

Abstract
Background
Genetic recombination maps provide important frameworks for comparative genomics, identifying gene functions, assembling genome sequences and for breeding. The molecular recombination map currently available for the model eudicot Antirrhinum majus is the result of a cross with Antirrhinum molle, limiting its usefulness within A. majus.
Results
We created a molecular linkage map of A. majus based on segregation of markers in the F2 population of two inbred lab strains of A. majus. The resulting map consisted of over 300 markers in eight linkage groups, which could be aligned with a classical recombination map and the A. majus karyotype. The distribution of recombination frequencies and distorted transmission of parental alleles differed from those of a previous inter-species hybrid. The differences varied in magnitude and direction between chromosomes, suggesting that they had multiple causes. The map, which covered an estimated of 95% of the genome with an average interval of 2 cM, was used to analyze the distribution of a newly discovered family of MITE transposons and tested for its utility in positioning seven mutations that affect aspects of plant size.
Conclusions
The current map has an estimated interval of 1.28 Mb between markers. It shows a lower level of transmission ratio distortion and a longer length than the previous inter-species map, making it potentially more useful. The molecular recombination map further indicates that the IDLE MITE transposons are distributed throughout the genome and are relatively stable. The map proved effective in mapping classical morphological mutations of A. majus.
Background
Antirrhinum majus, the garden snapdragon, has been used as a model system for genetics since the early 20th Century [1]. It is a member of a monophyletic group of about twenty five species that are native to the Mediterranean region share the same chromosome number (2n = 16) and are able to form fertile hybrids with each other [2]. The majority of species are allogamous, though cultivated A. majus lines and a few other wild species can self-fertilize.
A collection of A. majus mutants has been produced from some laboratory lines of A. majus selected for high transposon activity [3]. In several cases, these have been used to clone the corresponding genes by transposon tagging (e.g. [4-10]). In addition there is a collection of roughly four hundred classical mutants, mostly in an isogenic background (Sippe 50) [11,12]. The majority of these mutants does not show the genetic instability characteristic of transposon-induced mutations, and therefore have limited use for transposon tagging. The alternative approach of gene isolation by positional cloning is currently restricted by the availability of molecular recombination maps in Antirrhinum, though it has recently been successful in isolating the fistulata (fis) gene [13]. Though a classical fis mutation was genetically stable, it is caused by insertion of a miniature inverted-repeat transposable element (MITE), which is present in relatively low copy-number in all Antirrhinum species. Because the transposon family appeared relatively inactive it was called IDLE.
The existing molecular recombination map for Antirrhinum was built using the F2 of a cross between A. majus (line 165E) and a wild relative, A. molle [14]. The map identified eight linkage groups and use of common loci had allowed these to be related to a classical genetic map and to the A. majus chromosomes by in situ hybridization [15]. However, the majority of markers from the A. molle x A. majus hybrid showed significantly distorted transmission, which are likely to have affected the accuracy of the map, and the map also contained clusters of loci consistent with chromosome rearrangements between the species [12,15]. Such rearrangements were also suggested by observation of chromosomes [15]. These two factors would hinder attempts to map A. majus mutations in crosses to A. molle. A further disadvantage of using inter-species crosses to map A. majus mutations is that A. majus and A. molle differ in many morphological characters, including plant and organ size. Segregation of natural variation would therefore be likely to obscure the effects of mutations in hybrid mapping populations.
We therefore developed a linkage map of A. majus using the inbred lines Sippe50 and 165E. The map consists of 302 markers (protein coding sequences, AFLP and transposons), covering nearly 95% of the genome. As a proof of concept, we placed on the map six mutations affecting floral and overall size. We also mapped the distribution of IDLE transposons, revealing that they are allocated with coding genes in all Antirrhinum chromosomes.
Results & discussion
Construction of a molecular linkage map for A. majus
To construct a molecular linkage map for A. majus we crossed two inbred lines, 165E and Sippe 50. Line 165E originated from cultivated A. majus in the UK and is phenotypically distinct from Sippe 50, which was derived in Germany, possibly from a wild accession [11,16]. A single F1 progeny was self-pollinated to produce an F2 mapping population of 96 plants. This population therefore contains 192 recombinant copies of each chromosome, sufficient for mapping loci to a resolution of ~ 1 cM. The F2 population was genotyped at 377 loci. These included 90 protein-coding genes, in which polymorphisms were detected by sequencing the alleles from both parents. The identities of the protein-coding genes are given in Table 1. The remaining markers mainly comprised AFLP and insertions of the MITE transposon IDLE [13]. The genotype data were used to estimate a recombination map. Fifty-nine AFLP markers (28% of the total) and one dominant IDLE insertion were present in significantly more or fewer F2 plants than expected and could either not be mapped or mapped only by reducing support for linkage groups significantly. These markers were therefore rejected. The remaining markers formed a map comprising 90 protein-coding genes, 87 of which were mapped as co-dominant CAPS or size polymorphisms, 159 dominant AFLP and 53 IDLE insertions (10 with co-dominant alleles and 43 dominant markers). A complete list of primers for each marker and the corresponding map position can be found in Additional file 1. At nine loci AFLP bands from both parents showed complete linkage in repulsion and were subsequently treated as synthetic co-dominant markers.
Table 1.
