

Abstract
Benfotiamine (BFT) is a transketolase activator that directs glucose to the pentose phosphate pathway. The present study investigated whether BFT improves the recovery after myocardial infarction (MI) and explored underlying mechanisms of protection. Non-diabetic and streptozotocin-induced type 1 diabetic mice were supplemented with BFT (70 mg/kg/day in drinking water) for 4 weeks and then subjected to MI or sham operation. Cardiac function was monitored by echocardiography. At two weeks post-MI, intra-ventricular pressure was measured by Millar tip-catheter and hearts were collected for biochemical, immunohistochemical and expressional analyses. No treatment effect was observed in sham-operated mice. Post-MI mortality was higher in diabetic mice and hemodynamic studies confirmed the worsening effect of diabetes on functional recovery. Furthermore, diabetic mice demonstrated increased cardiomyocyte apoptosis, reduced reparative angiogenesis, larger scars, enhanced oxidative stress, and blunted activation of the pro-survival VEGF receptor-2/Akt/Pim-1 signaling pathway. BFT improved post-MI survival, functional recovery and neovascularization and reduced cardiomyocyte apoptosis and neurohormonal activation in diabetic as well as in non-diabetic mice. In addition, BFT stimulated the activity of pentose phosphate pathway enzymes, leading to reduction of oxidative stress, phosphorylation/activation of VEGF receptor-2 and Akt and increased Pim-1, pBad and Bcl-2 levels. These effects were contrasted on silencing glucose-6-phosphate dehydrogenase, the key enzyme in pentose phosphate pathway, or inhibiting Akt. BFT benefits post-MI recovery through stimulation of pro-survival mechanisms and containment of neurohormonal response. These results may have implications for the treatment of myocardial ischemia.
Keywords: Myocardial infarction, Type 1 diabetes mellitus, Benfotiamine, Oxidative stress, Pentose phosphate pathway, Glucose-6-phosphate dehydrogenase
1. Introduction
Myocardial infarction (MI) is the leading cause of death worldwide. Patients with diabetes mellitus (DM) have less favorable outcome after MI [1,2] or coronary artery revascularization [3,4], and are at high risk for developing heart failure compared to non-diabetic subjects [5,6]. Furthermore, DM is known to directly affect myocardial structure and function independently of coronary artery disease [7,8]. These considerations underscore the urgent need of mechanistic treatments for the cure of MI, especially in the high-risk population of diabetic patients.
Recent studies have highlighted the importance of the pentose phosphate pathway for preservation of cardiomyocyte contractility in ischemia. Under conditions of increased oxidative stress, the activity of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of pentose phosphate pathway, is rapidly increased in cardiomyocytes with consequent neutralization of free radical injury [9]. Furthermore, translocation of G6PD to the plasma membrane of endothelial cells reportedly induces the activation of vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2), protein kinase B (PKB/Akt) and endothelial nitric oxide synthase (eNOS), thereby leading to promotion of angiogenesis [10,11]. In DM, an impaired activity of G6PD and transketolase, the other pivotal enzyme that shunts glucose metabolites to the pentose pathway, results in depletion of reducing agents and accumulation of glycolysis end-products, which have deleterious effects for cardiovascular cells, such as endothelial cells, mural cells (pericytes and vascular smooth muscle cells) and cardiomyocytes [12,13]. Of note, similar to diabetic subjects, mice with partial deficiency of G6PD demonstrate impaired angiogenesis and increased myocardial dysfunction following ischemia–reperfusion [10,14]. However, no information exists on whether induction of G6PD-related anti-oxidative mechanism by ischemia translates in the activation of the VEGFR2/Akt signaling pathway in cardiomyocytes and whether this homeostatic response is maintained or disrupted in DM.
Benfotiamine (BFT), a vitamin B1 analogue and an activator of transketolase, reportedly ameliorates DM-induced vascular complications and healing of ischemic limbs [15–18]. Furthermore, we showed that BFT prevents DM-induced diastolic dysfunction and heart failure through activation of Akt and its downstream target, proviral integration site for Moloney murine leukemia virus-1 (Pim-1) [19].
The present study investigates whether treatment with BFT protects the heart from ischemic injury and explores cellular and molecular mechanisms of cardioprotection. Results indicate that BFT significantly aids post-MI functional recovery by protecting cardiomyocytes from apoptosis through phosphorylation of VEGFR2 and Akt and activation of Pim-1. Furthermore, BFT contained the excessive activation of neurohormonal systems in the infarcted heart.
2. Methods
2.1. Ethics
Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (the Institute of Laboratory Animal Resources, 1996) and with approval of the British Home Office and the University of Bristol.
2.2. Experimental protocol
As summarized in Supplemental Fig. 1, 8-week old male CD1 mice (Charles River, UK) were made diabetic using streptozotocin (STZ; 40 mg/kg body weight i.p. daily for 5 days), while age- and gender-matched controls were injected with STZ-vehicle [18]. DM was confirmed by measurements of glucose in peripheral blood (Supplemental Fig. 2). Four weeks following final STZ or vehicle injection, mice were randomly assigned to receive BFT (70 mg/kg body weight per day) or vehicle (1 mMol/L HCl) in drinking water until sacrifice. The BFT dosage used here reportedly produces a 4-fold increase in plasma thiamine [20]. Four weeks after entering the treatment, mice were randomized to MI or sham operation and then sacrificed 2 weeks later. MI was induced by permanent ligation of the left anterior descending coronary artery (LAD). In brief, with mice under anesthesia (2,2,2 tribromo ethanol, 0.3 g/kg, i.p.) and artificial ventilation, the chest cavity was opened and, after careful dissection of the pericardium, LAD was permanently ligated using a 7-0 silk suture. Animals were allowed to recover with aseptic precautions and received analgesic medication (buprenorphine, 0.1 mg/kg s.c.) to reduce post-operative pain. Sham-operated animals underwent a similar procedure without LAD ligation.
