

Abstract
We use Digital Holographic Microscopy to study dynamic responses of live cells to femtosecond laser cellular membrane photoporation. Temporal and spatial characteristics of morphological changes as well as dry mass variation are analyzed and compared with conventional fluorescent assays for viability and photoporation efficiency. With the latter, the results provide a new insight into the efficiency and toxicity of this novel optical method of drug delivery. In addition, quantitative phase maps reveal photoporation related sub-cellular dynamics of cytoplasmic vesicles.
OCIS codes: (090.1760) Computer holography, (120.5050) Phase measurement, (170.0170) Medical optics and biotechnology, (170.1530) Cell analysis, (999.9999) Photoporation
1. Introduction
Laser induced poration of the cell membrane has recently gained much attention [1]. It offers a selective non-contact sterile targeted method to introduce membrane impermeable molecules into the cell’s cytoplasm and emerges as an attractive alternative to the classical methods of intracellular drug delivery. It can be easily combined with microscopic imaging or optical tweezers [2], which opens the way to a variety of novel experiments in molecular biology.
Laser light irradiation has been used to optoinject a range of molecules and nanoparticles [3], as well as to transfect cells with foreign genetic material [1,4] or silence specific genes [5]. A good extensive review of the available laser based transfection techniques can be found in [1]. While many different types of lasers have been used for this purpose, femtosecond pulsed infrared lasers have emerged as a source of choice for their ability to target selectively single cells with a high degree of viability. The ultrashort pulsed beam, usually provided by a Titanium-Sapphire laser at a wavelength of approximately 800 nm and pulse duration of ~100fs, creates a low density plasma at the cellular membrane in a multiphoton process, which may lead to the appearance of a short lived cavitation bubble [6]. This induces a transient sub-micrometer pore in the membrane which rapidly seals leaving the cell viable.
The unrivaled accuracy of the treatment has already proven advantageous in a variety of challenging biological experiments such as the transfection of embryonic zebrafish [7] and stem cells [8,9]. It was also shown that a transcription factor Elk1 mRNA optoinjected into the soma of primary neurons causes a different cellular response to mRNA injected into the dendrites [10]. In another exciting application, the whole transcriptome was extracted from primary astrocyte cells and optoinjected into individual primary rat hippocampal neurons causing their phenotypical change into astrocytes [11].
Although this drug delivery technique has gained popularity, the dynamic cellular response to the femtosecond laser membrane poration has not, to date, been the subject of detailed investigation. Numerous questions present themselves including the physical response of the cell to the poration process and the correlation between the cell response and uptake of dye. It would be advantageous to develop label-free techniques to address these points. Previous studies have estimated that the relative volume exchanged in the process is 0.4 times the total cell volume [12] and that reactive oxygen species may contribute to the toxicity of femtosecond laser membrane poration [13]. However, no real-time volumetric study of the cellular dynamic response to photoporation has been shown to date. Further, the femtosecond membrane poration is very subtle in its nature and the resulting cellular response, except for the cavitation bubble itself, is minuscule and difficult to observe, in particular in brightfield imaging. In previous experiments, typically the efficiency of membrane poration was not verified directly during irradiation. Instead, membrane impermeable fluorescent dyes, like propidium iodide (PI) [12] or Lucifer Yellow (LY) [10] were used and their intracellular presence was checked post factum. Similarly, the cells’ viability was proven after a few hours of incubation using a fluorescent viability assay, e.g. Calcein AM [14] or in a fluorophore exclusion test [12]. However, fluorescence-based procedures are cumbersome, time consuming and depend on long-term tracking or identification of each cell. Moreover, fluorescent imaging carries the risk of cyto- and photo-toxicity, which may be disadvantageous in the case of sensitive cell types. In this work we explore the possibility of using label-free quantitative phase imaging to predict the uptake of biologically relevant compounds and subsequent cell viability of membrane photoporation.
Firstly, it is important to contrast this approach with other potential methods. Live cells are mostly optically transparent with minimal scattering occurring with the intracellular structures. As a consequence simple brightfield light microscopy is usually not suitable for detailed observation of cellular morphology. As an alternative, the variance of the refractive index of the intracellular structures, which results in optical phase shift of the transmitted light, can be used as an endogenous contrast technique. The two most popular methods of phase shift visualization, phase contrast (PC) and differential interference contrast (DIC) have been successfully used in live cell imaging [15]. However, both these techniques are based on a nonlinear relationship between the optical phase shift and the produced intensity distributions as such are inherently qualitative. As a result they do not offer the possibility to reconstruct the thickness of the cell from the recorded images. Moreover they produce artifacts, such as the halo in PC and directional loss of contrast in DIC.