List of EST-based markers and functional annotation.
| Sequence number | EST annotation | Length in bp | min. eValue |
|---|---|---|---|
| EM:AMA558924 | thioredoxin peroxidase | 723 | 1.0E-1.12106E-85 |
| EM:AJ801757 | proline-rich apg-like protein | 670 | 1.0E-2.50815E-86 |
| EM:AJ794773 | o-linked c-transferase | 758 | 1.0E-2.06855E-101 |
| EM:AJ804794 | histone h1 | 556 | 1.0E-1.27731E-27 |
| EM:AJ794598 | at4g20410-like protein | 736 | 1.0E-1.80515E-107 |
| EM:AJ805499 | stearoyl-acyl carrier protein desaturase | 481 | 1.0E-9.70457E-56 |
| EM:X57295 | TAP1 protein precursor | 5280 | 1.0E-1.98821E-44 |
| EM:AY072736 | HIRZINA KNOX protein | 1462 | 1.0E-0.0 |
| EM:AJ802708 | Eukaryotic translation initiation factor 6 | 630 | 1.0E-1.16373E-87 |
| EM:AJ794444 | mgc108135 protein | 675 | 1.0E-6.11236E-80 |
| EM:AJ802293 | translational inhibitor protein | 589 | 1.0E-1.37912E-65 |
| EM:AJ803243 | 60 s ribosomal protein l18 | 731 | 1.0E-2.80747E-92 |
| EM:AJ559267 | heavy-metal-associated domain-containing protein | 504 | 1.0E-2.98463E-13 |
| EM:AJ790658 | urease accessory protein g | 561 | 1.0E-3.52407E-97 |
| EM:AJ792971 | gtp-binding protein | 628 | 1.0E-1.89892E-82 |
| EM:AJ560201 | iron-sulfur cluster assembly protein | 768 | 1.0E-3.45238E-59 |
| EM:AJ620906 | STYLOSA co-repressor | 2865 | 1.0E-0.0 |
| EM:AJ806659 | atp-dependent clp protease proteolytic subunit | 711 | 1.0E-1.54692E-116 |
| EM:AJ560074 | leucine rich repeat protein | 754 | 1.0E-2.04947E-101 |
| EM:AM422773 | YABBY4 transcription factor | 1395 | 1.0E-7.9976E-177 |
| EM:X97639 | cyclin-dependent kinase, CDC2a | 1038 | 1.0E-2.78255E-170 |
| EM:AJ806654 | C2H2 zinc-finger protein | 530 | 1.0E-4.79832E-31 |
| EM:AM422772 | YABBY2 transcription factor | 1373 | 1.0E-3.22687E-114 |
| EM:AJ568130 | cyclic nucleotide-regulated ion channel | 620 | 1.0E-3.55048E-94 |
| EM:AJ568031 | amino acid permease familyexpressed | 1459 | 1.0E-1.38642E-78 |
| EM:AJ800340 | at3g04780 f7o18_27 | 721 | 1.0E-1.77924E-75 |
| EM:AJ568062 | chloroplast translation initiation factor 2 | 415 | 1.0E-4.51282E-45 |
| EM:S53900 | PLENA MADS-box transcription factor | 1073 | 1.0E-1.61386E-120 |
| EM:AJ801384 | actin associated protein | 538 | 1.0E-5.43416E-33 |
| EM:AJ800042 | multiple stress-responsive zinc-finger protein | 781 | 1.0E-1.45489E-60 |
| EM:AJ805889 | centromere microtubule binding protein cbf5 | 692 | 1.0E-3.39584E-97 |
| EM:AJ808934 | transcription factor | 884 | 1.0E-2.17962E-88 |
| EM:AJ568063 | polygalacturonase-inhibiting protein | 734 | 1.0E-1.19729E-66 |
| EM:AJ804237 | histidinol dehydrogenase | 634 | 1.0E-2.00488E-95 |
| EM:AJ795662 | flavonoid 3-o-glucosyltransferase | 750 | 1.0E-9.60942E-83 |
| EM:AJ800415 | erwinia induced protein 2 | 615 | 1.0E-2.67616E-49 |
| EM:AJ796551 | Frigida | 666 | 1.0E-5.06947E-39 |
| EM:AJ796122 | p-p-bond-hydrolysis-driven protein transmembrane transporter | 747 | 1.0E-3.07514E-73 |
| EM:AJ800197 | atp-dependent protease clp atpase subunit | 594 | 1.0E-3.10775E-69 |
| EM:AJ559052 | t-complex protein 1 epsilon tcp-1- | 690 | 1.0E-2.62699E-116 |
| EM:AJ803361 | monodehydroascorbate reductase | 730 | 1.0E-7.35389E-109 |
| EM:AJ802640 | isochorismatase hydrolase | 779 | 1.0E-1.51824E-94 |
| EM:AY223518 | LIP1 apetala2-like transcription factor | 1845 | 1.0E-0.0 |
| EM:AJ805150 | photoassimilate-responsive protein par-like protein | 530 | 1.0E-7.5899E-61 |
| EM:AJ620905 | STYLOSA1 co-repressor | 3123 | 1.0E-0.0 |
| EM:AJ801986 | at-rich element binding factor 3 | 590 | 1.0E-8.38628E-55 |
| EM:X68831 | GLOBOSA MADS-box transcription factor | 6108 | 1.0E-5.69779E-27 |
| EM:X76995 | polygalacturonase-inhibiting protein | 3545 | 1.0E-4.66003E-119 |
| EM:AJ558819 | psap psi-p ptac8 tmp14 (thylakoid membrane phosphoprotein of 14 kda) dna binding | 762 | 1.0E-2.07028E-40 |
| EM:AJ793550 | trna-methyltransferase subunit | 535 | 1.0E-7.89349E-61 |
| EM:AJ620909 | SEUSS3A co-repressor | 3683 | 1.0E-0.0 |
| EM:AJ800998 | Transcription factor lim | 710 | 1.0E-3.7918E-99 |
| EM:AJ620910 | seu3b protein | 2004 | 1.0E-2.36396E-82 |
| EM:AY451399 | CRABSCLAW-like YABBY transcription factor | 732 | 1.0E-1.78657E-70 |
| EM:AJ794665 | nuclear cap binding protein subunit 2 | 673 | 1.0E-1.25817E-85 |
| EM:AJ802365 | ---NA--- | 583 | 1.0E-1.68821E-23 |
| EM:AJ801224 | ---NA--- | 613 | 1.0E-2.33555E-13 |
| EM:AJ794216 | nuclear RNA binding | 758 | 1.0E-1.84607E-33 |
