

Abstract
Mammalian thioredoxin reductase is a homodimeric pyridine nucleotide disulfide oxidoreductase that contains the rare amino acid, selenocysteine (Sec) on a C-terminal extension. We previously have shown that a truncated version of mouse mitochondrial thioredoxin reductase missing this C-terminal tail will catalyze the reduction of a number of small molecules. Here we show that the truncated thioredoxin reductase will catalyze the reduction of methaneseleninic acid. This reduction is fast at pH 6.1 and is only 4-fold less than the full-length enzyme containing Sec. This finding suggested to us that if the C-terminal Sec residue in the holoenzyme became oxidized to the seleninic acid form (Sec-SeO2−) that it would be quickly reduced back to an active state by enzymic thiols, and further suggested to us that the enzyme would be very resistant to irreversible inactivation by oxidation. We tested this hypothesis by reducing the enzyme with NADPH and subjecting it to high concentrations of H2O2 (up to 50 mM). The results show that the enzyme strongly resisted inactivation by 50 mM H2O2. In order to determine the redox state of the C-terminal Sec residue we attempted to inhibit the enzyme with dimedone. Dimedone alkylates protein sulfenic acid residues and presumably will alklyate selenenic acid (Sec-SeOH) residues as well. The enzyme was not inhibited by dimedone even when 150-fold excess was added to the reaction containing enzyme and H2O2. We also tested the ability of the truncated enzyme to resist inactivation by oxidation as well and found that it also was resistant to high concentrations of H2O2. One assumption for the use of Sec in enzymes is that it is catalytically superior to the use of cysteine. We, and others have previously suggested that there are reasons for the use of Sec in enzymes that are unrelated to the conversion of substrate to product. The data presented here supports this assertion. The results also imply that the redox signaling function of the thioredoxin system can remain active under oxidative stress.
Mammalian thioredoxin reductase (mTR)1 is a homodimeric pyridine nucleotide disulfide oxidoreductase that contains the rare and interesting 21st amino acid, selenocysteine (Sec, U) (1, 2), in its enzyme active site as part of a C-terminal extension. In the oxidized state, the Sec residue forms a selenosulfide bond with an adjacent Cys residue as part of a conserved Gly-Cys1-Sec2-Gly tetrapeptide motif.2 Because the Cys and Sec residues of this motif are directly adjacent, formation of the S1–Se2 bond results in the formation of an unusual 8-membered ring motif (4, 5). The reduction of this selenosulfide by the N-terminal disulfide redox center results in the formation of a selenolate anion, which is presumed to attack the disulfide bond of its primary protein substrate, thioredoxin (Trx). However, a recent model of the TR-Trx complex calls this long held assumption into question (6). The reasons given for the use of selenium (Se) as Sec in TR and other enzymes as opposed to sulfur (S) as cysteine (Cys) have been: (i) the large rate enhancement with a “selenolate anion in dithiol-disulfide interchange reactions” (7), (ii) that the high reactivity of Sec confers broad substrate specificity to mTR relative to Cys-containing TRs (8–10 and references therein), (iii) the lower pKa of a selenol and the higher nucleophilicity of a selenolate confers a type of “catalytic advantage” over Cys when in the attacking position of thiol/disulfide interchange reactions (11) and, (iv) that Sec is needed for its superior leaving group ability in a thiol/selenosulfide interchange reaction (12).
The insertion of Sec into a protein requires complex machinery involving multiple accessory factors that are involved in the recoding of a UGA stop codon into a sense codon for Sec (13–16). The complexity of the recoding apparatus indicates that Se must be needed to perform a specialized chemical function that the S atom of Cys cannot. Being part of the same group of the periodic table, Se and S possess many similar chemical properties (17). However, Sec must confer to the enzyme a distinct biochemical advantage over Cys, otherwise evolution would not have continually selected for the energetically costly Sec-insertion machinery. The search for the identity of this unique chemical function has been the raison d’etre of seleno-enzymology for several decades.
One of the original ideas to explain the existence of Sec was that it was an “ancient relic of the anaerobic world” because Se is more sensitive to oxidation than S (14, 18). Recent work by Rhee and coworkers seem to offer support for this original idea as they showed that the selenoenzyme glutathione peroxidase-1 (Gpx-1) is partially inactivated by treatment with 1 mM H2O2. The inactivation of Gpx-1 by H2O2 was due to the oxidation of the Sec residue of Gpx-1 to a seleninic acid (Sec-SeO2−) residue with concomitant loss of H2SeO2 and formation of dehydroalanine (DHA) (19).