2.3. Hemodynamic measurements and collection of heart samples
Measurements of dimensional and functional parameters were performed at baseline (time 0), before mice entered the treatment (week 4), before induction of MI (week 8) and post-MI (week 10), using a high-frequency, high-resolution echocardiography system (Vevo 770, Visual Sonics, Toronto, Canada). Left ventricular (LV) chamber dimensions, LV ejection fraction (LVEF) and fractional shortening (LVFS) were determined as described [21].
At the occasion of the last echocardiography session, LV pressure measurements were performed using a high-fidelity 1.4F transducer tipped catheter [22]. Mice were then randomly allocated to 3 subgroups to be used for measurement of myocardial perfusion using fluorescent microspheres, immunohistochemistry (IHC) or assessment of gene expression. Hearts of mice undergoing IHC studies were stopped in diastole by intra-cardiac injection of cadmium chloride, followed by collection of the LV by perfusion fixation with 4% paraformaldehyde.
2.4. Biochemical measurements
Transketolase, G6PD and AGE activities in LV tissue were measured at sacrifice (n=5 mice per group) as described previously [12,19,23]. Levels of angiotensin II (SPI bio) and norepinephrine (IBL international) in plasma and LV tissue were measured as markers of neurohormonal activation [24] using commercially available ELISA kits (n=5 mice per group).
2.5. Immunofluorescence microscopy detection of microvascular density, apoptosis and oxidative stress
Mid-ventricular sections which include the middle part of the infarct, the peri-infarct zone (the normal myocardial area bordering the infarct zone), and the remote zone were used (n=5 mice per group). Vascular endothelial cells were identified by staining for isolectin B4, vascular smooth muscle cells by staining for α-smooth muscle actin, cardiomyocytes by α-sarcomeric actin and apoptotic cells by TUNEL assay (Roche). Levels of hydroxyl radicals was measured by immunostaining for 8-OHDG.
2.6. Morphometric evaluation of myocardial scar
Sections from three levels of each heart (apical, mid-ventricular, and basal) were stained by Azan Mallory to determine the scar size as described (n=5 mice per group) [25].
2.7. In vitro studies
HL-1 cardiomyocytes (a gift from Prof. William Claycomb, Louisiana State University Medical Center, New Orleans, USA) [26,27] were cultured in the presence of high d-glucose (HG, 30 mM) or normal glucose (5 mM) added with 25 mM d-mannitol as osmotic control. After 24 h, cells were supplemented with either BFT (150 μM) or vehicle (1 mM HCl) for further 48 h. Optimal BFT concentration was decided on the basis of earlier studies [18,19]. After 48 h, the cells were serum starved (0%) for 8 h and then exposed to hypoxic conditions (0.2% O2 and 5% CO2) for 18 h. At the end of the experiments, cardiomyocytes were immediately assessed for caspase 3/7 activity using a commercially available assay kit (Caspase-Glo® 3/7, Promega), which was performed in 6 wells per each condition and repeated 3 times. Additional samples were collected at the end of hypoxia experiments for expressional studies (n=4 biologic replicates per each group). Separate experiments were performed under normoxia to determine whether BFT’s action is independent of low oxygen levels.
To verify the mechanisms implicated in BFT’s action on cardiomyocytes, we silenced G6PD using small interfering RNA (siRNAG6PD, Dharmacon Technologies). To this aim, HG treated HL-1 cells were transfected with siRNAG6PD or the relevant scrambled siRNA (Dharmacon Technologies). Effective silencing was confirmed by measurement of G6PD activity (Fig. 8 and Supplemental Fig. 3(A)). Cells were then treated with BFT for 48 h and exposed to starvation and hypoxia as described above. Samples were finally assessed for caspase 3/7 activity (performed in 6 wells per each condition and repeated 3 times) and western blotting (n=4 biologic replicates per each group).
Fig. 8.


Silencing G6PD attenuates BFT-induced protection on cardiomyocytes exposed to hypoxia under normal or high glucose. Bar graphs show G6PD activity (A), levels of activated caspase 3/7 (B), pVEGFR2 (C), pAKt (D), Pim-1(E) and Bcl-2 (F) in cultured adult cardiomyocytes after silencing G6PD. Cardiomyocytes cultured in normal (NG) or high glucose (HG) were transfected with siRNAG6PD (NGSiG and HGSiG) and subjected to hypoxia with 0.2% O2 for 18 h after treating the cells with BFT (NGBSiG and HGBSiG) or vehicle (NGVSiG or HGVSiG). Values are mean±standard deviation and expressed as U/min/mg of protein for G6PD activity, relative units (RLU) for caspase 3/7 activity and n-fold changes toward vehicle-treated NG (NGV) for pVEGFR2, pAkt, Pim-1 and Bcl-2.§P<0.01, §§P<0.001 and §§§P<0.0001 versus NG or HG; *P<0.01 and **P<0.001 versus vehicle-treated NG (NGV) or HG (HGV); #P<0.01 and ##P<0.001 versus BFT-treated NG (NGB) or HG (HGB); δP<0.01 and δδP<0.001 versus corresponding treatment between NG or HG cultured cells. Each experiment was repeated four times in triplicate except caspase 3/7 activity, which was performed in 6 wells per each condition and repeated 3 times.
In separate experiments, we silenced Akt using a HA-tagged dominant negative mutated form of Akt (Ad.DN-Akt, K179M) or control Ad.Null (both at 100 MOI) [19]. After 24 h, the medium was replaced with a fresh one supplemented with either BFT or vehicle. After additional 24 h, cells were subjected to starvation and hypoxia as described above. Samples were then assessed for caspase 3/7 activity and western blotting.
2.8. Western blot
WB analyses were performed on the hearts (n=5 mice per group) or HL-1 cardiomyocytes (n=4 assays, each in triplicate) subjected respectively to MI/or hypoxia to verify the effects of BFT on total and pVEGFR2 (Tyr1175), total and pAkt (Ser473), Pim-1, total and pBad (Ser112), Bcl-2, cleaved caspase 3 and p38MAPK (Thr180/Tyr182).