| EM:AJ794472 | serine threonine protein kinase | 746 | 1.0E-1.35578E-75 |
| EM:AJ568099 | pgr5-like a | 1478 | 1.0E-9.46041E-75 |
| EM:AJ791655 | bzo2h3 (arabidopsis thaliana basic leucine zipper 63) dna binding transcription factor | 758 | 1.0E-9.80266E-27 |
| EM:AJ790549 | immunophilin | 566 | 1.0E-1.28799E-54 |
| EM:M55525 | FLORICAULA transcription factor | 1545 | 1.0E-0.0 |
| EM:AJ802861 | ---NA--- | 728 | 1.0E-1.22273E-47 |
| EM:AJ799233 | delta-12 oleate desaturase | 736 | 1.0E-8.2567E-116 |
| EM:AJ804300 | protein | 683 | 1.0E-6.22658E-88 |
| EM:AJ803800 | af319475_1 alpha-expansin 9 precursor | 590 | 1.0E-5.56035E-91 |
| EM:AJ803115 | protein | 654 | 1.0E-9.83593E-72 |
| EM:AJ790136 | tfIIb-related protein | 677 | 1.0E-1.59963E-96 |
| EM:AJ805759 | eukaryotic translation initiation factor 6 | 689 | 1.0E-1.55443E-110 |
| EM:AJ559280 | tic20-like protein | 710 | 1.0E-6.95853E-77 |
| EM:AJ620907 | SEUSS1 protein | 3495 | 1.0E-0.0 |
| EM:AJ789733 | ---NA--- | 675 | 1.0E-6.54566E-74 |
| EM:AJ800463 | tubulin alpha-6 chain | 605 | 1.0E-4.42202E-40 |
| EM:AJ791186 | Glutaredoxin-like protein | 684 | 1.0E-1.57483E-30 |
| EM:AJ790836 | b12 d protein | 564 | 1.0E-1.21222E-36 |
| EM:AJ794452 | duf1230-containing protein | 763 | 1.0E-1.97117E-91 |
| EM:AJ793362 | phosducin-like protein 3 | 578 | 1.0E-6.36625E-60 |
| EM:Y16313 | CYCLOIDEA TCP transcription factor | 861 | 1.0E-1.34443E-129 |
| EM:AY223519 | LIP2 apetala2-like transcription factor | 1914 | 1.0E-0.0 |
| EM:AJ800866 | polycomb protein ez1 | 507 | 1.0E-1.11628E-31 |
| EM:AJ805209 | rer1a protein | 582 | 1.0E-1.72796E-60 |
| EM:AJ568117 | phenylalanine ammonia-lyase | 619 | 1.0E-7.46548E-60 |
| EM:AJ791765 | microtubule motor | 745 | 1.0E-7.82606E-45 |
| EM:AJ808258 | flu (fluorescent in blue light) binding | 663 | 1.0E-2.25085E-39 |
| EM:AJ793379 | rpm1-interacting protein 4 | 580 | 1.0E-1.07793E-22 |
The ESTs were automatically annotated using BLAST2GO. The corresponding EST annotation was performed using a minimal threshold of e-6. Those genes with high homology to genes with unknown function were annotated as NA. Sequences with known mutant phenotypes in Antirrhinum are given their Antirrhinum gene names.
The resulting map comprised eight linkage groups with a total length of 562 cM that was estimated to cover 95% of the genome (Figure 1). At this level of coverage, the average interval between markers was 2.0 cM, with 88% of the genome estimated to lie within 2.0 cM of a marker and 99% within 5 cM. Although the average interval between co-dominant markers was 6.0 cM, a similar proportion of the genome (83%) was within 2.0 cM of the nearest co-dominant marker. Assuming a haploid genome size of 3.6 × 108 bp for A. majus [17], a marker interval of 2.0 cM represents on average 1.28 Mbp of DNA.
Figure 1.

A molecular linkage map for Antirrhinum majus Sippe 50 × 165E. The eight linkage groups are oriented, numbered and named as in previous Antirrhinum maps. Positions are given in centiMorgans (Kosambi). Protein-coding loci are named with their EMBL accession numbers as in Table 1 and with their Antirrhinum gene names in italics, where their functions are known from mutants. IDLE denotes a locus carrying an insertion of an IDLE transposon in one of the parents and loci with the suffix P are AFLP (see Materials & Methods for AFLP nomenclature). Loci with co-dominant alleles are shown in bold and those with dominant alleles in regular type.
Map comparison
A previous molecular map for Antirrhinum had been produced from the F2 progeny of a cross between A. majus (line 165E) and the wild species A. molle [14]. To allow identification and alignment of linkage groups in the two populations, the genotypes from the previous mapping population were used to reconstruct a map using the same parameters as for the A. majus x A. majus F2. Markers common to both maps allowed identification of corresponding linkage groups and their orientations (Additional file 2).
The total A. majus map was about 54% larger than for A. majus x A. molle. However the variations in length differed in magnitude and direction between chromosomes (Figure 2). Two linkage groups (5 and 3) were slightly smaller in map A, while the remainders were significantly longer, suggesting that the causes of length differences varied between chromosomes.
Figure 2.
