

Stabilized α-helices and nonpeptidic helix mimetics have emerged as powerful molecular scaffolds for the discovery of protein-protein interaction inhibitors.[1–8] Protein-protein interactions often involve large contact areas, which are often difficult for small molecules to target with high specificity.[9–10] The hypothesis behind the design of stabilized helices and helix mimetics is that these medium-sized molecules may pursue their targets with higher specificity because of a larger number of contacts. We recently introduced a new strategy for the preparation of stabilized α-helices, termed hydrogen bond surrogate (HBS) helices, which involves replacement of one of the main chain hydrogen bonds with a covalent linkage (Figure 1A).[11] The salient feature of the HBS approach is its ability to constrain very short peptides into highly stable α-helical conformation without blocking any molecular recognition surfaces. We have extensively analyzed the conformation adopted by HBS α-helices with 2D NMR, X-ray, and circular dichroism spectroscopies.[12–14] In addition, HBS helices have been shown to target their expected protein partners with high affinity in cell-free and cell culture assays.[15–17]
Figure 1.
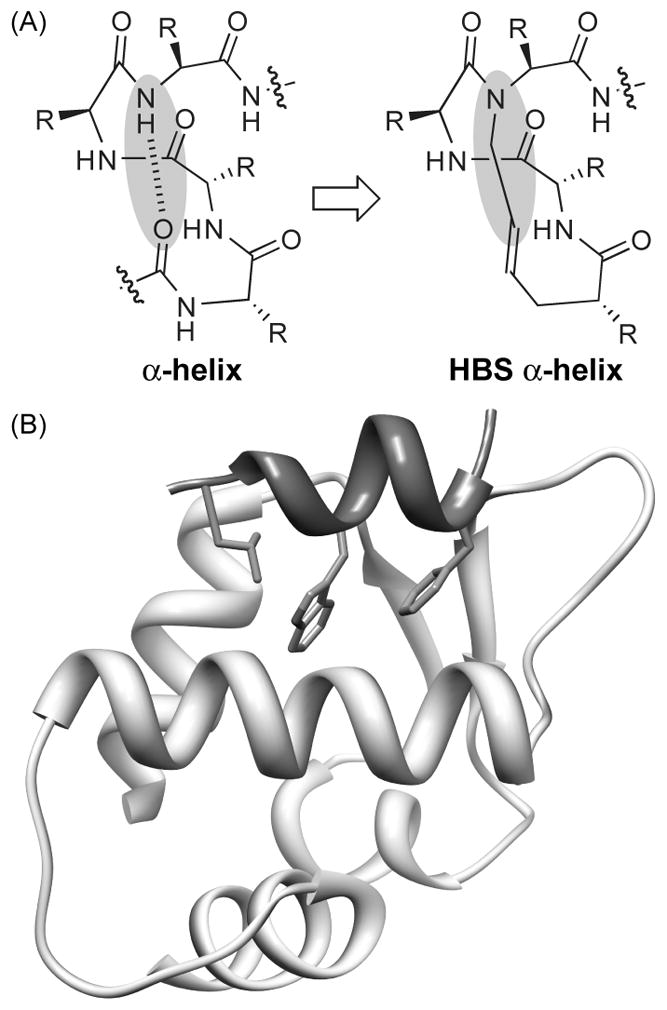
(a) Hydrogen-bond-surrogate (HBS) α-helices feature a carbon–carbon bond in place of an i and i+4 hydrogen bond. R=amino acid side chain. (b) A short helical segment (dark gray) from the p53 activation domain targets Mdm2 with three hydrophobic residues Phe-19, Trp-23, and Leu-26 forming key contacts.
A key argument for the development of larger ligands is the likely ability of these molecules to interact with protein surfaces with high specificity.[7, 15, 18] Herein we assess this hypothesis by testing the potential of HBS helices to target various protein interactions in cell-free split-protein reassembly assays.[19] In this proof-of-principle study, we utilized a well-studied model system – the complex between p53 activation domain and Murine Double Minute (MDM2). We evaluated the preference of HBS p53 α-helix to inhibit the p53/MDM2 interaction in relationship to several other helical protein interfaces, and compared its activity to a well-established small molecule inhibitor. Our results support the argument that peptide segments derived from protein interfaces are naturally optimized to interact with their cognate protein partner with high specificity.
