then these PCR products were amplified by nested PCR reaction. This reaction contained a mixture of gene-specific primers with M13 tails and the universal labelled M13 primers.
DNA samples were isolated from each B. napus accession (single plant, self-pollinated seeds harvested from this plant were used for erucic acid content test) and 50 ng DNA was used for 10 μl PCR reaction. B. napus cv. Huayou5 is a very old cultivar in China and it was speculated to have high divergence to the modern B. napus cultivars according to experiences of breeders. Aiming to detect more polymorphic sites on EcoTILLING gel, a replicate DNA sample of this accession was used as reference in this study. To test whether the two paralogues were heterozygous, these DNA samples were mixed 1:1 with the reference sample and also individually. The products of the first PCR which amplified by gene-specific primer pairs FAE1-A8 or FAE1-C3 were diluted 50-fold to carry out the nested PCR reaction. The screening protocol followed that described by Till et al [34]. CEL1 enzyme was extracted from celery according to Oleykowski et al [35]. To avoid PCR errors, all PCR reactions were amplified with proof-reading pfu taq polymerase (Tiangen Biotech, Inc., Beijing, China).
To confirm the polymorphisms identified by EcoTILLING, the accessions that showed polymorphism on a gel were randomly selected for amplification with the primer pairs FAE1-A8 or FAE1-C3 again with proof-reading pfu taq polymerase. The PCR products were electrophoresed on agarose gels to recover the target bands, and the purified products were sequenced with ABI 3730. In the analysis of the sequences obtained, chromatograms of each sample were checked to reduce PCR or sequencing errors.
4. Identify of FAE1 polymorphisms in B. rapa and B.oleracea
Three B. rapa sequences, AF400050, Y14975 and Y14974, and two B. oleracea sequences, Y14981 and AF05440, were retrieved from NCBI. For these sequences, which contained Bn.FAE1-A8 and Bn.FAE1-C3, a total of five primer pairs were designed to amplify the complete B. rapa and B. oleracea genomic FAE1 sequences (Table 2). Because of their high efficiency, FAE1-rapa and FAE1-oleracea were selected to amplify the seven B. rapa and nine B. oleracea accessions. The PCR products were electrophoresed on agarose gels to recover the target bands, and the purified products were sequenced with ABI 3730. Sequences of these cultivars were aligned using ClustalX 1.83 (downloaded from NCBI), and the polymorphisms were also determined by comparing them with their sequence traces to avoid negative SNPs.
5. Data analysis and phylogenetic tree construction
Associations between FAE1 polymorphisms of B. napus and seed erucic acid contents were analyzed by ANOVA (GLM model) and an LSD (Least Significant Difference) multi-comparison test (SAS 8.2 software).
To construct the phylogenetic tree for FAE1, six types of FAE1 sequences of B. napus (A8-H0, A8-H1, C3-H0, C3-H1, C3-H2 and C3-H3), three of B. rapa (B. rapa haplotypes 1 to 3), seven of B. oleracea (B. oleracea haplotypes 1 to 7) and the FAE1 sequence of Arabidopsis (AT4G34520) were collected, and phylogenetic and molecular evolutionary analyzes were conducted using Phylip 3.68 with a neighbour-joining (N-J) algorithm http://evolution.genetics.washington.edu/phylip.html[36]. To analyze genome-specific markers for the A and C genome, the above 17 sequences except the FAE1 sequence of Arabidopsis were aligned with ClustalX 1.83 with default parameters [37], and SNPs that differed between the A and C genomes were analyzed using the PARSESNP (Project Aligned Related Sequences and Evaluate SNPs) web tool for identifying restriction enzymes and predicting amino acid changes [38].
Authors' contributions
NW, FT and HN screened the polymorphisms with EcoTILLING and carried out all the sequencing validation and data analysis. FT extracted DNA of some of the 101 B. napus and determined the erucic acid contents of the seeds by GC for B. napus and B. oleracea. XW planted all these accessions of Brassica and provided the data about the erucic acid contents of B. rapa seeds. NW and LS wrote the paper and participated in editing it. YL, XW and JM edited the paper; JM and LS also supported the project. All authors read and approved the final manuscript.