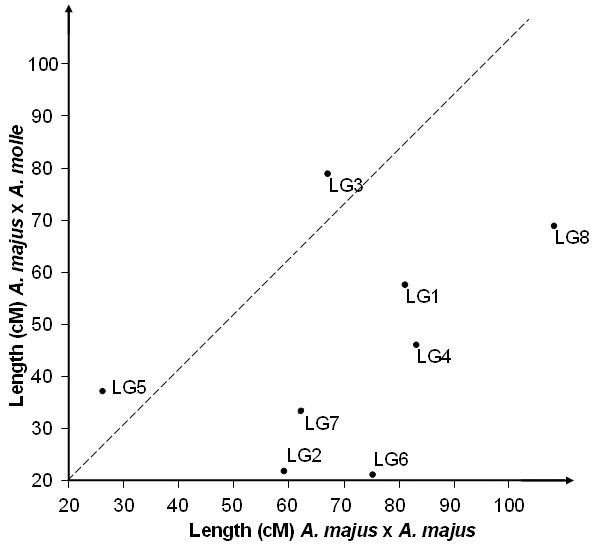
Comparative genetic lengths of chromosomes in the A. majus x A. molle and A. majus x A. majus maps. The estimated lengths of each of the eight linkage groups in the two maps are plotted against each other.
Previous studies have reported both smaller and larger maps for intra-specific crosses as compared to inter-specific crosses [18,19]. Several factors might contribute to variations in map lengths for Antirrhinum and might differ between chromosomes. Of particular relevance to the utility of the A. majus map is the possibility that the two marker sets cover different parts of the genome. However, this seems unlikely, because although the two maps contain a different number of loci (296 in map A and 227 in map B) they are estimated to cover a similar proportion of the genome (95% in A and 94% in B using Method 4 [20].) Moreover, randomly removing 69 markers from A, to make the numbers of markers the same in both populations, reduced the length of map A by an average of only 2% (Additional file 3). Similarly maps made only with dominant protein-coding genes from each F2 population showed the same trends in map length differences (data not shown), suggesting that they are not dependent on the number or types of markers used.
The insensitivity of map length to the type of marker also suggests that the 51 mapped IDLE transposons were relatively stable, because excision of an IDLE in members of the mapping population would result in the wrong parental origin being assigned to its locus and an over-estimation of recombination frequencies. The relative stability of IDLE markers was further supported by the finding that they were no more likely than other marker types to have an apparent recombination breakpoint immediately next to them, as would be expected if excision had resulted in an incorrect genotype.
Although many transposon families are predominant components of heterochromatin, MITE transposons have commonly been found associated with gene-rich regions [21,22]. This is consistent with the observed distribution of IDLE insertions in Antirrhinum, which are interspersed with protein-coding genes and do not appear to be clustered in centromeric or telomeric regions.
Transmission ratio distortion differences between inter and intra-specific maps
At least some of the length variation between maps might be attributed to transmission ratio distortion (TRD). This was more marked in the interspecies cross, in which loci representing most of the genome deviated significantly from their expected Mendelian ratios (Figure 3). It was most severe for LG6, in which A. molle carries a functional gametophytic self-incompatibility (S) locus. This prevented recovery of F2 plants homozygous for A. molle alleles at LG6 unless recombination had occurred between the marker and the S locus. In contrast, A. majus lacks a functional S locus and shows only mildly distorted transmission of markers from LG6 (Figure 3). TRD can lead to under-estimation of map distances [23] and loss of marker information, for example no dominant markers closely linked to the A. molle S allele were identified in map B. It can also lead to markers being wrongly assigned to linkage groups. The lower level of TRD in map A therefore justifies the use of mapping populations of A. majus rather than inter-species hybrids.
Figure 3.

Transmission of parental alleles to F2 mapping populations. For the A. majus x A. majus population (a) the proportion of Sippe 50 homozygotes (crosses), 165E homozygotes (triangles) and half the proportion of heterozygotes (diamonds) is plotted for each locus according to its position in the eight linkage groups (LG). The solid horizontal line represents the expected average proportion (0.25) of each genotype class that is expected in the absence of distorted transmission. The solid and broken grey lines represent the approximate thresholds for significantly distorted genotype frequencies at the 0.95 and 0.99 levels, respectively. Genotype frequencies for the A. majus x A. molle population are shown in (b). The genotype frequencies and significance levels are represented as in a), except that crosses denote A. molle homozygotes.
It was previously suggested that the clustering of markers in map B may be caused by chromosome inversions that distinguished A. majus from A. molle [14], preventing mapping of loci that lie within inversions. However, there is significant clustering (p < 0.0001) of markers in both maps and significantly more clustering in map A than map B. Since fewer rearrangements are expected between two A. majus lines than between A. majus and A. molle clusters of markers appear unlikely to represent inversions.
Map validation by mapping mutations affecting size
One possible use of a molecular map is to determine the chromosomal positions of loci that have been identified by mutation. This can, for example, classify mutations that are potentially allelic, which is particularly useful for dominant mutations, and allows isolation of the affected genes on the basis of their positions. We therefore tested the utility of the A. majus map in determining the position of six classical mutations affecting aspects of plant size (Figure 4). All six mutations were in the Sippe 50 mutant background and therefore crossed to wild-type 165E to generate F2 mapping populations.
Figure 4.

Phenotypic characteristics of the mutants used to validate the map. Phenotypes of compacta (a), formosa (b), compacta ähnlich (c) and nana (d), Pictures show wild type on the left and mutant on the right. The mutant heroina (e) above and wild type below. Stems of hero correspond to same internode in siblings. The mutant Nitida (f) wild type left, mutant right.