The p53 tumor suppressor protein, generally considered the guardian of the genome, plays a fundamental role in apoptotic signaling and cell cycle arrest.[20] In response to DNA damage or cellular stress, phosphorylation of p53 signals for the expression of genes that activate apoptosis and prevent the proliferation. MDM2 and the human homolog (HDM2) have been shown to bind the activation domain (AD) of p53 and repress its activity; accordingly, the p53/MDM2 interaction has become a target for drug discovery.[21] The p53 AD adopts an α-helical conformation when bound to MDM2 (Figure 1B),[22] and several classes of stabilized helices and helix mimetics have been shown to target this interaction.[23–30] In addition, several potent small molecule inhibitors of this interaction are known.[21, 31–32] The p53/MDM2 complex, is thus an ideal model system to test both the efficacy of the designed ligands and the suitability of the split-protein assay.
We began design of HBS helices that target MDM2 by synthesizing and characterizing an unconstrained peptide (1) that closely mimics the wild-type sequence of p53 AD, HBS helix analogs (2 and 4) and a negative control (3) (Table 1). HBS helices were synthesized as described.[33–35] We utilized a previously described fluorescence polarization assay to determine the binding affinity of p53 mimetics for His-tagged MDM2,[36] and circular dichroism spectroscopy to assess their solution conformation. Peptide 1 features the wild type p5317-31 sequence with Thr-18, a non-interfacial residue, mutated to alanine to facilitate synthesis of HBS helices. This unconstrained peptide bound MDM2 with a dissociation constant of 340 nM, consistent with previous reports, suggesting that the T→A mutation does not affect binding of 1 to the target receptor (Table 1 and Figure 2B). Circular dichroism spectroscopy shows that 1 is essentially unstructured in 10% TFE in phosphate-buffered saline (Figure 2A). The constrained mimic of 1, HBS 2, is more helical (Table 1 and Supporting Information) but binds MDM2 with a seven-fold lower affinity. HBS 3 is a negative control of 2, with two key residues Phe-19 and Leu-26 mutated to alanine residues. As expected, HBS 3 fails to target the receptor with reasonable affinity (>125 μM). Zondlo and coworkers have shown that Pro-27 disrupts the binding of p53 to MDM2;[37] in agreement with their results, we find that the CD spectrum of 2 suggests a high degree of polyproline conformation (Supporting Information). Accordingly, we prepared HBS 4 in which this proline residue was mutated to an alanine. The CD spectrum of HBS 4 displays double minima at 208 and 222 nm and maxima near 190 nm consistent with those observed for canonical α-helices (Figure 2A). However, the binding affinity of 4 for MDM2 is only slightly improved compared to that of 2 (Table 1 and Figure 2B).
Table 1.
Sequence, biophysical data and binding affinities of p53 HBS α-helices designed to target MDM2.
| peptide | sequence[a] | % helicity[b] | KD (nM)[c] |
|---|---|---|---|
| 1 (p5317-31T18A) | AcEAFSDLWKLLPENNV | 14 | 340 ± 160 |
| HBS 2 | 53 | 2300 ± 210 | |
| HBS 3 | 21 | > 125000 | |
| HBS 4 | 63 | 1800 ± 170 | |
| HBS 5 | 25 | 2300 ± 110 | |
| HBS 6 | 55 | 2200 ± 400 | |
| HBS 7 | 31 | 160 ± 80 | |
| HBS 8 | 34 | > 400000 |
X and Ac denote pentenoic acid residue and acetyl group, respectively.
Values obtained from circular dichroism spectroscopy studies.
Binding affinity for MDM2 as determined by a competitive fluorescence polarization assay.
Figure 2.
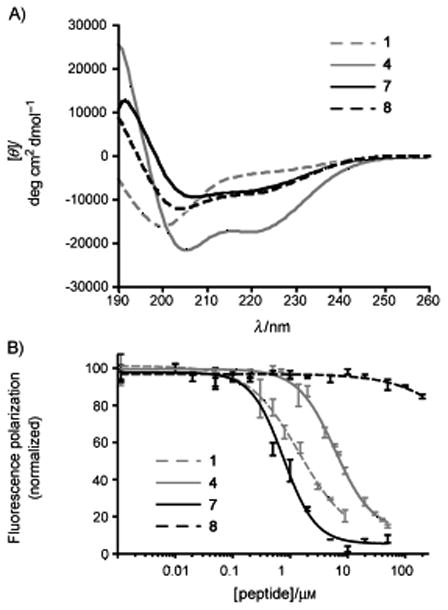
a) Circular dichroism spectra of 1, 4, 7, and 8 in 10% trifluoroethanol (TFE) in phosphate-buffered saline (PBS). b) Determination of peptide binding to His-tagged MDM2 by a fluorescence polarization assay. Circular dichroism spectra and fluorescence anisotropy binding curves for 2, 3, 5 and 6 are included in the Supporting Information.