Quantitative phase microscopy, has recently gained attention as it provides high quality cytometric data on live cells without any additional sample preparation [16–19]. Among other applications Quantitative Phase Imaging (QPI) has been used to investigate a cell’s response to hypotonic shock [20,21]; the cellular membrane dynamics in living red blood cells [22–24]; as well as the beating motion of cardiac myocytes [17]. QPI has also been used to evaluate the impact of nanosecond laser cell microsurgery where a change in optical thickness of a cell was observed along the ablation path [25,26]. The mechanism of laser surgery depends strongly on the pulse duration and focusing of the beam, and in the case of loosely focused nanosecond pulses, the main effect is photothermal [27]. In contrast, our studies use a tightly focused femtosecond laser beam to create low density plasma with minimal heating so that the damage to the cell is related mainly to laser induced chemical decomposition. Also, optical breakdown is achieved at a much lower mean power than with a nanosecond laser which limits the affected area. Here, we show that as a consequence of this significant difference in the laser-cell interaction, the cellular response to light is transient and more localized when using femtosecond laser membrane poration as compared to nanosecond irradiation [25]. In our study we use the cytometric data from both quantitative phase and fluorescence imaging to verify viability and cellular membrane integrity.
In this paper, we use QPI obtained with a single frame off-axis transmission Digital Holographic Microscopy (DHM) [28] to analyze the cell’s response to transient poration of the cellular membrane caused by the multiphoton absorption of a femtosecond near infrared beam. Off-axis DHM provides high spatial resolution comparable to brightfield diffraction limited imaging, while the temporal resolution is limited solely by the frame rate of the camera owing to single frame recording. Moreover, the cell thickness can be reconstructed with sub-wavelength precision [20,21]. These features make DHM particularly well suited for quantitative phase observation of dynamic processes in live cells. In addition it offers the possibility of numerical refocusing, which enables three dimensional in-focus reconstruction of the whole sample and facilitates simultaneous observation of events in multiple image planes. Also, the optical phase shift can be translated into the physical thickness of a cell if the refractive indices of the cell and of the surrounding medium are known.
To our knowledge this is the first time that the cellular response to femtosecond laser membrane photoporation has been quantified using a label-free technique. We present two examples of a typical dynamic morphological reaction resulting in either a viable or non-viable cell and quantify these changes. Next, we relate the temporal and spatial scale of the observed swelling that occurs under laser irradiation to well established fluorescence-based efficiency and viability assays. We also show how intracellular dynamics can be revealed in such a quantitative phase map.
2. Experimental procedure
2.1 Photoporation system with DHM
An off-axis transmission DHM system was developed and incorporated within a NIKON TE-2000E inverted microscope as shown in Fig. 1 .
Fig. 1.
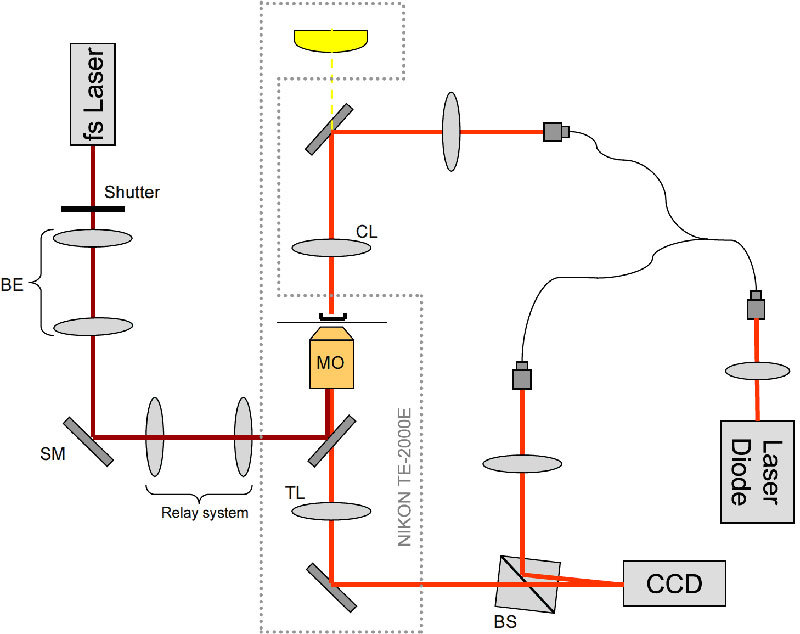
DHM (implemented with a laser diode) integrated with an inverted research microscope and photoporation beam (fs laser). CL-condenser lens (NA 0.3), MO – microscope objective (60x, NA = 1.3, oil), BS – cube beam splitter, SM – steering mirror for the poration beam, TL - tube lens, BE - telescopic beam expander. Shutter controls the irradiation time and number of doses.
Light from a laser diode (Hitachi HL6344G, λ = 635 nm, coherence length lc≈90 mm) is coupled into a single mode optical fiber splitter. The beam from one of the arms is combined with the microscope’s brightfield illumination and focused through a long-working distance condenser (NA = 0.3) onto the sample. Light transmitted through the sample in a Petri dish, is collected by an objective (60x, NA = 1.3) and the image is created in a side port of the microscope. Light from the second arm of the fiber splitter is used as a reference beam in the Mach-Zehnder interferometer configuration. The two beams are recombined by a cube beam-splitter at a small angle to produce a typical off-axis hologram on the CCD camera (Imaging Source DMK31BU03 1024x768, 8 bit, 30 fps). The complex wavefront in the object plane is reconstructed from the holograms using the Fourier space filtering technique [29] either in real time using LabView or in MATLAB post-processing. When necessary the reconstructed phase is unwrapped and the quadratic curvature of the background is fitted and subtracted [30].