The nana (na) mutation described at the end of the XIXth century in the Vilmorin catalog [24], reduces plant size in a recessive fashion and flowers early irrespective of photoperiod. The na mutant phenotype segregated as expected for a recessive mutation in the F2 generation of the cross to 165E. However, a second allele nanalargiflora, which caused a somewhat weaker phenotype in the Sippe 50 genetic background [12], could not be distinguished from wild-type in F2 populations produced by crossing to 165E line. This highlights a potential problem arising from suppression of a weak mutant phenotype in a cross between two lines that differ, albeit slightly, in morphology. Another difficulty was identified in the case of the recessive muscoides (mus) mutation, which causes dwarfism. No mus mutants were initially identified in the F2 of the cross with 165E. However, mus mutants were recovered at a low frequency (2 out of 60 plants) when F2 seeds were germinated in Petri dishes, suggesting that the mus mutation can be lethal in the 165E genetic background.
The mutant hero affected stem thickness, a trait that seems to be partly controlled by genes affecting floral size in Arabidopsis like Bigbrother and Kluh [25,26]. However hero did not show a statistical difference from wild-type in lateral organ size, either in the original Sippe 50 background or in F2 populations, and segregated as expected of a recessive mutation in the F2 (data not shown).
Four mutations affecting floral size also segregated as expected in the F2 populations produced by crossing to 165E with compacta (co), compacta-ähnlich (coan) and formosa (fo) mutations appearing fully recessive, and Nitida (Ni) as semi-dominant [11]. In the case of Ni and co mutants, their phenotypes in the F2 were similar to those of the original background while coan, and fo mutants showed slightly larger differences from wild-type.
The mutants affect floral size in different ways, coan decreases flower size without affecting vegetative body size [27], while the co mutation reduces both flower and lateral organ size. The fo mutation increases floral size, [28] while the na mutation reduces plant height and leaf width without significantly affecting flower size while the Ni mutation reduces the sizes of flowers, leaves and internodes in a dosage-dependent fashion.
As an initial approach, the mutations were mapped by bulk-segregant analysis [29]. DNA was extracted from several pools of four plants that shared the same phenotype and screened with a CAPS marker located in a middle region of each chromosome arm (a total of 16 markers). Markers that χ2 tests suggested were not linked to the size mutation were rejected. Where evidence for linkage was found, additional CAPS markers from the same chromosome regions were used to analyze individual F2 plants to refine map positions. Statistically significant linkage was found between the additional markers and the mutations in all cases (Table 2). The distance between a mutation and the closet marker ranged from the coan locus and the marker AJ790136 in LG3, which showed no recombination in 43 homozygous mutants, to na and AJ568062 in LG2 which were separated by about 26 cM.
Table 2.
Map position of six mutants.
| Mutant | Closest Marker | LG | Marker position (cM) | Kosambi distance (cM) | n | significance |
|---|---|---|---|---|---|---|
| coan | AJ790136 | LG3 | FLO | 0 | 43 | * |
| co | AJ568117 | LG6 | 30 | 3.1 | 49 | * |
| Fo | SEU3A | LG7 | 34 | 7.2 | 21 | * |
| hero | SEU3A | LG7 | 34 | 4.6 | 22 | * |
| na | AJ568062 | LG2 | 52 | 25.9 | 21 | * |
| Ni | PLE (S53900) | LG2 | 55 | 9.1 | 50 | * |
Mutants were originally obtained in the Sippe 50 background and mapped by crossing with 165E. Markers were considered significantly linked for χ2 tests of p < 0.05
We have therefore shown that it is feasible to map mutations in crosses between these two Antirrhinum lines, even mutations with relatively subtle effects on plant size. Extending this approach to map based cloning should become more feasible as the density of molecular markers in Antirrhinum increases. However, the ability to map with even moderate resolution can be used to identify potentially allelic mutations, including natural variants. One of the major features of Antirrhinum species is that they differ widely from each other in size. Several genes underlying this size variation have been mapped as quantitative trait loci (QTL), e.g. [30]. Like the size mutants analyzed here, the QTL can affect a single type of organ or have more pleiotropic effects. It should now be possible to identify whether any classical size mutations might correspond to size QTL on the basis of map positions and so select candidate mutations for more allelism tests. A corresponding classical mutation can facilitate QTL isolation and the understanding of QTL gene function.
Conclusions
We have constructed a molecular linkage map using two inbred lines of Antirrhinum majus, 165E and Sippe 50. The newly developed map has eight linkage groups and a total length of 562 cM with an estimated coverage of 95% of the genome. There is an average interval of 2 cM between codominant markers in 88% of cases and 5 cM in 99%, and assuming a genome size of 3.6 × 108 bp, an interval of 2 cM represents on average 1.28 Mbp of DNA.
The new map is 54% longer than the previously published map of A.majus x A. molle, and this difference is caused by increased length of the different linkage groups, except 3 and 5 that were slightly shorter indicating that map length differences were the result of differences between chromosomes in the two crosses.
We have mapped 51 IDLE transposons that are interspersed with EST-based markers indicating that MITE transposons, like in other plants, are found in gene-rich regions. Determination of EST-based markers will allow future use of the A.majus map for comparative genomic studies with other plants.
The new map has fewer regions of TRD reinforcing its usefulness to determine genome positions with higher accuracy. This has been achieved by validating the map with six classic mutants affecting floral size (Ni, co, coah and fo), body size (Hero and na) and flowering time (na). We have been able to obtain map positions for each mutant using F2 mapping populations.
Methods
Plant material
Seeds of Antirrhinum majus L. were germinated and grown as described by [31].
The A. majus line Sippe 50 [11] was obtained originally from the IPK Gatersleben and maintained by self-pollination while the second wild-type line 165E was produced by several generations of self-pollination from line JI.98 [16,32]. An F2 population (n = 96) for mapping molecular markers was selected at random from the progeny of a single F1 hybrid of Sippe 50 × 165E. The mutants compacta (co) [33], compacta ähnlich (coan) [12], formosa (fo), Grandiflora (Graf), heroina (Hero), Nitida (Ni) [11] and nana (na) [24] were obtained from the IPK Gatersleben collection. All the mutants are in the Sippe 50 genetic background. Mutations were mapped in F2 populations produced by crossing mutants to the 165E wild-type.