2.9. Statistical analysis
Results are represented as mean±standard deviation. Data from different groups were compared by the use of two-way ANOVA with or without repeated measures, as appropriate. This was followed by pair-wise comparison using the Holm–Sidak method (for multiple comparisons) or t-test (paired or unpaired as appropriate). Survival curves were analyzed by the Kaplan–Meier method and comparisons were made with the Gehan–Breslow log-rank test using SigmaStat statistical software. A P value of <0.05 was considered statistically significant for all parameters.
3. Results
3.1. BFT improves post-MI survival
At 14 days post-MI, only 25% of the diabetic mice in the vehicle group survived as compared to 50% of non-diabetic controls (P<0.0001, Fig. 1). Treatment with BFT markedly improved the survival of both non-diabetic (80%) and diabetic mice (50%) as compared with the respective vehicle-treated group (P<0.001).
Fig. 1.
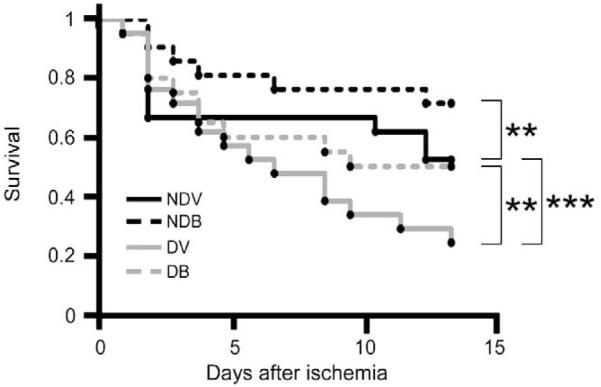
BFT improves post-MI survival. Vehicle-treated diabetic (DV) mice showed a reduced survival rate (***P<0.0001 versus age-matched vehicle-treated non-diabetic mice, NDV). BFT significantly improved survival in both non-diabetic (NDB) and diabetic (DB) groups (**P<0.001 versus corresponding vehicle-treated group).
3.2. BFT accelerates post-MI functional recovery
As shown in Table 1, echocardiography measurements before MI induction showed the presence of diastolic dysfunction in diabetic mice, as denoted by significantly decreased E/A ratio (P<0.01 versus non-diabetic mice), whereas indices of systolic function, LVEF and LVFS, were preserved. BFT improved diastolic function of diabetic mice (P<0.01 versus vehicle-treated diabetics).
Table 1.
Echocardiographic parameters.
| Pre-MI |
Post-MI |
|||||||
|---|---|---|---|---|---|---|---|---|
| NDV | NDB | DV | DB | NDV | NDB | DV | DB | |
| LVAWs (mm) | 1.50±0.05 | 1.43±0.04 | 1.36±0.04 | 1.40±0.07 | 0.33±0.07δδ | 0.46±0.07δδ* | 0.28±0.05δδ | 0.42±0.05δδ# |
| LVAWd (mm) | 0.90±0.04 | 0.89±0.04 | 0.90±0.04 | 0.91±0.04 | 0.32±0.05δδ | 0.38±0.05δδ* | 0.26±0.03δδ* | 0.35±0.06δδ# |
| LVPWs (mm) | 1.27±0.08 | 1.33±0.04 | 1.30±0.10 | 1.30±0.09 | 1.07±0.31δ | 1.36±0.21* | 1.04±0.12δ | 1.33±0.11# |
| LVPWd (mm) | 0.95±0.07 | 0.95±0.07 | 0.95±0.06 | 0.90±0.04 | 0.8±0.22 | 0.93±0.11 | 0.80±0.14 | 1.06±0.13δ# |
| LVESD (mm) | 2.30±0.44 | 2.31±0.44 | 2.40±0.19 | 2.33±0.26 | 5.28±0.45δδ | 4.78±0.31δδ* | 5.90±0.29δδ | 5.06±0.19δδ# |
| LVEDD (mm) | 3.30±0.30 | 3.30±0.31 | 3.20±0.30 | 3.40±0.10 | 5.69±0.37δδ | 5.71±0.31δδ | 6.20±0.26δδ | 5.44±0.29δδ# |
| LVESV (μl) | 15.8±1.4 | 15.4±1.9 | 16.8±2.8 | 16.3±1.2 | 123.7±33.1δδ | 92.8±10.7δδ* | 164.4±20.7δδ* | 110.3±13.7δδ# |
| LVEDV (μl) | 59.3±2.7 | 56.0±2.0 | 60.3±3.7 | 61.2±2.3 | 157.9±26.1δδ | 148.4±17.2δδ | 185.0±19.5δδ* | 150.9±14.0δδ# |
| LVSV (μl) | 43.5±2.7 | 40.6±3.5 | 43.5±4.1 | 44.8±2.6 | 34.3±11.6δ | 55.6±17.2δ* | 20.6±7.6δδ* | 40.6±6.7# |
| LVEF (%) | 70.5±6.6 | 70.9±4.5 | 70.8±3.0 | 70.5±4.1 | 22.7±9.5δδ | 36.9±9.3δδ* | 11.9±4.3δδ* | 25.3±4.2δδ# |
| LVFS (%) | 38.7±2.2 | 38.3±2.7 | 38.4±2.4 | 38.5±1.7 | 11.3±5.6δδ | 16.5±4.1δδ* | 7.7±3.2δδ | 11.6±2.3δδ# |
| HR (bpm) | 497±13 | 498±13 | 482±31 | 488±22 | 532±19δ | 544±20δ | 489±19* | 504±12 |
| CO (mL/min) | 21.7±1.9 | 22.9±1.8 | 20.9±2.3 | 21.9±1.6 | 18.1±5.7 | 24.2±9.3* | 10.2±3.9δδ* | 20.5±3.6# |
| E (mm/s) | 941±77 | 943±77 | 769±52γ | 862±59δ† | 638±131δδ | 680±93δδ | 598±83δδ | 638±88δδ |
| A (mm/s) | 438±33 | 440±21 | 680±65γγ | 500±56δ† | 804±91δδ | 698±53δδ* | 944±86δδ* | 901±66δδ |
| E/A | 2.15±0.19 | 2.17±0.17 | 1.12±0.11γγ | 1.73±0.24† | 0.80±0.17δδ | 0.95±0.14δδ* | 0.64±0.11δδ* | 0.71±0.09δδ |
Data are presented as mean±SD. NDV, non-diabetic vehicle-treated; NDB, non-diabetic BFT-treated; DV, diabetic vehicle-treated; DB, diabetic BFT-treated; LVAWs, LV anterior wall, end-systole; LVAWd, LV anterior wall, end-diastole; LVPWs, LV posterior wall, end-systole; LVPWd, LV posterior wall, end-diastole; LVESD, LV end-systolic diameter; LVEDD, LV end-diastolic diameter; LVESV, LV end-systolic volume; LVEDV, LV end-diastolic volume; LVSV, LV stroke volume; LVEF, LV ejection fraction; LVFS, LV fractional shortening; HR, heart rate; CO, cardiac output; E, mitral valve velocity during early diastolic filling; A, mitral valve velocity during atrial contraction
P<0.01 and
P<0.001 versus NDV pre-MI;
P<0.01 versus DV pre-MI.