The optical phase shift φ can be translated into the physical thickness of a cell d = λ(φ/2π)/(ncell-nmedium) if the refractive indices of the cell ncell and of the surrounding medium nmedium are known. For the CHO-K1 cells and culture medium used in this work, we assume ncell = 1.39 [31] and nmedium = 1.34 when the physical thickness is discussed, which means that 1 rad of phase shift translates to 2 μm of cell thickness. However, it is important to note that the refractive index of a cell may vary significantly at the sub-cellular level, in particular during laser photoporation and the related swelling. Since the implementation of DHM used in the presented work does not allow for an independent measurement of the refractive index for clarity of presentation we discuss changes in terms of optical rather than physical thickness. We verified a good temporal stability of the system with a standard deviation of the reconstructed phase signal fluctuations of STD = 0.082 rad over a period of 30 minutes, which corresponds with an axial resolution of approximately 165 nm.
The photoporation infrared beam (800 nm, 180 fs @ 80 MHz generated by Coherent MIRA900F) is coupled into the microscope objective using a dichroic mirror placed in the epifluorescence turret of the microscope. The beam expanding telescope (BE) and the steering mirror (SM) placed in a plane conjugate to the back focal plane of the objective, are used to focus the beam precisely on the top of the membrane of the cell. The back aperture of the objective is overfilled in order to obtain a diffraction limited focal spot.
2.2 Cell preparation
Chinese hamster ovary (CHO-K1) cells were cultured at 37 °C and 5% CO2 in Modified Eagles Medium (Sigma, UK) with 10% Foetal Bovine Serum (Sera Laboratories International), L-Glutamine (2 mM, Sigma), streptomycin (100 μg/ml, Sigma) and penicillin (100 units/ml, Sigma). Cells were routinely passaged three times a week. CHO-K1 cells were seeded at a density of 2.4 x104 cells per ml onto 35mm glass-bottomed culture grade dishes (World Precision Instruments) to achieve 40-50% confluency. Before the experiments, the cells were incubated at 37 °C and 5% CO2 for 48 h to allow cell attachment to the bottom of the glass dishes.
For the fluorophore optoinjection experiments, the cell monolayer was washed twice with 1 ml OptiMEM before the addition of 3 μM of Propidium Iodide (PI, Invitrogen). The fluorescent signal from PI was obtained at least 5 min after irradiation. Cells were then washed twice with 1 ml of OptiMEM and fresh medium was added to the cells before further incubating for at least 90 min. Prior fluorescence imaging for cell viability, cells were washed twice with 1 ml of Hanks’ Balanced Salt Solution (HBSS, Sigma), and then 2 μM of Calcein AM (CAM, Invitrogen) in HBSS solution was added and the cells further incubated for 15 min. Non-fluorescent CAM is cell membrane permeant and is converted to green fluorescent Calcein after hydrolysis of intracellular cell esterases. Once inside, it is retained by the cells that have intact plasma membranes. However, in damaged or dead cells both unhydrolyzed and fluorescent products can leak out of the cell. Therefore healthy cells have characteristically bright green fluorescence while minimal and punctuate signal is indicative of cell death or compromised viability.
Fluorescence imaging was performed using an EMCCD camera (Andor iXon + ) with the mercury lamp as the excitation source. FITC and TRITC filter sets were used for CAM and PI imaging, respectively.
3. Results
In the literature, typically two different fs laser dosages have been used to date to achieve optoinjection and phototransfection. In one approach, a single irradiation dose is used with the power and irradiation time optimized experimentally [12], while in the other, three doses are applied in a sequence, usually using lower laser fluence, possibly profiting from accumulation of laser induced chemical effects [9]. In both cases the trade-off between membrane poration efficiency and cell viability is crucial and has to be determined using fluorescence imaging.
In our work, the off-axis DHM system was used to acquire a series of time lapse recordings of the membrane poration events at the full frame rate of the camera (30 fps) using various sets of irradiation parameters with both single and triple dosages. The quantitative phase and amplitude maps were reconstructed and analyzed. In the case of out-of-focus images, the reconstructed complex amplitude was numerically re-propagated using a convolution implementation of the Kirchhoff-Fresnel diffraction formula [32] to a plane in which the details of interest, such as edge of the cell, cavitation bubble or intracellular organelles, were in focus. Figure 2 illustrates how this distinctive feature of DHM can be used to obtain at will, a focused intensity image of the photoporated cell (plane in which the diffraction pattern around the outer edge of the cell disappears, Fig. 2c) and simultaneously, a sharp-edged image of the cavitation bubble which forms 8.5 μm above the surface of the dish (Fig. 2b). As the cavitation bubble is formed on the top surface of the cell membrane, the distance between the two planes approximates to the thickness of the cell. Similar transient cavitation bubbles were previously imaged by brightfield microscopy by Vogel et al. [33] but without the added advantage of simultaneous in-focus imaging of the irradiated cell. Although the intensity images shown in Fig. 2 suffer from minor laser speckle noise originating from the coherent illumination, one can clearly determine the actual diameter of the cavitation bubble. In this example a manual technique based on analysis of the diffraction pattern around the scatterer has been used, but in a more automated manner, an autofocus criterion could be used [34,35] or an extended focused image could even be created [36].