DNA was extracted using a NucleoSpin® kit (Macherey-Nagel) from 100 mg of frozen leaf samples that had been ground to a powder in liquid N2.
Mapping transcribed genes
Sequence tagged sites (STS) were generated using primers able to amplify regions from a collection of A. majus EST sequences that showed differences between Sippe50 and 165E [34]. The identities of PCR products were confirmed by sequencing. For six genes, both parental alleles could be distinguished by differences in the sizes of their amplified products in agarose gels without digestion. A further three loci amplified from only one parent and were therefore treated as dominant markers. For the remaining genes, restriction site polymorphisms were identified by comparing sequences of amplified products and the loci scored as co-dominant CAPS resolved in agarose gels. The ESTs used to develop markers were annotated automatically using the BLAST2GO program [35,36].
AFLP analysis
AFLP were amplified from DNA that had been digested with PstI and MseI using eight combinations of selective primers. Primers for the PstI ends of fragments had 3' selective di-nucleotides AA (P11), AC (P12) or AT (P14) and were labeled with one of four different fluorescent dyes (6-FAM, VIC, NED or PET) while those at the MseI ends had 3' extensions of ACA, AGC, AGT, CAC or CAT. Products were separated with the LIZ-500 internal size standard (ABI) using an ABI 3730 DNA Analyzer. Output files were converted to fsa format using the program obtained from http://dna.biotech.wisc.edu/ABRF/3730toGSconverter.exe, processed using Genescan software (ABI) and the presence or absence of bands scored from virtual gels created using a version of the program Genographer http://hordeum.oscs.montana.edu/genographer/ that had been modified by its authors to accommodate the five different color channels. AFLPs were scored as dominant markers. They were named according to the primers used to generate them, their size and their parent of origin - e.g. locus 11AGA141J amplified with selective primers P11 and Mse-AGA as a band of 141 nt and originated from parent 165E.
Mapping MITE transposons
Different copies of the IDLE transposon were identified by homology to the insertion in the fistulata-2 mutation in A. majus [13] either by hybridization to genomic clones or comparison to A. majus BAC clones. The host sequences to both sides of 10 IDLE insertions were identified. In these cases, primers from the two flanking regions were used to distinguish the presence or absence of the transposon on the basis of size polymorphism allowing these loci to be scored as co-dominant markers. For 43 insertions only one flanking sequence was obtained and a flanking primer was used with an IDLE primer to detect the presence of an insertion, which was treated as a dominant marker.
Map construction
To construct the molecular recombination map for the F2 population, co-dominant markers were scored as one of three allelic states (homozygous 165E, homozygous Sippe 50 or heterozygous) while dominant markers were assigned to one or other parent and scored for the presence or absence in F2 individuals. A map was estimated from the genotype data at 377 loci using Joinmap 3.0 [37], using a minimum LOD score of 6.0 to identify potential linkage groups. Maps of each linkage group were then established using the default thresholds for elimination of markers and establishing marker order and the Kosambi mapping function [38] to calculate genetic distances. Transmission ratio distortion was represented for loci with co-dominant alleles by plotting the frequencies of each homozygote and half the frequency of heterozygotes and for dominant loci by the frequency of homozygotes lacking the dominant allele. Each class was expected with a frequency of 0.25 and significant deviations from this expectation were assessed with χ2 tests.
Total genome size was estimated using Method 4 from [20] or by adding twice the average marker spacing to each chromosome, with both methods providing very similar estimates. The percentage of the genome within a particular map distance of the nearest molecular marker was estimated with the method used by [39]. To analyze whether markers showed non-random clustering, the number of 1 cM intervals expected to contain a particular number of markers was calculated from the total number of markers and map length, assuming that the markers were distributed randomly (i.e. that the number of markers per 1 cM interval followed a Poisson distribution). This null hypothesis was tested against the observed frequency distribution of markers, using a χ2 test. The frequency distributions of marker densities for the two maps were also compared directly, using a χ2 test.
Mutant mapping
To map mutations, F2 plants were selected for genotyping on the basis of their phenotype. Four pools, each containing a similar amount of DNA from four homozygous plants, were first used to scan for linkage to one of 16 markers-representing both arms of all eight chromosomes. Linkage to a marker locus was suggested by an enrichment of one of its parental alleles in more than one of the pools (i.e. enrichment of the Sippe 50 allele in pools of recessive mutations or the 165E allele in wild-type pools in the case of dominant mutants). Suspected linkage was investigated further by genotyping between 20 and 60 F2 individuals for the original locus and at additional loci linked to it. Linkage was assessed using χ2 tests to identify significant deviations from random segregation in the mutant population and the Kosambi function used to estimate map distances between mutations and markers from recombination frequencies.
List of abbreviations
AFLP: Amplified fragment length polymorphism; cM: centiMorgan; Co: Compacta; Coan Compacta ähnlich; Fo: Formosa; Hero: Heroina; MITE: Miniature Inverted Repeat Transposable element; Na: Nana; Ni: Nitida; TRD: transmission ratio distortion
Authors' contributions
ZsSS, TG, AH and MEC designed experiments. ZsSS developed the EST and IDLE markers, TG and AH did the AFLP markers, TG, ZsSS and AH made the map. MEC did the bioinformatic analysis of EST annotation. PGC, LDG and JW mapped the mutants. PGC, LDG, JW and MEC grew the F2 populations and scored the phenotypes. MEC did the mutant pictures. LDG, JW and MEC did the phenotypic analysis of the mutants. AW and MEC wrote the draft and all the authors except ZsSS read, corrected and approved it.