P<0.01 and
P<0.001 versus corresponding group pre-MI;
P<0.01 versus NDV post-MI;
P<0.01 versus DV post-MI.
MI deteriorated diastolic function and contractility and caused LV chamber dilatation. Cardiac output (CO) was partially maintained in non-diabetic mice through increased heart rate (HR). Expectedly, DM worsened the consequences of MI as reflected by remarkably reduced LVEF and CO. BFT significantly improved the functional deterioration of infarcted hearts in both non-diabetic and diabetic mice. Furthermore, no reactive tachycardia was observed in vehicle-treated infarcted diabetic mice, with this response being restored by BFT.
In addition, pressure tip-catheter analyses demonstrated a significant improvement in LV end-systolic and end-diastolic pressures and maximal rates of LV pressure (dP/dtmax and dP/dtmin) in both non-diabetic and diabetic animals supplemented with BFT (Figs. 2(A–C)). Of note, BFT caused a leftward shift of PV loops, confirming the benefit of treatment on LV performance (Fig. 2(D)).
Fig. 2.

BFT improves cardiac function post-MI. (A–C) Bar graphs showing the effect of BFT on LV end-diastolic pressure (LVEDP), LV end-systolic pressure (LVESP) and maximum and minimum rates of developed pressure (dP/dt) at 2 weeks post-MI (n=at least 12 mice in each group). (D) Representative pressure-volume loops obtained by integrated measurement of LV pressure (Millar catheter) and volume (echocardiography). Values are mean±standard deviation. *P<0.01 and **P<0.001 versus vehicle-treated non-diabetic (NDV) mice; #P<0.01 and ##P<0.001 and ###P<0.0001 versus vehicle-treated diabetic (DV) mice.
Following sham operation, no difference in echocardiography indices of cardiac function as well as pressure and volume parameters was observed between vehicle and BFT-treated mice within the diabetic or non-diabetic subgroups (Supplemental Fig. 4).
3.3. BFT improves myocardial perfusion and promotes reparative neovascularization in peri-infarct zone
As shown in Fig. 3(A), myocardial perfusion was significantly lower in diabetic compared to non-diabetic mice after MI (P<0.01), with BFT improving LV perfusion in both non-diabetic and diabetic animals (P<0.01 versus respective vehicle-treated groups for both comparisons). We then analyzed the impact of DM and BFT on reparative angiogenesis (Figs. 3(B and C)). Vehicle-treated diabetic hearts showed less capillaries and arterioles in the peri-infarct zone as compared to non-diabetic hearts (P<0.01). BFT significantly improved the peri-infarct vascularization at capillary and arteriole level as compared to vehicle (P<0.01). No difference in vascular density was observed between treatment groups at the level of the remote zone, although BFT-treated mice tended to have more capillaries than vehicle-treated mice (Supplemental Fig. 5).
Fig. 3.

BFT improves cardiac perfusion and neovascularization. (A) Bar graphs show the effect of BFT on myocardial perfusion at 2 weeks after MI. (B–C) Representative immunohistochemistry images of microvasculature and bar graphs showing the effect of BFT on LV capillary (B) and arteriole densities (C) at 2 weeks after MI. Scale bars are 50 μm for capillary density and 100 μm for arteriole density. The white dotted line delimits the border zone. Values are mean±standard deviation. *P<0.01 and **P<0.001 versus vehicle-treated non-diabetic (NDV) mice; #P<0.01 and ##P<0.001 versus vehicle-treated diabetic (DV) mice. Each group consisted of 5 mice.
3.4. BFT reduces oxidative stress, cardiomyocyte apoptosis and scar size
As shown in Fig. 4, DM increased the myocardial levels of 8-OHDG (P<0.01 versus non-diabetic) and BFT reduced oxidative stress in the peri-infarct zone of diabetic and non-diabetic mice (P<0.01 versus vehicle). Similarly, BFT reduced 8-OHDG levels in the remote zone (Supplemental Fig. 6). As shown in Fig. 5(A), BFT also reduced the number of TUNEL positive apoptotic cardiomyocytes in the peri-infarct zone of both diabetic and non-diabetic hearts (P<0.01 for both comparisons).
Fig. 4.

BFT reduces oxidative stress in infarcted hearts. Representative immunohistochemistry images and quantitative analysis of the levels of 8-OHDG in myocardium at 2 weeks after MI. Arrowheads point 8-OHDG-positive cardiomyocytes. Scale bars are 50 μm. The white dotted line delimits the border zone. Values are mean±standard deviation. *P<0.01 versus vehicle-treated non-diabetic (NDV) mice; #P<0.01 versus vehicle-treated diabetic (DV) mice. Each group consisted of 5 mice.