Fig. 2.

Intensity image of a cell during photoporation (with the cavitation bubble visible in the center of the cell) reconstructed from a recorded hologram: (a) in the original recorded plane, (b) after numerical refocusing to the plane in which the bubble is focused, (c) after numerical refocusing to the plane in which the cell is focused. The distance between the image planes in (b) and (c) is 8.5 μm. The process of numerical refocusing between the two planes can be seen in Media 1 (2.3MB, AVI) . The speckle noise visible in these images is related to the laser source used for the illumination. Scale bars 5 μm.
3.1 Time-lapse quantitative phase recordings
The time-lapse recordings of a series of membrane poration events showed that the cellular response strongly depends not only on the parameters of irradiation but also on the location of the photoporation site on the cell, as well as on the morphology, size and shape of the cell. Figure 3 shows two distinctive examples of the observed cellular dynamic response. They illustrate the typical responses with a cell either remaining viable or suffering from irreversible damage. Both cells were irradiated using a moderate power of P = 75 mW at the sample and an irradiation time of T = 40 ms. The cell shown in Fig. 3a–3f was dosed once leading to a localized swelling within a 4 μm radius that reached its maximum of 0.68 rad, which corresponds to a change of approximately 1.4 μm. The swelling retracted 45 seconds after irradiation with the cell recovering to its pre-treatment state. No change in optical thickness in other parts of the cell was observed apart from that coming from its natural movement. Notably, irradiation triggered dislocation of a submicron sized vesicle-like intracellular object that could be resolved in the phase map (Media 2 (3.5MB, AVI) and Fig. 3b). The vesicle seen in Fig. 3b was initially attracted towards the poration spot and then repelled during recovery, which suggests hydrodynamic flow within the cytoplasm.
Fig. 3.
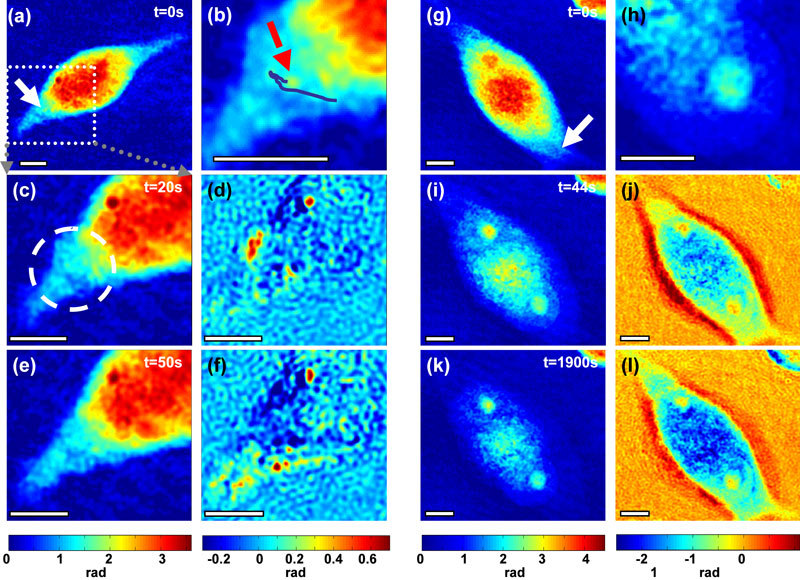
Optical thickness of cells reveals various responses of CHO-K1 cells depending on the irradiation dose. A cell dosed once (a-f) swells up locally and recovers to its original state after about 50 seconds (Media 2 (3.5MB, AVI) ), b) shows the trajectory of a vesicle-like body. A similar cell dosed three times (g-l) (Media 4 (3.7MB, AVI) ) exhibits swelling of the whole cell body; d),f) (Media 3 (3.6MB, AVI) ),j), and l) (Media 5 (3.9MB, AVI) ) show the quantitative change in optical thickness compared to time t = 0s, h) magnified region of the easily discernable nucleus with distinct nucleolus. White arrows point to the irradiation spot. All scale bars 5 μm.
The same irradiation dose (P = 75 mW, T = 40 ms) but with three consecutive doses, caused global swelling of the cell as shown in Fig. 3g–3l. The whole body of the cell expanded laterally while the optical thickness in the center of the cytoplasm decreased significantly (by up to 58%). A similar behavior has been reported previously in neurons during hypotonic shock [20] where the decrease in optical thickness is related to a significant influx of water into the cell and a consequent decrease in the refractive index of the diluted cytoplasm, which dominates over the increase in the axial dimension of the cell. Interestingly, initially the nuclear membrane remained intact preventing the drop in refractive index of the nucleus (Fig. 3g, 3i, 3k), which increased the contrast between the nucleus and cytoplasm (Fig. 3h). This revealed the position and shape of the nucleus which was undistinguishable before the photoporation.