These preliminary results suggested that perhaps one of the key residues might not be contacting the protein pocket appropriately, leading to the poor binding affinities of HBS 2 and 4. Specifically, we conjectured that Phe-19 residue which lies within the HBS macrocycle in 2 and 4 might be inaccessible even though our previous studies with HBS helices and HIV gp41 targeting did not reveal that residues within the macrocycle have trouble contacting the intended protein surfaces.[17] Nevertheless, we designed a second-generation HBS p53 mimetics in which the placement of Phe-19 was varied (Scheme 1). As part of these efforts, we also sought to develop shorter stabilized helices that target MDM2 with high affinity but consist of the minimal recognition epitope of p53 encompassing the three important residues. HBS 5 is a shorter analog of 2 but binds the protein with similar affinity to 2 (and 4), confirming the limited role played by residues 27-31 in MDM2 binding. The Phe-19 residue is retained at the N-alkyl position in HBS 5, but moved to inside or outside of the macrocycle in 6 and 7, respectively (Scheme 1). HBS 6 and 7 also feature a serine residue at position 27, in place of the proline residue. This mutation has been suggested to improve the binding of p53 peptides.[37]
Scheme 1.

Placement of Phe-19 residue, from left to right, at the N-alkyl residue, inside and outside the HBS macrocycle.
Relative placement of the phenylalanine residue results in a dramatic change in the binding affinity of the compounds for MDM2. The affinity of 6 for the target is similar to that of 5 but HBS 7 binds the receptor with a 14-fold improved binding constant (Kd = 160 nM). Importantly, the negative control 8, in which Phe-19 and Leu-26 have been mutated to alanine residues, is a poor binder (Table 1 and Figure 2B). MDM2 can, thus, be targeted with sequence selectivity by HBS helices, and HBS 7 is a high affinity ligand for MDM2.
We next evaluated the preference of HBS 7 for MDM2 in comparison to various other proteins that are known to accommodate helical peptides,[38] including the Bcl-2 family proteins,[39] TAZ1 domain of CBP,[15, 40–41] and MDM4 (or MDMX), which is a p53 binding protein closely related to MDM2 (Figure 3).[42] We utilized a cell-free split-protein luciferase assay to screen for protein complexes that can be inhibited by peptide 1, HBS 7, and HBS 8.[19] This assay examines the ability of two protein partners, each conjugated to a different half of the luciferase protein, to reassemble and impart luminescence in the presence of luciferin (Figure 4A).[43–44] Inhibition of the protein complex formation reduces relative bioluminescence and allows assessment of potential protein-protein interaction inhibitors in a concentration-dependent manner. The results of the split-protein assays performed at 10 μM inhibitor concentrations are shown in Figure 4B. The optimized MDM2 ligand, HBS 7, targets the complex with high specificity; whereas, the negative control, HBS 8, does not inhibit any of the complexes tested in this study. The wild type p53 peptide, 1, is marginally active for MDM2 in this assay. To gauge the suitability of the split-protein assay, we compared the specificity of HBS 7 for the various complexes with nutlin-3, which is a well-characterized agonist for p53/MDM2.[21] Both nutlin-3 and HBS 7 displayed remarkably similar and high specificities for MDM2. It should be noted that nutlin-3 is a highly engineered lead from the pharmaceutical industry, whereas HBS 7 is a direct mimic of a naturally optimized peptide sequence whose specificity and affinity may nevertheless be further improved.
Figure 3.