Supplementary Material
Collection of B. napus for determining seed erucic acid contents and their polymorphisms in Bn.FAE1-A8 and Bn.FAE1-C3.
Contributor Information
Nian Wang, Email: nian.wang1982@gmail.com.
Lei Shi, Email: Leish@mail.hzau.edu.cn.
Fang Tian, Email: fangtian@webmail.hzau.edu.cn.
Huicai Ning, Email: nhc830818@webmail.hzau.edu.cn.
Xiaoming Wu, Email: wuxm@oilcrops.cn.
Yan Long, Email: longren0807@gmail.com.
Jinling Meng, Email: jmeng@mail.hzau.edu.cn.
Acknowledgements
The authors thank Prof. Dr. Christian Jung (Plant Breeding Institute, University of Kiel, Germany) for supporting the repeated experiments and thank Dr. Bob Redden (Australian Temperate Field Crops Collection, ATFCC) for providing the seeds of two B. napus cv. Liho and Oro. We also thank Prof. Zhibiao Ye (Horticulture Department of HAU) for providing the seeds of the nine B. oleracea cultivars, and Miss Xiuli Sun (College of Plant Science & Technique in HAU) for GC analysis of erucic acid content. Financial support for this work was provided by the National Basic Research and Development Program (2006CB101600), National Nature Science Foundation of China (30600373), and National 863 High Technology Program in China (2008AA10Z152).
References
- U.N. Genome-analysis in Brassica with special reference to the experimental formation of B. napus and its peculiar mode of fertilization. Japanese Journal of Botany. 1935;7:389–452. [Google Scholar]
- Town CD, Cheung F, Maiti R, Crabtree J, Haas BJ, Wortman JR, Hine EE, Althoff R, Arbogast TS, Tallon LJ. et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss fragmentation, and dispersal after polyploidy. Plant Cell. 2006;18(6):1348–1359. doi: 10.1105/tpc.106.041665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TJ, Kim JS, Kwon SJ, Lim KB, Choi BS, Kim JA, Jin M, Park JY, Lim MH, Kim HI. et al. Sequence-Level Analysis of the Diploidization Process in the Triplicated FLOWERING LOCUS C Region of Brassica rapa . Plant Cell. 2006;18(6):1339–1347. doi: 10.1105/tpc.105.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Koo DH, Hong CP, Lee SJ, Jeon JW, Lee SH, Yun PY, Park BS, Kim HR, Bang JW. et al. Physical mapping and microsynteny of Brassica rapa ssp. pekinensis genome corresponding to a 222 kbp gene-rich region of Arabidopsis chromosome 4 and partially duplicated on chromosome 5. Mol Genet Genomics. 2005;274(6):579–588. doi: 10.1007/s00438-005-0041-4. [DOI] [PubMed] [Google Scholar]
- Metzger JO, Bornscheuer U. Lipids as renewable resources: current state of chemical and biotechnological conversion and diversification. Appl Microbiol Biotechnol. 2006;71(1):13–22. doi: 10.1007/s00253-006-0335-4. [DOI] [PubMed] [Google Scholar]
- Vles RO, Bijster GM, Timmer WG. Nutritional evaluation of low-erucic-acid rapeseed oils. Arch Toxicol Suppl. 1978. pp. 23–32. [DOI] [PubMed]
- James DW Jr, Lim E, Keller J, Plooy I, Ralston E, Dooner HK. Directed tagging of the Arabidopsis FATTY ACID ELONGATION1 (FAE1)) gene with the maize transposon activator. Plant Cell. 1995;7(3):309–319. doi: 10.1105/tpc.7.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassner MW, Lardizabal K, Metz JG. A jojoba beta-Ketoacyl-CoA synthase cDNA complements the canola fatty acid elongation mutation in transgenic plants. Plant Cell. 1996;8(2):281–292. doi: 10.1105/tpc.8.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Luhs W, Sonntag K, Zahringer U, Borchardt DS, Wolter FP, Heinz E, Frentzen M. Functional characterization of beta-ketoacyl-CoA synthase genes from Brassica napus L. Plant Mol Biol. 2001;46(2):229–239. doi: 10.1023/A:1010665121980. [DOI] [PubMed] [Google Scholar]