The time scale of the global swelling process was significantly longer than the local swelling that occurred in the first cell, as this second cell reached its maximum volume after 800 seconds. Figure 4 compares the different time scales and recovery rates in the cytoplasm and swelling region in both cells. The second cell was clearly not able to recover from the swelling process. Moreover, after about 600 seconds the refractive index of the nucleus gradually started to decrease indicating the breakdown of the nuclear membrane. At the same time the optical thickness of the first healthy cell returns to its original value with only a small decrease of 0.12 rad (or 240 nm) in the poration spot which most likely was related to the natural movement of the cell.
Fig. 4.
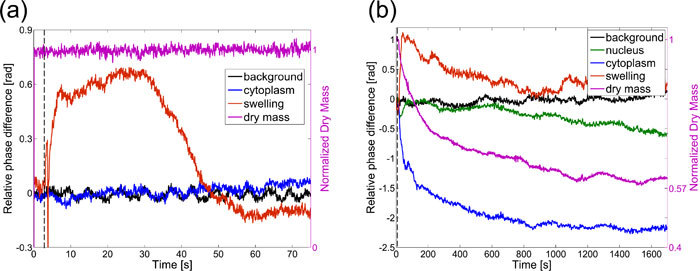
Time trace of optical thickness change in various places in the cell. a) cell dosed once recovers from local swelling after 45 seconds, b) cell dosed three times swells up without recovery. Dry mass is normalized to the average value before photoporation (right axis). The rapid variation in phase shift of the swelling region (red curve) in graph (a) is related to the transient cavitation bubble created in this spot (signal acquired at 30 fps). This is not visible in graph (b) as the sampling rate in this acquisition (1 fps) is lower than the life-time of the bubble. Dashed vertical lines indicate the irradiation onset. “Background” – region of background remote from the cell; “Nucleus”-region within the nucleus; “Cytoplasm” – region of cytoplasm remote from the poration site within the original boundaries of the cell; “Swelling” – (a) small region including the poration site, (b) region originally outside of the cell into which the cell expanded due to swelling.
The irreversible damage to the cellular membrane is substantiated further by the behavior of the cellular dry mass. This is one of the key cytometric parameters that can be retrieved from the quantitative phase map of the cell [18] and is proportional to the surface integral of the phase shift φ(x,y):
| (1) |
where A is the area of the cell. As the intracellular refractive index and consequently φ(x,y) and M depend mainly on the concentration of proteins, in healthy cells M is constant under cellular volume changes. However, if the cellular membrane remains open for a significant period of time, the mobile contents of the cytoplasm may diffuse out of the cell resulting in a drop of the total dry mass. Indeed, comparison of the dry mass variation in the two discussed cells shows that while there was no noticeable dry mass change in the viable cell a significant exponential decrease is clearly observed in the permanently damaged one. This decrease saturates at approximately 60% of the original dry mass which most likely corresponds with the amount of the immobile protein content in cytoplasm. We have not observed any noticeable increase in the refractive index of the surrounding medium due to the dry mass leakage, which is most probably a result of the high diffusion rate of proteins in water (in the order of 10−6 cm2/s [37]) and a large volume of the surrounding medium. Strikingly, the optical thickness in the cytoplasm (blue curve in Fig. 4b) decays at a similar rate to that of the dry mass, which suggests that cytoplasmic dry mass loss is the main cause of the rapid drop in the optical thickness of the cytoplasm in the second cell. It must be noted that a qualitatively similar local decrease in cytoplasmic optical thickness, observed in viable cells under hypotonic shock, results purely from water uptake during swelling [20]. However, in the non-viable photoporated cell, the loss of dry mass confirms an irreversible response and indicates necrosis. Importantly, this significant difference would pass unnoticed for such cells observed in brightfield imaging while PC and DIC would show the qualitative temporal change in the cell’s image but would not enable dry mass determination and decoupling of these two inherently different mechanisms.
3.2 Relation of dynamic phase map information to viability and efficiency fluorescent assays
To further investigate the relationship between the spatial and temporal cellular reaction, as revealed in the acquired quantitative phase maps, to the efficiency and toxicity of photoporation we compared the phase information with a traditionally used fluorescent viability assays. The efficiency of cellular membrane poration can be most directly assessed by optoinjection of a fluorophore whose molecules are small enough to diffuse freely through the transient opening created in the membrane. We used propidium iodide (PI), which binds to nucleic acids within the cell and becomes fluorescent within a few minutes. PI is membrane impermeable in healthy cells and at the concentration used it is completely excluded from viable cells for the whole duration of the experiment. We also used Calcein AM as a viability assay since its intensity is related to two cell viability indicators – concentration of esterases and integrity of the membrane.