Supplementary Material
Map positions, primers and restriction enzymes used to detect IDLE transposons and EST-based markers.
Anchoring of linkage groups in map B (on the left) to those of the newly created map A (right).
{kind=link}
The effects of marker number on the length of map A. Map B contained 69 fewer markers than map A. To investigate whether a larger number of markers was responsible for the longer length of map A, 69 markers were removed at random and map A recalculated. This was repeated 1,000 times with removal of a different set of randomly selected markers each time. The frequency distribution of total map lengths obtained in the simulations is shown. The average length was 552 cM, a reduction of only 2% from the map estimated with all markers. Therefore a larger number of markers does not account for map A being 54% longer than map B
Contributor Information
Zsuzsanna Schwarz-Sommer, Email: marcos.egea@upct.es.
Thomas Gübitz, Email: Thomas.Guebitz@dfg.de.
Julia Weiss, Email: julia.weiss@upct.es.
Perla Gómez-di-Marco, Email: perla.gomez@upct.es.
Luciana Delgado-Benarroch, Email: luciana.delgado@upct.es.
Andrew Hudson, Email: andrew.hudson@ed.ac.uk.
Marcos Egea-Cortines, Email: marcos.egea@upct.es.
Acknowledgements
This work is dedicated to the memory of Zsuzsanna Schwarz-Sommer. We would like to express our deepest sorrow for the loss of Zsuzsanna-Schwarz-Sommer while this work was under way. Work in the lab of MEC was supported by grant AGL2007-61384 and Bananasai (BioCARM). Work in the lab of AH was supported by the BBSRC.
References
- Schwarz-Sommer Z, Davies B, Hudson A. An everlasting pioneer: the story of Antirrhinum research. Nature Reviews Genetics. 2003;4(8):657–666. doi: 10.1038/nrg1127. [DOI] [PubMed] [Google Scholar]
- Vargas P, Carrió E, Guzmán B, Amat E, Güemes J. A geographical pattern of Antirrhinum (Scrophulariaceae) speciation since the Pliocene based on plastid and nuclear DNA polymorphisms. Journal of Biogeography. 2009;36(7):1297–1312. doi: 10.1111/j.1365-2699.2008.02059.x. [DOI] [Google Scholar]
- Carpenter R, Coen ES. Floral Homeotic Mutations Produced by Transposon-Mutagenesis in Antirrhinum-Majus. Genes and Dev. 1990;4(9):1483–1493. doi: 10.1101/gad.4.9.1483. [DOI] [PubMed] [Google Scholar]
- Bonas U, Sommer H, Saedler H. The 17-kb Tam1 element of Antirrhinum majus induces a 3-bp duplication upon integration into the chalcone synthase gene. EMBO J. 1984;3(5):1015–1019. doi: 10.1002/j.1460-2075.1984.tb01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Carpenter R, Sommer H, Saedler H, Coen ES. Molecular analysis of instability in flower pigmentation of Antirrhinum majus, following isolation of the pallida locus bt transposon tagging. EMBO J. 1985;4(7):1625–1630. doi: 10.1002/j.1460-2075.1985.tb03829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D, Carpenter R, Sommer H, Hartley N, Coen E. Complementary floral homeotic phenotypes result from opposite orientations of a transposon at the plena locus of Antirrhinum. Cell. 1993;72(1):85–95. doi: 10.1016/0092-8674(93)90052-R. [DOI] [PubMed] [Google Scholar]
- Waites R, Selvadurai HR, Oliver IR, Hudson A. The PHANTASTICA gene encodes a MYB transcription factor involved in growth and dorsoventrality of lateral organs in Antirrhinum. Cell. 1998;93(5):779–789. doi: 10.1016/S0092-8674(00)81439-7. [DOI] [PubMed] [Google Scholar]
- Luo D, Carpenter R, Vincent C, Copsey L, Coen E. Origin of floral asymmetry in Antirrhinum. Nature. 1996;383(6603):794–799. doi: 10.1038/383794a0. [DOI] [PubMed] [Google Scholar]
- Hudson A, Carpenter R, Doyle S, Coen ES. Olive: a key gene required for chlorophyll biosynthesis in Antirrhinum majus. EMBO J. 1993;12(10):3711–3719. doi: 10.1002/j.1460-2075.1993.tb06048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Glover BJ, Linstead P, Martin C. Flower colour intensity depends on specialized cell shape controlled by a Myb-related transcription factor. Nature. 1994;369(6482):661–664. doi: 10.1038/369661a0. [DOI] [PubMed] [Google Scholar]
- Stubbe H. Genetik und Zytologie von Antirrhinum L. sect. Antirrhinum. Jena: Veb Gustav Fischer Verlag; 1966. [Google Scholar]
- Stubbe H. New mutants of Antirrhinum majus. Kultupflanze. 1974;22:189–213. doi: 10.1007/BF02070669. [DOI] [Google Scholar]
- Cartolano M, Castillo R, Efremova N, Kuckenberg M, Zethof J, Gerats T, Schwarz-Sommer Z, Vandenbussche M. A conserved microRNA module exerts homeotic control over Petunia hybrida and Antirrhinum majus floral organ identity. Nature Genetics. 2007;39:901–905. doi: 10.1038/ng2056. [DOI] [PubMed] [Google Scholar]
- Schwarz-Sommer Z, Silva ED, Berndtgen R, Lonnig WE, Muller A, Nindl I, Stuber K, Wunder J, Saedler H, Gubitz T. et al. A linkage map of an F-2 hybrid population of Antirrhinum majus and A-molle. Genetics. 2003;163(2):699–710. doi: 10.1093/genetics/163.2.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DF, Yang QY, Bao WD, Zhang Y, Han B, Xue YB, Cheng ZK. Molecular cytogenetic characterization of the Antirrhinum majus genome. Genetics. 2005;169(1):325–335. doi: 10.1534/genetics.104.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison BJ, Carpenter R. Resurgence of Genetic Instability in Antirrhinum-Majus. Mutat Res. 1979;63(1):47–66. [Google Scholar]