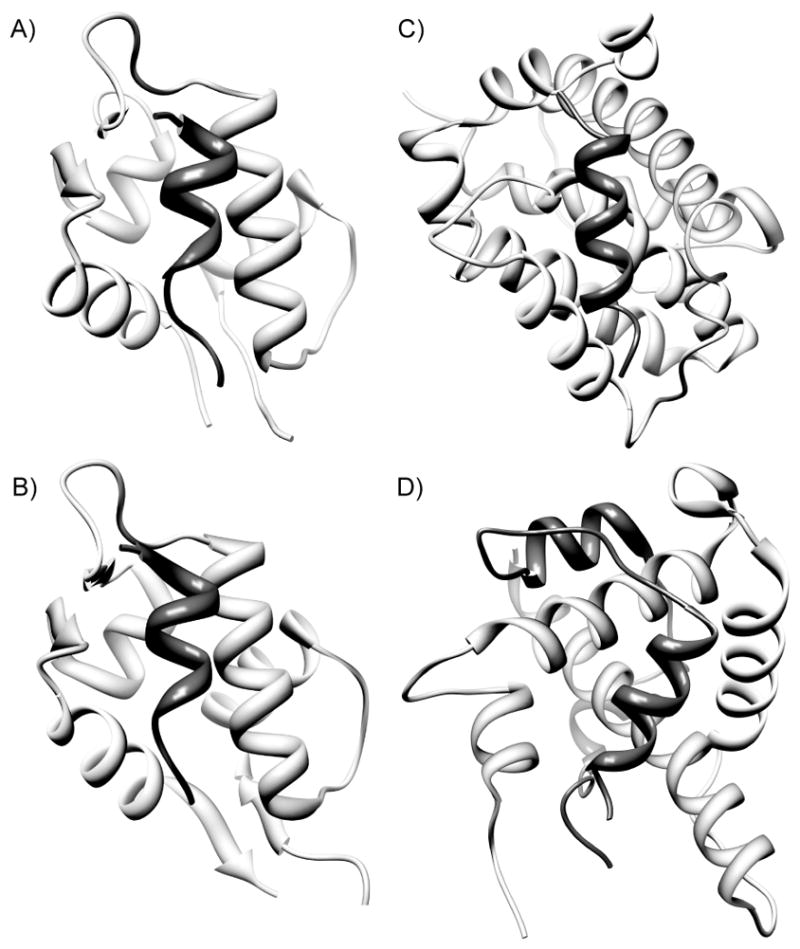
Examples of helical protein interfaces evaluated in the current study: (a) p53 (dark gray)/MDM2 (light gray), (b) p53 (dark gray)/MDM4 (light gray), (c) Bak BH3 (dark gray)/Bcl-xL (light gray), and Hif-1α (dark gray)/CBP (light gray). PDB codes: 1YCR, 2DAB, 1BXL and (d) 1L8C.
Figure 4.

Schematic for the cell-free interrogation of protein-protein interaction inhibitors. Reassembly of split-luciferase fusions of the interaction of interest results in luminescence. Inhibition of the protein complex formation results in abolishment of luciferase activity. Luciferase assays performed with 10 μM peptide, HBS helices and nutlin-3.
The last decade has seen enormous progress in the design of peptidic and nonpeptide helix mimetic. A number of these mimetics have been shown to bind chosen protein targets with high affinity including several for MDM2; however, with a few exceptions, the specificity of the designed ligands has rarely been rigorously examined. We find that the split-protein assay provides a rapid approach to assess the specificity of synthetic inhibitors of protein-protein interactions. The present work with a stabilized helix supports the hypothesis that peptide sequences adapted from protein interfaces are naturally evolved to interact with cognate protein surfaces with high specificity.
Supplementary Material
Acknowledgments
We thank Drs. Susan and Neal Zondlo (University of Delaware) for the His-MDM2 construct. I.G. thanks the NSF (CHE-0548264) for financial support of this work. P.S.A. is grateful to the NIH for financial support of this work (GM073943), and thanks the NSF for equipment Grant CHE-0958457.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
Contributor Information
Prof. Indraneel Ghosh, Email: ghosh@email.arizona.edu.
Prof. Paramjit S. Arora, Email: arora@nyu.edu.