- Wang N, Wang Y, Tian F, King GJ, Zhang C, Long Y, Shi L, Meng J. A functional genomics resource for Brassica napus: development of an EMS mutagenized population and discovery of FAE1 point mutations by TILLING. New Phytol. 2008;180(4):751–765. doi: 10.1111/j.1469-8137.2008.02619.x. [DOI] [PubMed] [Google Scholar]
- Qiu D, Morgan C, Shi J, Long Y, Liu J, Li R, Zhuang X, Wang Y, Tan X, Dietrich E. et al. A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet. 2006;114(1):67–80. doi: 10.1007/s00122-006-0411-2. [DOI] [PubMed] [Google Scholar]
- Rahman M, Sun Z, McVetty PB, Li G. High throughput genome-specific and gene-specific molecular markers for erucic acid genes in Brassica napus (L.) for marker-assisted selection in plant breeding. Theor Appl Genet. 2008;117(6):895–904. doi: 10.1007/s00122-008-0829-9. [DOI] [PubMed] [Google Scholar]
- Katavic V, Barton DL, Giblin EM, Reed DW, Kumar A, Taylor DC. Gaining insight into the role of serine 282 in B. napus FAE1 condensing enzyme. FEBS Lett. 2004;562(1-3):118–124. doi: 10.1016/S0014-5793(04)00198-X. [DOI] [PubMed] [Google Scholar]
- Katavic V, Mietkiewska E, Barton DL, Giblin EM, Reed DW, Taylor DC. Restoring enzyme activity in nonfunctional low erucic acid Brassica napus fatty acid elongase 1 by a single amino acid substitution. Eur J Biochem. 2002;269(22):5625–5631. doi: 10.1046/j.1432-1033.2002.03270.x. [DOI] [PubMed] [Google Scholar]
- Nath UK, Goswami G, Clemens R, Becker HC, Möllers C. Inheritance and variation of erucic acid content in a transgenic rapeseed (Brassica napus L) doubled haploid population. Molecular Breeding. 2009;23(1):125–138. doi: 10.1007/s11032-008-9220-4. [DOI] [Google Scholar]
- Wu G, Wu Y, Xiao L, Li X, Lu C. Zero erucic acid trait of rapeseed (Brassica napus L) results from a deletion of four base pairs in the fatty acid elongase 1 gene. Theor Appl Genet. 2008;116(4):491–499. doi: 10.1007/s00122-007-0685-z. [DOI] [PubMed] [Google Scholar]
- Wu Y, Xiao L, Wu G, Lu C. Cloning of fatty acid elongase1 gene and molecular identification of A and C genome in Brassica species. Sci China C Life Sci. 2007;50(3):343–349. doi: 10.1007/s11427-007-0042-0. [DOI] [PubMed] [Google Scholar]
- Comai L, Young K, Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR. et al. Efficient discovery of DNA polymorphisms in natural populations by Ecotilling. Plant J. 2004;37(5):778–786. doi: 10.1111/j.0960-7412.2003.01999.x. [DOI] [PubMed] [Google Scholar]
- Gilchrist EJ, Haughn GW, Ying CC, Otto SP, Zhuang J, Cheung D, Hamberger B, Aboutorabi F, Kalynyak T, Johnson L. et al. Use of Ecotilling as an efficient SNP discovery tool to survey genetic variation in wild populations of Populus trichocarpa. Mol Ecol. 2006;15(5):1367–1378. doi: 10.1111/j.1365-294X.2006.02885.x. [DOI] [PubMed] [Google Scholar]
- Nieto C, Piron F, Dalmais M, Marco CF, Moriones E, Gomez-Guillamon ML, Truniger V, Gomez P, Garcia-Mas J, Aranda MA. et al. EcoTILLING for the identification of allelic variants of melon eIF4E, a factor that controls virus susceptibility. BMC Plant Biol. 2007;7:34. doi: 10.1186/1471-2229-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhang C, Liu L, Yu L. Development of Relationship Between A, B and C Genomes in Brassica genera. Hereditas (Beijing) 2005;27(4):671–676. [PubMed] [Google Scholar]