Fig. 5.

BFT prevents cardiomyocyte apoptosis and reduces fibrotic remodelling. (A) Representative microphotographs and bar graphs showing the effect of BFT on cardiomyocyte apoptosis after MI. Cardiomyocytes were identified by their positivity for α-sarcomeric actin. Scale bars are 50 μm. The white dotted line delimits the border zone. (B) Representative microphotographs and bar graphs showing scar size assessed by Azan Mallory staining (mid-ventricular sections) at 2 weeks after MI. Fibrotic tissue stains blue. Values are mean±standard deviation. *P<0.01 and **P<0.001 versus vehicle-treated non-diabetic (NDV) mice; #P<0.01 versus vehicle-treated diabetic (DV) mice. Each group consisted of 5 mice.
Histological assessment of fibrosis by Azan Mallory revealed larger scars in vehicle-treated diabetic hearts compared to non-diabetic hearts (P<0.001, Fig. 5(B)). BFT significantly reduced scar dimensions in both non-diabetic and diabetic hearts (P<0.01 versus respective vehicle-treated groups).
3.5. BFT boosts the pentose phosphate pathway in infarcted hearts
We then investigated whether BFT activates key enzymes of the pentose phosphate pathway (Figs. 6(A–C)). We found that G6PD is activated in ischemic hearts compared to sham-operated hearts and, importantly, this effect was significantly dampened by DM (Fig. 6(B)). Of note, BFT increased transketolase and G6PD activities in non-diabetic hearts and rescued the impaired pentose phosphate pathway in diabetic hearts (Figs. 6(A and B)) thereby reducing the formation of AGE (Fig. 6(C)).
Fig. 6.


BFT boosts the pentose phosphate pathway and moderates AGE and neurohormonal activation in infarcted hearts. Bar graphs showing the effect of MI and BFT on transketolase (A) and G6PD activity (B) and AGE levels (C) in myocardium of diabetic and non-diabetic mice. Plasma and myocardial levels of angiotensin II (D) and norepinephrine (E) are also shown (n=5 mice per group, each assay performed in triplicate). Values expressed as U/min/mg of protein for transketolase and G6PD activity and absorbance (OD) for AGE levels. Angiotensin II and epinephrine levels were expressed as pg/ml or pg/gm of protein for plasma and heart respectively. All the values are mean±standard deviation. *P<0.01 and **P<0.001 versus vehicle-treated sham-operated mice and #P<0.01 and ##P<0.001 versus vehicle-treated MI mice within non-diabetic or diabetic group. δP<0.01 and δδP<0.01 versus corresponding treatment between non-diabetic and diabetic group. NDS — vehicle-treated sham-operated non-diabetic; NDV — vehicle-treated non-diabetic with MI; NDB — BFT-treated- non-diabetic with MI; DS — vehicle-treated sham-operated diabetic; DV — vehicle-treated diabetic with MI; DB — BFT-treated diabetic with MI.
3.6. BFT moderates neurohormonal activation after MI
MI induced a remarkable increase in angiotensin II (Fig. 6(D)) and norepinephrine levels (Fig. 6(E)) in both peripheral blood and myocardium. Although the levels of angiotensin II were comparable between diabetic and non-diabetic mice, norepinephrine was significantly increased in DM (P<0.01 at both basal and post-MI). Treatment with BFT markedly reduced the levels of both factors in diabetic and non-diabetic mice (P<0.001 for both comparisons).
3.7. BFT activates pro-survival signaling mechanism in infarcted hearts
In non-diabetic mice, the ischemia-induced activation of G6PD was associated to a significant increase in VEGFR2 phosphorylation, Akt phosphorylation and activity, Pim-1 levels and target pBad and Bcl-2 proteins, all these changes being negated by DM (Figs. 7(A–F)). Furthermore, MI-induced increase in cleaved caspase 3 levels was enhanced in the hearts of diabetic mice (Fig. 7(G)). Importantly, BFT boosted the Akt/Pim-1 signaling pathway in infarcted hearts of diabetic and non-diabetic mice.
Fig. 7.


BFT stimulates survival signaling. Representative blots and bar graphs showing the levels of pVEGFR2 (A), pAkt (B), Akt activity (C), Pim-1 (D), pBad (E), Bcl-2 (F), and cleaved caspase 3 (G) and phospho p38MAPK (H) in total LV lysates (n=5 per group). Values are expressed as n-fold changes toward vehicle-treated sham-operated non-diabetic (NDS) mice and are mean±standard deviation. *P<0.01, **P<0.001 and ***P<0.0001 versus NDS and #P<0.01, ##P<0.001 and ###P<0.001 versus vehicle-treated MI mice within non-diabetic and diabetic group. δP<0.01 and δδP<0.001 versus corresponding treatment (vehicle or BFT) between non-diabetic and diabetic groups.
The Akt effector Pim-1 is known to limit the pro-apoptotic Bad within the cytosol thus leaving the Bcl-XL and Bcl-2 free to inhibit apoptosis [28,29]. In line with this, we found that activation of Pim-1 by BFT is associated with increased pBad and Bcl-2 (Figs. 7(E and F)) and decreased cleaved caspase 3 levels in ischemic hearts (Fig. 7(G)). Of note, BFT did not exhibit any effect on the phosphorylation levels of p38MAPK (Fig. 7(H)), another major mechanism involved in cardiomyocyte survival.
To confirm the direct effect of BFT, cardiomyocytes were exposed to hypoxia in the presence of normal or high glucose levels. High glucose enhanced caspase 3/7 activity and inhibited VEGFR2/Akt/Pim signaling in hypoxic cardiomyocytes (Supplemental Fig. 7). BFT significantly reduced caspase 3/7 activity and preserved VEGFR2/Akt/Pim-1 signaling under normal or high glucose. Under normoxic conditions, BFT did not exhibit any effect on cells treated with normal glucose (Supplemental Fig. 8), however, it significantly prevented the high glucose-induced activation of caspase 3/7 activity and the downregulation of Akt/Pim-1 signaling (Supplemental Fig. 8).