In direct contrast to the work of Rhee and coworkers, we have recently presented the novel hypothesis that Sec-containing enzymes are resistant to irreversible inactivation, in part due to the ease in which an oxidized Sec residue in the form of seleninic acid (Sec-SeO2−) can be converted back to a selenol in comparison to the very slow chemical conversion of cysteine-sulfinic acid (Cys-SO2−) to cysteine-thiol (Cys-SH) in a Cys-containing enzyme (20). It is known from the chemical literature that the reduction of Cys-SO2− to Cys-SH is a slow reaction, requiring the reaction to be conducted in refluxing H2O for complete conversion (21, 22). In contrast, mild reduction conditions for benzeneselenic acid have been reported by Kice and Lee (23). Hilvert and colleagues have also observed that conversion of RSeO2− to RSeH occurs rapidly and under mild conditions (24,25).
The reason for the much more facile reduction of RSeO2− in comparison to RSO2− is due to the much higher electrophilicity of Se in comparison to S. Work by Reich has shown that Se is ~104 more electrophilic than S as measured by butyllithium exchange reactions (26). The importance of the high electrophilic character of Se in comparison to S has largely been ignored in the biochemical literature, while the high nucleophilicity of a selenolate has been emphasized as a rationale for its use in enzymes (27 and references therein). In mTR, it is commonly supposed that Se is important for the enzymatic reduction of Trx due to its superior ability to attack the disulfide bond of Trx relative to S of Cys. This implies that the rate-limiting step in the reaction is the nucleophilic attack step governed by kNuc-Se as shown in Figure 1. In contrast, we have supported the idea that the thiol/selenosulfide exchange step (with rate constant kex) between N- and C-terminal redox centers is the rate-limiting step (12, 20).
Figure 1.

Two important ways in which Se is involved in the catalytic mechanism. Se is involved in accepting electrons from the N-terminal redox center (governed by rate constant kex – top panel) as well as donating electrons to the protein substrate Trx (governed by rate constant kNuc-Se – middle panel). The Sec residue of mammalian TR is part of the conserved Gly-Cys1-Sec2-Gly motif found on a flexible C-terminal extension of the enzyme. The N-terminal redox center (green) is on the opposite subunit of the homodimer. Reducing equivalents originate from NADPH and are passed onto the flavin, which in turn reduces the N-terminal disulfide. The top and middle panels only show two discrete steps in the catalytic cycle. For a complete description of the enzymatic mechanism of high Mr TRs, please see (28). Here, and previously, we have produced a truncated mTR missing the C-terminal redox center (bottom panel). This truncated mitochondrial mTR can reduce a number of small molecule substrates (SM) such as DTNB, lipoic acid, and selenite (10, 12). The use of the truncated mTR allows us to isolate and study the exchange step in greater detail. Note that CysIC denotes the interchange Cys residue that attacks either the selenosulfide bond of the C-terminal redox center or the small molecule substrate.
There are two possible mechano-enzymatic ways in which Se could accelerate the flow of electrons from the thiolate of CysIC in the exchange step to the C-terminal redox center. Previously, we have cited the superior leaving group ability (12) of Se in this step of the mechanism as the manner in which rate acceleration is achieved relative to S in the Cys-mutant (kcat is ~500-fold slower). However, our study of the ability of the truncated enzyme missing the Sec-containing C-terminal tail (Gly-Cys1-Sec2-Gly-OH) to reduce a variety of small molecule substrates has caused us to question our original model of rate acceleration by Se in the exchange step of the TR reaction mechanism (and other selenoenzymes as well). In this report we show that the truncated TR (mTRΔ8) can reduce methaneseleninate (CH3SeO2−) at significant rates, especially at acidic pH, but cannot reduce an analogous S-compound (hydroxymethanesulfinate – HOCH2SO2−). This finding supports the idea that Se accelerates the exchange step through its superior ability to accept electrons (electrophilicity) in comparison to S. This observation led us to investigate how the superior electrophilicity of Se could aid TR’s ability to resist inactivation by resisting irreversible oxidation, possibly via facile reduction of an oxidized Sec-SeO2− residue to a Sec-SeH residue (our original hypothesis presented in 20). The data in this paper provides the first experimental support for this hypothesis.