- Lysak MA, Koch MA, Pecinka A, Schubert I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005;15(4):516–525. doi: 10.1101/gr.3531105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum CM, Comai L, Greene EA, Henikoff S. Targeted screening for induced mutations. Nat Biotechnol. 2000;18(4):455–457. doi: 10.1038/74542. [DOI] [PubMed] [Google Scholar]
- Heckmann AB, Lombardo F, Miwa H, Perry JA, Bunnewell S, Parniske M, Wang TL, Downie JA. Lotus japonicus nodulation requires two GRAS domain regulators one of which is functionally conserved in a non-legume. Plant Physiol. 2006;142(4):1739–1750. doi: 10.1104/pp.106.089508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade AJ, Fuerstenberg SI, Loeffler D, Steine MN, Facciotti D. A reverse genetic nontransgenic approach to wheat crop improvement by TILLING. Nat Biotechnol. 2005;23(1):75–81. doi: 10.1038/nbt1043. [DOI] [PubMed] [Google Scholar]
- Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L. Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 2007;7:19. doi: 10.1186/1471-2229-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-X, Imaizumi T, Li WS, Hiromasa, Terauchi R, Ohsako T, Tominaga T. Self-EcoTILLING to identify single-nucleotide mutations in multigene family. Pesticide Biochemistry and Physiology. 2008;92(1):24–29. doi: 10.1016/j.pestbp.2008.05.001. [DOI] [Google Scholar]
- Downey RK, Craig BM. Genetic control of fatty acid biosynthesis in rapeseed (Brassica napus L) Journal of the American Oil Chemists' Society. 1964;41(7):475–478. doi: 10.1007/BF02670026. [DOI] [Google Scholar]
- Chen X, Li M, Shi J, Fu D, Qian W, Zou J, Zhang C, Meng J. Gene expression profiles associated with intersubgenomic heterosis in Brassica napus. Theor Appl Genet. 2008;117(7):1031–1040. doi: 10.1007/s00122-008-0842-z. [DOI] [PubMed] [Google Scholar]
- Qian W, Chen X, Fu D, Zou J, Meng J. Intersubgenomic heterosis in seed yield potential observed in a new type of Brassica napus introgressed with partial Brassica rapa genome. Theor Appl Genet. 2005;110(7):1187–1194. doi: 10.1007/s00122-005-1932-9. [DOI] [PubMed] [Google Scholar]
- Li MT, Li ZY, Zhang CY, Qian W, Meng JL. Reproduction and cytogenetic characterization of interspecific hybrids derived from crosses between Brassica carinata and B.rapa. Theor Appl Genet. 2005;110(7):1284–1289. doi: 10.1007/s00122-005-1965-0. [DOI] [PubMed] [Google Scholar]
- Parkin IA, Sharpe AG, Lydiate DJ. Patterns of genome duplication within the Brassica napus genome. Genome. 2003;46(2):291–303. doi: 10.1139/g03-006. [DOI] [PubMed] [Google Scholar]
- Kaushik N, Agnihotri A. GLC analysis of Indian rapeseed-mustard to study the variability of fatty acid composition. Biochem Soc Trans. 2000;28(6):581–583. doi: 10.1042/BST0280581. [DOI] [PubMed] [Google Scholar]
- Till BJ, Zerr T, Comai L, Henikoff S. A protocol for TILLING and Ecotilling in plants and animals. Nat Protoc. 2006;1(5):2465–2477. doi: 10.1038/nprot.2006.329. [DOI] [PubMed] [Google Scholar]
- Oleykowski CA, Bronson Mullins CR, Godwin AK, Yeung AT. Mutation detection using a novel plant endonuclease. Nucleic Acids Res. 1998;26(20):4597–4602. doi: 10.1093/nar/26.20.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. An alternating least-squares approach to inferring phylogenies from pairwise distances. Syst Zool. 1997;46(1):101–111. doi: 10.1093/sysbio/46.1.101. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NE, Greene EA. PARSESNP: A tool for the analysis of nucleotide polymorphisms. Nucleic Acids Res. 2003;31(13):3808–3811. doi: 10.1093/nar/gkg574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Collection of B. napus for determining seed erucic acid contents and their polymorphisms in Bn.FAE1-A8 and Bn.FAE1-C3.