We further confirmed the signaling pathway involved in BFT-induced effects by silencing G6PD. This resulted in remarkable reduction of G6PD activity (Fig. 8(A)) and attenuation of BFT-mediated inhibition of cell apoptosis under hypoxia with or without HG (Fig. 8 (B)). Silencing G6PD also markedly reduced the activation of pro-survival signaling cascade by BFT (Figs. 8(C–F)). No effect was instead induced by scrambled siRNA (Supplemental Fig. 3). Inhibition of Akt activity using HA-tagged AD.DN-Akt also attenuated the activation of pro-survival signaling cascade by BFT (Fig. 9) without affecting G6PD activity (data not shown).
Fig. 9.

Silencing Akt attenuates BFT-induced protection on cardiomyocytes exposed to hypoxia under normal or high glucose. Bar graphs show levels of activated caspase 3/7 (A), pAKt (B), Pim-1(C) and Bcl-2 (D) in cultured adult cardiomyocytes after silencing Akt. Cardiomyocytes cultured in normal (NG) or high glucose (HG) were transfected with Ad. DN-Akt (NGdnAkt and HGdnAkt) and subjected to hypoxia with 0.2% O2 for 18 h after treating the cells with BFT (NGBdnAkt and HGBdnAkt) or vehicle (NGVdnAkt or HGVdnAkt). Values are mean±standard deviation and expressed as relative units (RLU) for caspase 3/7 activity and n-fold changes toward vehicle-treated NG (NGV) for pAkt, Pim-1 and Bcl-2.§P<0.01 and §§P<0.001versus NG or HG; *P<0.01 and **P<0.001 versus vehicle-treated NG (NGV) or HG (HGV); #P<0.01 and ##P<0.001 versus BFT-treated NG (NGB) or HG (HGB); δP<0.01 and δδP<0.001 versus corresponding treatment between NG or HG cultured cells. Each experiment was repeated four times in triplicate except caspase 3/7 activity, which was performed in 6 wells per each condition and repeated 3 times.
4. Discussion
Post-MI mortality and morbidity are remarkable in patients with DM [30,31]. Different mechanisms have been proposed for the worsened recovery of diabetic infarcted hearts. Data from both diabetic patients [32] and streptozotocin-induced diabetic mice [33] suggest that a consistent loss of myocytes occurs as a consequence of oxidative stress-triggered apoptosis in the area at risk, leading to larger scars and maladaptive remodelling [34]. The present study newly shows that vitamin B1 analogue BFT improves the recovery from MI by reducing oxidative stress, correcting the impairment of adaptive pro-survival mechanism and moderating neurohormonal activation.
Our study confirms that transketolase and G6PD, the rate-limiting enzymes of the pentose phosphate pathway, are inhibited in the diabetic heart under basal conditions [35,36]. Furthermore, G6PD activity increases during ischemia–reperfusion in isolated hearts [14], which is in line with present results in non-diabetic mice with acute MI. Upregulation of G6PD in ischemic heart may reflect a compensatory response attempting to contrast oxidative stress. In fact, genetically modified mice carrying a mutation that reduces myocardial G6PD activity by 80% exhibited greater sensitivity to ischemia/reperfusion induced LV dysfunction, with depletion of intracellular thiols and impairment of redox homeostasis [14,37]. This phenotype closely resembles the situation of diabetic mice, which also show a remarkably blunted response of G6PD to myocardial ischemia, increased oxidative stress and profound deterioration of contractile function and cardiomyocyte survival. Therefore, restoring proper G6PD levels may represent a therapeutic target to prevent excessive cardiomyocyte damage in DM. Accordingly, BFT markedly increased G6PD and transketolase activity, reduced oxidative stress and preserved cardiomyocyte viability and LV function in diabetic mice subjected to acute MI. Interestingly, BFT also benefited the post-MI recovery of non-diabetic mice, suggesting that there is a potential advantage from boosting the pentose phosphate pathway independently of its baseline activity state.
Our results indicate that BFT improves the vascularization of the peri-infarct zone, an effect that may have contributed in limiting the extension of myocardial damage. Leopold et al. showed that antisense knockdown of G6PD is associated with a significant decrease in in vitro angiogenesis, whereas forced G6PD expression by gene transfer promoted this process through a VEGFR2/Akt/eNOS mediated mechanism [10]. Our results newly highlight the implication of a similar signaling mechanism in the maintenance of cardiomyocyte viability. We found that ischemia induces the G6PD-dependent activation of VEGFR2/Akt and that this response is blunted in DM. BFT restored this reaction in diabetic hearts and cardiomyocytes exposed to high glucose and also enhanced its spontaneous activation in non-diabetic hearts. Silencing studies strongly support the link between G6PD and Akt in maintaining cardiomyocyte survival under ischemia, however they do not clarify the direct mechanism by which G6PD regulates Akt activity. Further studies addressing this aspect are therefore needed.
Forced expression of Akt reportedly inhibits cardiomyocyte apoptosis in infarcted wild-type mice but not in Pim-1 deficient mice, thus demonstrating the essential role of Pim-1 in Akt-induced cardioprotection [38]. Pim-1 exerts a protective effect of the myocardium through induction of Bcl-2 and Bcl-xL protein levels and phosphorylation/inactivation of Bad. Our results indicate for the first time that DM dampens Pim-1 expression after MI, thereby jeopardizing endogenous mechanisms of protection against apoptosis. Importantly, BFT restored Pim-1 in diabetic hearts and enhanced Pim-1 activation in non-diabetic hearts, resulting in significant preservation of cardiomyocyte viability. Thus, modulation of Pim-1 produces functional gains independent of its basal state of expression.