MATERIALS AND METHODS
Materials
NADPH was purchased from AppliChem (Darmstadt, Germany). Methaneseleninic acid, sodium hydroxymethanesulfinate hydrate, dithionitrobenzoic acid (DTNB), and DL-α-lipoic acid were all obtained from Sigma-Aldrich (St. Louis, MO). Hydrogen peroxide (30% solution) was purchased from Fisher Scientific (Fair lawn, NJ). 5,5-dimethyl-1,3-cyclohexanedione (dimedone) was obtained from Acros Organics (Morris Plains, New Jersey). Enzyme kinetic assays were performed on a Cary50 UV-Vis spectrophotometer (Walnut Creek, CA). Microcon Ultracel YM-50 centrifugal filters by Millipore (Billerica, MA) were used for desalting of enzyme samples. The production and purification of the recombinant and semisynthetic enzymes used in this study have been previously reported (12, 29). The Se content of the semisynthetic enzyme is 91% as reported in (29). All enzymatic assays were conducted at room temperature unless otherwise noted.
Activity Assays and H2O2 Concentration
Activity assays following NADPH consumption were monitored by the decrease in absorbance at 340 nm (A340) using an extinction coefficient of 6220 M−1•cm−1(30). Activity assays following the consumption of DTNB were monitored by the increase in absorbance at 412 nm (A412), corresponding to the formation of the TNB anion (31), and were calculated using an extinction coefficient of 13.6 mM−1•cm−1. Stock solutions of H2O2 were freshly prepared before each experiment with the concentration of H2O2 determined spectrophotomerically using an extinction coefficient (43.6 M−1•cm−1) at 240 nm (32).
Preparation of Potassium Methaneseleninate
A 1 M stock solution of potassium methaneseleninate (CH3SeO2K) was prepared by the addition of 127 mg of methaneseleninic acid (CH3SeO2H) to 500 μL of 2 M potassium hydroxide. To this solution, 500 μL of T buffer (100 mM potassium phosphate, 2 mM EDTA, pH 7.4) was added to produce a final concentration of 1 M. For activity assays, working solutions were prepared by diluting aliquots of the 1 M stock.
Potassium Methaneseleninate Reductase Activity of mTR and Mutant Enzymes
CH3SeO2K was assayed against the full-length wild type (WT) mTR (mTR-GCUG) and two mutant mTR enzymes (mTRΔ8 and mTR-GCCG).2 The methaneseleninic acid reductase activity was measured by following the consumption of NADPH at A340. Assays contained 200 μM NADPH in 1 mL of either a standard assay buffer (50 mM potassium phosphate buffer, 2 mM EDTA, pH 7.0), or T buffer. Enzyme assays to determine activity as a function of pH were performed with 1 – 34 nM enzyme and 300 – 500 μM substrate over a pH range of 5.0 to 8.0 with 100 mM citrate, 100 mM sodium phosphate, and 100 mM Tris buffers. For the determination of kinetic parameters at the optimum pH, assays were performed with 100 mM sodium phosphate at pH 6.1 (mTRΔ8 – 34 nM), or in 500 mM potassium phosphate at pH 6.3 (mTR-GCUG – 1.2 nM), or in 250 mM potassium phosphate at pH 6.5 (mTR-GCCG – 11 nM).
Activity Knockdown Assay for mTRΔ8
Here we describe an assay in which we first reduced the truncated mTR with NADPH then attempted to inactivate it by treatment with H2O2. The sample was then desalted and assayed for its ability to reduce DTNB or lipoic acid. Because we are attempting to inactivate the enzyme, we refer to this assay as an “activity knockdown assay”. Fresh DTNB (63 mM) or lipoic acid (242 mM) in ethanol was prepared before each experiment. To fully reduce the N-terminal redox center a 154-fold excess of NADPH (0.2 μmol) was added to 100 μL aliquots of mTRΔ8 (1.3 nmol) and allowed to incubate for one min. Increasing amounts of H2O2 were then pipetted into the sample of reduced enzyme and incubated for 30 min at 37 °C. Following incubation, the oxidized enzyme was desalted using Microcon YM-50 centrifugal filters and buffer exchanged with 50 mM potassium phosphate, 300 mM NaCl, 1 mM EDTA, pH 8.0 (repeated twice) to remove excess H2O2. After desalting, the concentration of the enzyme samples was determined spectrophotometrically using the absorbance of flavin at 460 nm with an extinction coefficient of 22.6 mM−1 •cm−1 (30). The desalted enzyme solution was then diluted to make a 1 or 2 μM working stock for use in the DTNB or lipoic acid assays.