N = 40 cells were photoporated with a single laser dose using the average power of P = 75 mW and T = 40 ms irradiation time. The appropriate fluorescent images and quantitative phase time-lapse sequences were recorded and analyzed. Figure 5 shows a typical set of acquired images. Each cell was checked for viability and optoinjection and the time-scale and extent of swelling was determined. The observed response depended significantly on the cell size, shape, morphology and the location of the photoporation site. However, it was possible to identify a range of typical values. Viably optoinjected cells showed transient localized swelling of no more than 0.83 rad (or 1.66 μm) in the 1-4.5 μm radius around the photoporation site with a typical retraction time of 24-73 s. In the cases when the swelling radius was larger than 11 μm, or when recovery lasted longer than 95 s, the cells proved to be necrotic after 90 min from irradiation. Characteristically, in all viable cells no noticeable dry mass loss was observed.
Fig. 5.
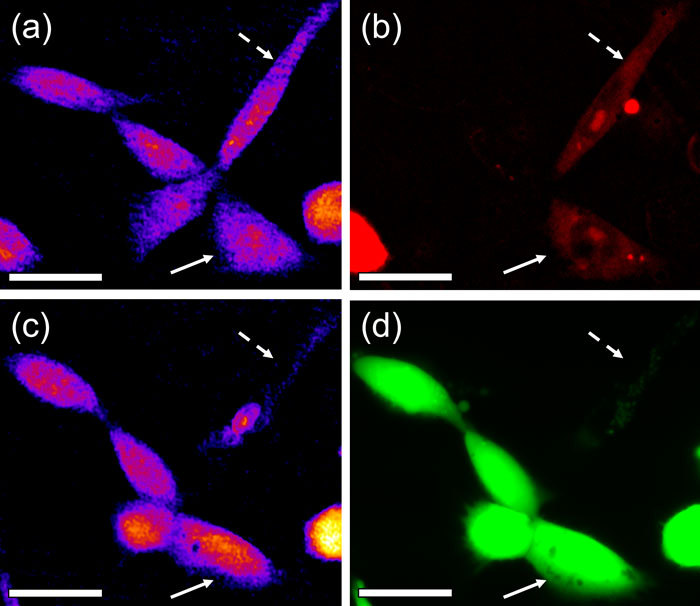
Comparison of quantitative phase maps with fluorescent assays during an optoinjection experiment: a) phase map and b) propidium iodide fluorescence (optoinjection assay) 5 min after irradiation ; c) phase map and d) Calcein AM fluorescence (viability assay) after 90 min incubation. Two cells were successfully optoinjected - one proved viable (solid arrow) while the other (dashed arrow) was necrotic after 90 min. Note the significant decrease in the optical thickness of the non-viable cell. Scale bars 20 μm.
4. Conclusion
In this study, we have demonstrated that DHM provides a new insight into the cell’s response to cellular membrane poration with femtosecond near-infrared lasers. In photoporated cells that remain viable, the observed morphological changes are minimal and characterized by transient localized swelling. Interestingly, dynamics of this swelling may lead to intracellular vesicle movement.
We showed that the temporal dynamics of the cellular optical thickness can be related to the toxicity of the treatment and give a viability indication without fluorescent staining. We have confirmed this relation using a commonly used viability assay based on fluorescent imaging. The quantitative phase map also revealed the loss of the cellular dry mass which is related to the leakage of intracellular vesicles and cytoplasmic material through the injured cellular membrane. This effect constitutes another indication of potential irreversible damage to the cellular membrane. The efficiency of membrane poration has been verified by using a membrane impermeable fluorescent dye (PI). No dry mass loss has been observed in viably optoinjected cells, while the observed irradiation-triggered swelling was confined to the poration site and typically fully retracted within the time-scale of about one minute.
Although the observed changes depended on individual properties of each given cell and poration site, it was possible to identify a typical spatial and temporal scale of changes characteristic to viably optoinjected cells. The additional cytology data provided by the quantitative phase map, in particular cellular dry mass, gives further insight into the extent of membrane damage and proves membrane repair in viable cells. This could be used as a direct indication of the toxicity and success of optoinjection experiments in which fluorescent imaging has to be avoided and a signal provided by a label-free technique would be advantageous.
Acknowledgment
We thank the Scottish University Life Sciences Alliance (SULSA) and the UK Engineering and Physical Sciences Research Council (EPSRC) for funding. MLTM acknowledges the support of a SUPA Prize Studentship. KD is a Royal Society Wolfson Merit Award holder. FGM and KD contributed equally to this work.