An intriguing aspect of our study consists of the parallel effects of BFT between non-diabetic and diabetic groups. One possible explanation is that high dosage of BFT may have overpowered the impaired pentose phosphate activity in diabetes. Alternatively, BFT may interfere with mechanisms independent of the pentose phosphate pathway, as supported by the counter-regulatory effect of BFT on the neurohormonal response of infarcted mice. Importantly, in diabetic mice, BFT-induced neurohormonal modulation was associated to restoration of adrenergic sensitivity, which accounts for reactive tachycardia and improved inotropic and lusitropic responses. In the CONSENSUS study, high levels of angiotensin II, atrial natriuretic peptide and norepinephrine were strong predictors of mortality in post-MI patients, regardless of treatment randomization [24]. Furthermore, it has been demonstrated that the activation of neurohormonal systems could help in maintaining the circulatory homeostasis in the early stage post-MI, but excessive or sustained activation has deleterious effects on cardiac function, contributing to the progression to chronic heart failure [39–41]. These data reinforce the potential therapeutic value of BFT for prevention of cardiac dysfunction.
Taken together, supplementation of BFT benefits post-MI outcome through multiple mechanisms involving neurohormonal regulation, control of redox state and AGE formation, stimulation of angiogenesis and promotion of pro-survival G6PD/Akt/Pim-1 pathway (summarized in Supplemental Fig. 9). Importantly, BFT also exhibited protective effects on non-diabetic hearts, thus calling for its clinical application in both diabetic and non-diabetic patients.
Supplementary Material
Acknowledgments
We thank Professor Takayuki Sato, Kochi Medical School, Japan for his kind scientific advice on markers of neurohormonal activation and Mr. Paul Salvage for his kind technical assistance in immunohistochemical analyses.
Funding sources: This study was supported by a project grant from UK Diabetes Research Foundation (RD06/0003413) and Resolve (Resolve chronic inflammation and achieve healthy ageing, 202047). CE holds a British Heart Foundation (BHF) Basic Science fellowship (BS/05/01).
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yjmcc.2010.05.014.
References
- [1].Granger CB, Califf RM, Young S, Candela R, Samaha J, Worley S, et al. Outcome of patients with diabetes mellitus and acute myocardial infarction treated with thrombolytic agents. The Thrombolysis and Angioplasty in Myocardial Infarction (TAMI) Study Group. J Am Coll Cardiol. 1993 Mar 15;21(4):920–5. doi: 10.1016/0735-1097(93)90348-5. [DOI] [PubMed] [Google Scholar]
- [2].Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998 Jul 23;339(4):229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- [3].Ashfaq S, Ghazzal Z, Douglas JS, Morris DC, Veledar E, Weintraub WS. Impact of diabetes on five-year outcomes after vein graft interventions performed prior to the drug-eluting stent era. J Invasive Cardiol. 2006 Mar;18(3):100–5. [PubMed] [Google Scholar]
- [4].Flaherty JD, Davidson CJ. Diabetes and coronary revascularization. Jama. 2005 Mar 23;293(12):1501–8. doi: 10.1001/jama.293.12.1501. [DOI] [PubMed] [Google Scholar]
- [5].Stone PH, Muller JE, Hartwell T, York BJ, Rutherford JD, Parker CB, et al. The effect of diabetes mellitus on prognosis and serial left ventricular function after acute myocardial infarction: contribution of both coronary disease and diastolic left ventricular dysfunction to the adverse prognosis. The MILIS Study Group. J Am Coll Cardiol. 1989 Jul;14(1):49–57. doi: 10.1016/0735-1097(89)90053-3. [DOI] [PubMed] [Google Scholar]
- [6].Ishihara M, Inoue I, Kawagoe T, Shimatani Y, Kurisu S, Nishioka K, et al. Impact of acute hyperglycemia on left ventricular function after reperfusion therapy in patients with a first anterior wall acute myocardial infarction. Am Heart J. 2003 Oct;146(4):674–8. doi: 10.1016/S0002-8703(03)00167-4. [DOI] [PubMed] [Google Scholar]
- [7].Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. 2006 Feb 21;47(4):693–700. doi: 10.1016/j.jacc.2005.09.050. [DOI] [PubMed] [Google Scholar]
- [8].Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006 Mar 17;98(5):596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- [9].Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, et al. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003 Jul 25;93(2):e9–e16. doi: 10.1161/01.RES.0000083489.83704.76. [DOI] [PubMed] [Google Scholar]
- [10].Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang YY, Loscalzo AJ, et al. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem. 2003 Aug 22;278(34):32100–6. doi: 10.1074/jbc.M301293200. [DOI] [PubMed] [Google Scholar]
- [11].Pan S, World CJ, Kovacs CJ, Berk BC. Glucose 6-phosphate dehydrogenase is regulated through c-Src-mediated tyrosine phosphorylation in endothelial cells. Arterioscler Thromb Vasc Biol. 2009 Jun;29(6):895–901. doi: 10.1161/ATVBAHA.109.184812. [DOI] [PubMed] [Google Scholar]
- [12].Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am J Physiol Ren Physiol. 2005 Nov;289(5):F1040–7. doi: 10.1152/ajprenal.00076.2005. [DOI] [PubMed] [Google Scholar]
- [13].Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem. 2000 Dec 22;275(51):40042–7. doi: 10.1074/jbc.M007505200. [DOI] [PubMed] [Google Scholar]
- [14].Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, et al. Increased myocardial dysfunction after ischemia–reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation. 2004 Feb 24;109(7):898–903. doi: 10.1161/01.CIR.0000112605.43318.CA. [DOI] [PubMed] [Google Scholar]