- Bennett MD, Bhandol P, Leitch IJ. Nuclear DNA Amounts in Angiosperms and their Modern Uses--807 New Estimates. Ann Bot. 2000;86(4):859–909. doi: 10.1006/anbo.2000.1253. [DOI] [Google Scholar]
- Hall MC, Willis JH. Transmission ratio distortion in intraspecific hybrids of Mimulus guttatus: implications for genomic divergence. Genetics. 2005;170(1):375–386. doi: 10.1534/genetics.104.038653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht E, Chetelat RT. Comparative genetic linkage map of Solanum sect. Juglandifolia: evidence of chromosomal rearrangements and overall synteny with the tomatoes and related nightshades. Theor Appl Genet. 2009;118(5):831–847. doi: 10.1007/s00122-008-0943-8. [DOI] [PubMed] [Google Scholar]
- Chakravarti A, Lasher LK, Reefer JE. A maximum likelihood method for estimating genome length using genetic linkage data. Genetics. 1991;128(1):175–182. doi: 10.1093/genetics/128.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Initiative IB. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463(7282):763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Arbuckle J, Wessler SR. Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions of maize. Proc Natl Acad Sci USA. 2000;97(3):1160–1165. doi: 10.1073/pnas.97.3.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B-H. Statistical genomics. Cleveland/Boca Ratón, Fl.: CRC Press; 1998. [Google Scholar]
- Vilmorin. Vilmorin's, Blumengärtnerei. Bd.1 Antirrhinum majus, S.756. Vol. 1. Berlin: Paul Parey; 1896. [Google Scholar]
- Anastasiou E, Kenz S, Gerstung M, MacLean D, Timmer J, Fleck C, Lenhard M. Control of Plant Organ Size by KLUH/CYP78A5-Dependent Intercellular Signaling. Dev Cell. 2007;13:843–856. doi: 10.1016/j.devcel.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Disch S, Anastasiou E, Sharma VK, Laux T, Fletcher JC, Lenhard M. The E3 ubiquitin ligase BIG BROTHER controls Arabidopsis organ size in a dosage-dependent manner. Curr Biol. 2006;16(3):272–279. doi: 10.1016/j.cub.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Delgado-Benarroch L, Weiss J, Egea-Cortines M. The mutants compacta ahnlich, Nitida and Grandiflora define developmental compartments and a compensation mechanism in floral development in Antirrhinum majus. J Plant Res. 2009;122:559–569. doi: 10.1007/s10265-009-0236-6. [DOI] [PubMed] [Google Scholar]
- Delgado-Benarroch L, Causier B, Weiss J, Egea-Cortines M. FORMOSA controls cell division and expansion during floral development in Antirrhinum majus. Planta. 2009;229:1219–1229. doi: 10.1007/s00425-009-0910-x. [DOI] [PubMed] [Google Scholar]
- Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA. 1991;88(21):9828–9832. doi: 10.1073/pnas.88.21.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Wilson Y, Bowers J, Kennaway R, Bangham A, Hannah A, Coen E, Hudson A. Evolution of Allometry in Antirrhinum. Plant Cell. 2009;21(10):2999–3007. doi: 10.1105/tpc.109.069054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayo-Canha A, Delgado-Benarroch L, Weiss J, Egea-Cortines M. Artificial decrease of leaf area affects inflorescence quality but not floral size in Antirrhinum majus. Sci Hort. 2007;113:383–386. doi: 10.1016/j.scienta.2007.04.015. [DOI] [Google Scholar]
- Sommer H, Saedler H. Structure of the Chalcone Synthase Gene of Antirrhinum-Majus. Molecular & General Genetics. 1986;202(3):429–434. doi: 10.1007/BF00338392. [DOI] [PubMed] [Google Scholar]
- Kuckuck H, Schick R. Die Erbfaktoren bei Antirrhinum majus und ihre Bezeichnung. Zf Induck Abst-u Vererbungsl. 1930;56:51–83. doi: 10.1007/BF01739798. [DOI] [Google Scholar]
- Bey M, Stuber K, Fellenberg K, Schwarz-Sommera Z, Sommer H, Saedler H, Zachgo S. Characterization of Antirrhinum petal development and identification of target genes of the class B MADS box gene DEFICIENS. Plant Cell. 2004;16(12):3197–3215. doi: 10.1105/tpc.104.026724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucl Acids Res. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Van Ooijen JW, Voorrips RE. JoinMap® 3.0, Software for the calculation of genetic linkage maps. Wageningen, the Netherlands: Plant Research International; 2001. [Google Scholar]
- Kosambi DD. The estimation of map distances from recombination values. Ann Eugen. 1944;12:172–175. [Google Scholar]
- Fishman L, Kelly AJ, Morgan E, Willis JH. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics. 2001;159(4):1701–1716. doi: 10.1093/genetics/159.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Map positions, primers and restriction enzymes used to detect IDLE transposons and EST-based markers.
Anchoring of linkage groups in map B (on the left) to those of the newly created map A (right).
The effects of marker number on the length of map A. Map B contained 69 fewer markers than map A. To investigate whether a larger number of markers was responsible for the longer length of map A, 69 markers were removed at random and map A recalculated. This was repeated 1,000 times with removal of a different set of randomly selected markers each time. The frequency distribution of total map lengths obtained in the simulations is shown. The average length was 552 cM, a reduction of only 2% from the map estimated with all markers. Therefore a larger number of markers does not account for map A being 54% longer than map B