References and links
- 1.Stevenson D. J., Gunn-Moore F. J., Campbell P., Dholakia K., “Single cell optical transfection,” J. R. Soc. Interface 7(47), 863–871 (2010). 10.1098/rsif.2009.0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown C. T. A., Stevenson D. J., Tsampoula X., McDougall C., Lagatsky A. A., Sibbett W., Gunn-Moore F. J., Dholakia K., “Enhanced operation of femtosecond lasers and applications in cell transfection,” J Biophotonics 1(3), 183–199 (2008). 10.1002/jbio.200810011 [DOI] [PubMed] [Google Scholar]
- 3.McDougall C., Stevenson D. J., Brown C. T. A., Gunn-Moore F., Dholakia K., “Targeted optical injection of gold nanoparticles into single mammalian cells,” J Biophotonics 2(12), 736–743 (2009). 10.1002/jbio.200910030 [DOI] [PubMed] [Google Scholar]
- 4.Tirlapur U. K., König K., “Targeted transfection by femtosecond laser,” Nature 418(6895), 290–291 (2002). 10.1038/418290a [DOI] [PubMed] [Google Scholar]
- 5.Torres-Mapa M. L., Angus L., Ploschner M., Dholakia K., Gunn-Moore F. J., “Transient transfection of mammalian cells using a violet diode laser,” J. Biomed. Opt. 15(4), 041506 (2010). 10.1117/1.3430730 [DOI] [PubMed] [Google Scholar]
- 6.Vogel A., Noack J., Huttman G., Paltauf G., “Mechanisms of femtosecond laser nanosurgery of cells and tissues,” Appl. Phys. B 81(8), 1015–1047 (2005). 10.1007/s00340-005-2036-6 [DOI] [Google Scholar]
- 7.Kohli V., Robles V., Cancela M. L., Acker J. P., Waskiewicz A. J., Elezzabi A. Y., “An alternative method for delivering exogenous material into developing zebrafish embryos,” Biotechnol. Bioeng. 98(6), 1230–1241 (2007). 10.1002/bit.21564 [DOI] [PubMed] [Google Scholar]
- 8.Uchugonova A., König K., Bueckle R., Isemann A., Tempea G., “Targeted transfection of stem cells with sub-20 femtosecond laser pulses,” Opt. Express 16(13), 9357–9364 (2008). 10.1364/OE.16.009357 [DOI] [PubMed] [Google Scholar]
- 9.Mthunzi P., Dholakia K., Gunn-Moore F., “Photo-transfection of mammalian cells using femtosecond laser pulses: optimisation and applicability to stem cell differentiation,” J. Biomed. Opt. 15(4), 041507 (2010). 10.1117/1.3430733 [DOI] [PubMed] [Google Scholar]
- 10.Barrett L. E., Sul J. Y., Takano H., Van Bockstaele E. J., Haydon P. G., Eberwine J. H., “Region-directed phototransfection reveals the functional significance of a dendritically synthesized transcription factor,” Nat. Methods 3(6), 455–460 (2006). 10.1038/nmeth885 [DOI] [PubMed] [Google Scholar]
- 11.Sul J. Y., Wu C. W., Zeng F., Jochems J., Lee M. T., Kim T. K., Peritz T., Buckley P., Cappelleri D. J., Maronski M., Kim M., Kumar V., Meaney D., Kim J., Eberwine J., “Transcriptome transfer produces a predictable cellular phenotype,” Proc. Natl. Acad. Sci. U.S.A. 106(18), 7624–7629 (2009). 10.1073/pnas.0902161106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumgart J., Bintig W., Ngezahayo A., Willenbrock S., Murua Escobar H., Ertmer W., Lubatschowski H., Heisterkamp A., “Quantified femtosecond laser based opto-perforation of living GFSHR-17 and MTH53 a cells,” Opt. Express 16(5), 3021–3031 (2008). 10.1364/OE.16.003021 [DOI] [PubMed] [Google Scholar]
- 13.Baumgart J., Kuetemeyer K., Bintig W., Ngezahayo A., Ertmer W., Lubatschowski H., Heisterkamp A., “Repetition rate dependency of reactive oxygen species formation during femtosecond laser-based cell surgery,” J. Biomed. Opt. 14(5), 054040 (2009). 10.1117/1.3253382 [DOI] [PubMed] [Google Scholar]
- 14.Antkowiak M., Torres-Mapa M. L., Gunn-Moore F., Dholakia K., “Utilising dynamic diffractive optics for enhanced femtosecond laser based cell transfection,” J. Biophoton. in press) [DOI] [PubMed] [Google Scholar]
- 15.Stephens D. J., Allan V. J., “Light microscopy techniques for live cell imaging,” Science 300(5616), 82–86 (2003). 10.1126/science.1082160 [DOI] [PubMed] [Google Scholar]
- 16.Kemmler M., Fratz M., Giel D., Saum N., Brandenburg A., Hoffmann C., “Noninvasive time-dependent cytometry monitoring by digital holography,” J. Biomed. Opt. 12(6), 064002 (2007). 10.1117/1.2804926 [DOI] [PubMed] [Google Scholar]
- 17.Shaked N. T., Rinehart M. T., Wax A., “Dual-interference-channel quantitative-phase microscopy of live cell dynamics,” Opt. Lett. 34(6), 767–769 (2009). 10.1364/OL.34.000767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popescu G., Park Y., Lue N., Best-Popescu C., Deflores L., Dasari R. R., Feld M. S., Badizadegan K., “Optical imaging of cell mass and growth dynamics,” Am. J. Physiol. Cell Physiol. 295(2), C538–C544 (2008). 10.1152/ajpcell.00121.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann C. J., Yu L., Kim M. K., “Movies of cellular and sub-cellular motion by digital holographic microscopy,” Biomed. Eng. Online 5(1), 21 (2006). 10.1186/1475-925X-5-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rappaz B., Marquet P., Cuche E., Emery Y., Depeursinge C., Magistretti P., “Measurement of the integral refractive index and dynamic cell morphometry of living cells with digital holographic microscopy,” Opt. Express 13(23), 9361–9373 (2005). 10.1364/OPEX.13.009361 [DOI] [PubMed] [Google Scholar]