- [15].Marchetti V, Menghini R, Rizza S, Vivanti A, Feccia T, Lauro D, et al. Benfotiamine counteracts glucose toxicity effects on endothelial progenitor cell differentiation via Akt/FoxO signaling. Diabetes. 2006 Aug;55(8):2231–7. doi: 10.2337/db06-0369. [DOI] [PubMed] [Google Scholar]
- [16].Balakumar P, Chakkarwar VA, Singh M. Ameliorative effect of combination of benfotiamine and fenofibrate in diabetes-induced vascular endothelial dysfunction and nephropathy in the rat. Mol Cell Biochem. 2009 Jan;320(1–2):149–62. doi: 10.1007/s11010-008-9917-z. [DOI] [PubMed] [Google Scholar]
- [17].Beltramo E, Berrone E, Tarallo S, Porta M. Different apoptotic responses of human and bovine pericytes to fluctuating glucose levels and protective role of thiamine. Diab/Metab Res Rev. 2009 Sep;25(6):566–76. doi: 10.1002/dmrr.996. [DOI] [PubMed] [Google Scholar]
- [18].Gadau S, Emanueli C, Van Linthout S, Graiani G, Todaro M, Meloni M, et al. Benfotiamine accelerates the healing of ischaemic diabetic limbs in mice through protein kinase B/Akt-mediated potentiation of angiogenesis and inhibition of apoptosis. Diabetologia. 2006 Feb;49(2):405–20. doi: 10.1007/s00125-005-0103-5. [DOI] [PubMed] [Google Scholar]
- [19].Katare RG, Caporali A, Oikawa A, Meloni M, Emanueli C, Madeddu P. Vitamin B1 analogue benfotiamine prevents diabetes-induced diastolic dysfunction and heart failure through Akt/Pim-1 Mediated survival pathway. Circ Heart Fail. 2010 Mar;3(2):294–305. doi: 10.1161/CIRCHEARTFAILURE.109.903450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. 2003 Aug;52(8):2110–20. doi: 10.2337/diabetes.52.8.2110. [DOI] [PubMed] [Google Scholar]
- [21].de Simone G, Wallerson DC, Volpe M, Devereux RB. Echocardiographic measurement of left ventricular mass and volume in normotensive and hypertensive rats. Necropsy validation. Am J Hypertens. 1990 Sep;3(9):688–96. doi: 10.1093/ajh/3.9.688. [DOI] [PubMed] [Google Scholar]
- [22].Spillmann F, Graiani G, Van Linthout S, Meloni M, Campesi I, Lagrasta C, et al. Regional and global protective effects of tissue kallikrein gene delivery to the peri-infarct myocardium. Regen Med. 2006 Mar;1(2):235–54. doi: 10.2217/17460751.1.2.235. [DOI] [PubMed] [Google Scholar]
- [23].Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Investig. 2003 Oct;112(7):1049–57. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation. 1990 Nov;82(5):1730–6. doi: 10.1161/01.cir.82.5.1730. [DOI] [PubMed] [Google Scholar]
- [25].Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, et al. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol. 2007 Jun;102(6):2104–11. doi: 10.1152/japplphysiol.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998 Mar 17;95(6):2979–84. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol. 2004 Mar;286(3):H823–9. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- [28].Muraski JA, Fischer KM, Wu W, Cottage CT, Quijada P, Mason M, et al. Pim-1 kinase antagonizes aspects of myocardial hypertrophy and compensation to pathological pressure overload. Proc Natl Acad Sci USA. 2008 Sep 16;105(37):13889–94. doi: 10.1073/pnas.0709135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997 Oct 17;91(2):231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- [30].Logstrup BB, Hofsten DE, Christophersen TB, Moller JE, Botker HE, Pellikka PA, et al. Influence of abnormal glucose metabolism on coronary microvascular function after a recent myocardial infarction. Jacc. 2009 Oct;2(10):1159–66. doi: 10.1016/j.jcmg.2009.06.012. [DOI] [PubMed] [Google Scholar]
- [31].Hofsten DE, Logstrup BB, Moller JE, Pellikka PA, Egstrup K. Abnormal glucose metabolism in acute myocardial infarction: influence on left ventricular function and prognosis. Jacc. 2009 May;2(5):592–9. doi: 10.1016/j.jcmg.2009.03.007. [DOI] [PubMed] [Google Scholar]
- [32].Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, et al. Myocardial cell death in human diabetes. Circ Res. 2000 Dec 8;87(12):1123–32. doi: 10.1161/01.res.87.12.1123. [DOI] [PubMed] [Google Scholar]
- [33].Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002 Jun;51(6):1938–48. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- [34].Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D, et al. Oxidative damage to DNA in diabetes mellitus. Lancet. 1996 Feb 17;347(8999):444–5. doi: 10.1016/s0140-6736(96)90013-6. [DOI] [PubMed] [Google Scholar]
- [35].Jermendy G. Evaluating thiamine deficiency in patients with diabetes. Diab Vasc Dis Res. 2006 Sep;3(2):120–1. doi: 10.3132/dvdr.2006.014. [DOI] [PubMed] [Google Scholar]
- [36].Sudnikovich EJ, Maksimchik YZ, Zabrodskaya SV, Kubyshin VL, Lapshina EA, Bryszewska M, et al. Melatonin attenuates metabolic disorders due to streptozotocin-induced diabetes in rats. Eur J Pharmacol. 2007 Aug 27;569(3):180–7. doi: 10.1016/j.ejphar.2007.05.018. [DOI] [PubMed] [Google Scholar]
- [37].Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008 Jan 5;371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- [38].Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007 Dec;13(12):1467–75. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- [39].Chatterjee K. Neurohormonal activation in congestive heart failure and the role of vasopressin. Am J Cardiol. 2005 May 2;95(9A):8B–13B. doi: 10.1016/j.amjcard.2005.03.003. [DOI] [PubMed] [Google Scholar]
- [40].Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol. 1992 Jul;20(1):248–54. doi: 10.1016/0735-1097(92)90167-l. [DOI] [PubMed] [Google Scholar]
- [41].Moe GW, Rouleau JL, Charbonneau L, Proulx G, Arnold JM, Hall C, et al. Neurohormonal activation in severe heart failure: relations to patient death and the effect of treatment with flosequinan. Am Heart J. 2000 Apr;139(4):587–95. doi: 10.1016/s0002-8703(00)90035-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.