References
- 1.Henchey LK, Jochim AL, Arora PS. Curr Opin Chem Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garner J, Harding MM. Org Biomol Chem. 2007;5:3577–3585. doi: 10.1039/b710425a. [DOI] [PubMed] [Google Scholar]
- 3.Davis JM, Tsou LK, Hamilton AD. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 4.Yin H, Hamilton AD. Angew Chem Int Ed. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 5.Horne WS, Gellman SH. Acc Chem Res. 2008;41:1399–1408. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seebach D, Gardiner J. Acc Chem Res. 2008;41:1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- 7.Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 10.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 11.Patgiri A, Jochim AL, Arora PS. Acc Chem Res. 2008;41:1289–1300. doi: 10.1021/ar700264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Wang D, Zheng Q, Lu M, Arora PS. J Am Chem Soc. 2008;130:4334–4337. doi: 10.1021/ja077704u. [DOI] [PubMed] [Google Scholar]
- 13.Wang D, Chen K, Dimartino G, Arora PS. Org Biomolec Chem. 2006;4:4074–4081. doi: 10.1039/b612891b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Chen K, Kulp JL, III, Arora PS. J Am Chem Soc. 2006;128:9248–9256. doi: 10.1021/ja062710w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. J Am Chem Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, Liao W, Arora PS. Angew Chem Int Ed. 2005;44:6525–6529. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Lu M, Arora PS. Angew Chem Int Ed. 2008;47:1879–1882. doi: 10.1002/anie.200704227. [DOI] [PubMed] [Google Scholar]
- 18.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Proc Natl Acad Sci U S A. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porter JR, Stains CI, Jester BW, Ghosh I. J Am Chem Soc. 2008;130:6488–6497. doi: 10.1021/ja7114579. [DOI] [PubMed] [Google Scholar]
- 20.Chene P. Nat Rev Cancer. 2003;3:102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 21.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 22.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 23.Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew Chem Int Ed. 2005;44:2704–2707. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- 24.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. J Am Chem Soc. 2007;129:2456–2457. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fasan R, Dias RL, Moehle K, Zerbe O, Vrijbloed JW, Obrecht D, Robinson JA. Angew Chem Int Ed Engl. 2004;43:2109–2112. doi: 10.1002/anie.200353242. [DOI] [PubMed] [Google Scholar]
- 26.Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J Am Chem Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- 27.Murray JK, Gellman SH. Biopolymers. 2007;88:657–686. doi: 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]
- 28.Plante JP, Burnley T, Malkova B, Webb ME, Warriner SL, Edwards TA, Wilson AJ. Chem Commun. 2009:5091–5093. doi: 10.1039/b908207g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakurai K, Schubert C, Kahne D. J Am Chem Soc. 2006;128:11000–11001. doi: 10.1021/ja063102j. [DOI] [PubMed] [Google Scholar]
- 30.Shaginian A, Whitby LR, Hong S, Hwang I, Farooqi B, Searcey M, Chen J, Vogt PK, Boger DL. J Am Chem Soc. 2009;131:5564–5572. doi: 10.1021/ja810025g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shangary S, Wang S. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu S, Qin D, Shangary S, Chen J, Wang G, Ding K, McEachern D, Qiu S, Nikolovska-Coleska Z, Miller R, Kang S, Yang D, Wang S. J Med Chem. 2009;52:7970–7973. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman RN, Arora PS. Org Lett. 2006;8:5825–5828. doi: 10.1021/ol062443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimartino G, Wang D, Chapman RN, Arora PS. Org Lett. 2005;7:2389–2392. doi: 10.1021/ol0506516. [DOI] [PubMed] [Google Scholar]
- 35.Patgiri A, Witten MR, Arora PS. Org Biomol Chem. 2010;8:1773–1776. doi: 10.1039/c000905a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knight SM, Umezawa N, Lee HS, Gellman SH, Kay BK. Anal Biochem. 2002;300:230–236. doi: 10.1006/abio.2001.5468. [DOI] [PubMed] [Google Scholar]
- 37.Zondlo SC, Lee AE, Zondlo NJ. Biochemistry. 2006;45:11945–11957. doi: 10.1021/bi060309g. [DOI] [PubMed] [Google Scholar]
- 38.Jochim AL, Arora PS. Mol BioSyst. 2009;5:924–926. doi: 10.1039/b903202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petros AM, Olejniczak ET, Fesik SW. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 40.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Proc Natl Acad Sci U S A. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Proc Natl Acad Sci U S A. 2002;99:5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, Mills N, Smithson DC, Regni CA, Bashford D, Cicero SA, Schulman BA, Jochemsen AG, Guy RK, Dyer MA. J Biol Chem. 2010;285:10786–10796. doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luker KE, Smith MCP, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Proc Natl Acad Sci U S A. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Nat Biotech. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.