- 21.Shaked N. T., Finan J. D., Guilak F., Wax A., “Quantitative phase microscopy of articular chondrocyte dynamics by wide-field digital interferometry,” J. Biomed. Opt. 15(1), 010505 (2010). 10.1117/1.3290242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rappaz B., Barbul A., Hoffmann A., Boss D., Korenstein R., Depeursinge C., Magistretti P. J., Marquet P., “Spatial analysis of erythrocyte membrane fluctuations by digital holographic microscopy,” Blood Cells Mol. Dis. 42(3), 228–232 (2009). 10.1016/j.bcmd.2009.01.018 [DOI] [PubMed] [Google Scholar]
- 23.Park Y., Best C. A., Badizadegan K., Dasari R. R., Feld M. S., Kuriabova T., Henle M. L., Levine A. J., Popescu G., “Measurement of red blood cell mechanics during morphological changes,” Proc. Natl. Acad. Sci. U.S.A. 107(15), 6731–6736 (2010). 10.1073/pnas.0909533107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popescu G., Park Y., Choi W., Dasari R. R., Feld M. S., Badizadegan K., “Imaging red blood cell dynamics by quantitative phase microscopy,” Blood Cells Mol. Dis. 41(1), 10–16 (2008). 10.1016/j.bcmd.2008.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu L., Mohanty S., Zhang J., Genc S., Kim M. K., Berns M. W., Chen Z., “Digital holographic microscopy for quantitative cell dynamic evaluation during laser microsurgery,” Opt. Express 17(14), 12031–12038 (2009). 10.1364/OE.17.012031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu L., Mohanty S., Liu G., Genc S., Chen Z., Berns M. W., “Quantitative phase evaluation of dynamic changes on cell membrane during laser microsurgery,” J. Biomed. Opt. 13(5), 050508 (2008). 10.1117/1.2997375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quinto-Su P. A., Venugopalan V., “Mechanisms of laser cellular microsurgery,” Methods Cell Biol. 82, 113–151 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Cuche E., Bevilacqua F., Depeursinge C., “Digital holography for quantitative phase-contrast imaging,” Opt. Lett. 24(5), 291–293 (1999). 10.1364/OL.24.000291 [DOI] [PubMed] [Google Scholar]
- 29.Kreis T., “Digital holographic interference-phase measurement using the Fourier-transform method,” J. Opt. Soc. Am. A 3(6), 847–855 (1986). 10.1364/JOSAA.3.000847 [DOI] [Google Scholar]
- 30.Ferraro P., De Nicola S., Finizio A., Coppola G., Grilli S., Magro C., Pierattini G., “Compensation of the inherent wave front curvature in digital holographic coherent microscopy for quantitative phase-contrast imaging,” Appl. Opt. 42(11), 1938–1946 (2003). 10.1364/AO.42.001938 [DOI] [PubMed] [Google Scholar]
- 31.Takagi M., Kitabayashi T., Ito S., Fujiwara M., Tokuda A., “Noninvasive measurement of three-dimensional morphology of adhered animal cells employing phase-shifting laser microscope,” J. Biomed. Opt. 12(5), 054010 (2007). [DOI] [PubMed] [Google Scholar]
- 32.U. Schnars, and W. Jueptner, Digital holography: digital hologram recording, numerical reconstruction, and related techniques (Springer, Berlin, 2005), pp. ix, 164 p. [Google Scholar]
- 33.Vogel A., Linz N., Freidank S., Paltauf G., “Femtosecond-laser-induced nanocavitation in water: implications for optical breakdown threshold and cell surgery,” Phys. Rev. Lett. 100(3), 038102 (2008). 10.1103/PhysRevLett.100.038102 [DOI] [PubMed] [Google Scholar]
- 34.Langehanenberg P., Kemper B., Dirksen D., von Bally G., “Autofocusing in digital holographic phase contrast microscopy on pure phase objects for live cell imaging,” Appl. Opt. 47(19), D176–D182 (2008). 10.1364/AO.47.00D176 [DOI] [PubMed] [Google Scholar]
- 35.Dubois F., Schockaert C., Callens N., Yourassowsky C., “Focus plane detection criteria in digital holography microscopy by amplitude analysis,” Opt. Express 14(13), 5895–5908 (2006). 10.1364/OE.14.005895 [DOI] [PubMed] [Google Scholar]
- 36.Antkowiak M., Callens N., Yourassowsky C., Dubois F., “Extended focused imaging of a microparticle field with digital holographic microscopy,” Opt. Lett. 33(14), 1626–1628 (2008). 10.1364/OL.33.001626 [DOI] [PubMed] [Google Scholar]
- 37.Verkman A. S., “Solute and macromolecule diffusion in cellular aqueous compartments,” Trends Biochem. Sci. 27(1), 27–33 (2002). 10.1016/S0968-0004(01)02003-5 [DOI] [PubMed] [Google Scholar]