Activity Knockdown Assay for mTRΔ8 measured by DTNB
For the DTNB “knockdown” assay the reaction contained 200 μM NADPH, 10 mM EDTA and 0.5 mM DTNB in 0.5 mL of 100 mM potassium phosphate, pH 7.0. The reaction was initiated with the addition of either H2O2-treated or untreated 4 nM mTRΔ8. The activity was measured by monitoring the increase in A412. The activity of the enzyme was corrected for background hydrolysis of DTNB by performing control reactions lacking enzyme. This background slope was then subtracted from the slope of the reaction that contained enzyme. The activities of desalted H2O2-treated and desalted untreated (control) enzymes were compared.
Activity Knockdown Assay for mTRΔ8 measured by Lipoic Acid
The lipoic acid “knockdown” assay measured lipoic acid reductase activity by monitoring NADPH consumption. The reaction mixture contained 4.5 mM lipoic acid, 150 μM NADPH, 1 mM EDTA, and 50 nM mTRΔ8 (H2O2-treated and untreated) in 0.5 mL of 100 mM potassium phosphate buffer, pH 6.1. The background was corrected by performing control assays in which either lipoic acid or enzyme was omitted.
General Procedure for mTRΔ8 Activity Knockdown Assay in the presence of H2O2
The assays described above were designed to inhibit the enzyme by first attempting its oxidation with H2O2 and then determining the amount of remaining activity. The assay described in this section was designed to measure the activity of the truncated enzyme in the presence of H2O2 by determining its DTNB reductase and lipoic acid reductase activities. The procedures are generally the same as those described prior, except mTRΔ8 was not pretreated with H2O2 but rather H2O2 was added to the assay reaction mixture in a range from 0 (control) to 10 mM.
mTRΔ8 DTNB Reductase Activity Knockdown Assay in the presence of H2O2
For the DTNB reductase activity assay, the reaction contained H2O2 in the range from 0 (control) to 10 mM as well as 200 μM NADPH, 10 mM EDTA and 0.5 mM DTNB in 0.5 mL of 100 mM potassium phosphate buffer, pH 7.0. The reaction was initiated with the addition of 2 nM mTRΔ8 and the activity was measured by monitoring the increase in A412. To account for non-enzymatic background hydrolysis of DTNB, a control was performed where H2O2 was added to the reaction mixture in the absence of enzyme. The background slope was then subtracted from the reaction that contained enzyme. An additional control experiment was carried out to measure the H2O2-mediated TNB anion oxidation back to DTNB in the reaction cuvette. In this experiment 10 μM dithiothreitol (DTT) was reacted with 0.5 mM DTNB in a 0.5 mL reaction mixture also containing 1 mM EDTA and 200 μM NADPH in 100 mM potassium phosphate buffer, pH 7.0. The reaction cuvette was initially zeroed with 0.5 mM DTNB and the A412 was followed for 2 min (background hydrolysis of DTNB) upon which 10 μM DTT was pipetted into the cuvette. After an additional 2 min, 10 mM H2O2 was added to the reaction and the decrease in A412 was continually monitored.
mTRΔ8 Lipoic Acid Reductase Activity Knockdown Assay in the Presence of H2O2
The lipoic acid reductase activity was measured by monitoring the consumption of NADPH. The reaction mixture contained 4.5 mM lipoic acid, 150 μM NADPH, 1 mM EDTA, 50 nM truncated TR, and H2O2 in the range from 0 (control) to 10 mM H2O2, in 0.5 mL of 100 mM potassium phosphate buffer, pH 6.1. A control reaction to measure the non-enzymatic reduction of lipoic acid was performed in the presence of the same concentration of H2O2 in which enzyme was present. This background slope was subtracted from the experiment in which enzyme was present in the assay.
Measuring Trx-Reductase Activity of WT mTR After Incubation With NADPH and H2O2
TR has hydrogen peroxidase activity due to the presence of the Se atom of Sec (27). This assay was designed to determine whether TR’s Trx-reductase activity was sensitive to H2O2 inactivation, or conversely resistant to inactivation by H2O2. This was accomplished by determining TR’s ability to reduce Trx after first being exposed to high concentrations of H2O2 in the presence of NADPH. First, WT mTR was incubated with NADPH and H2O2 and the peroxidase activity was determined by measuring the ΔA340 for long time periods (~ 20 min). Subsequently, Trx was added to the assay cuvette. Since Trx is a much better substrate for TR than H2O2 (as judged by Km values), the consumption of NADPH should increase (resulting in a more negative slope as A340 is monitored over time) if Trx is reduced by TR in the presence of H2O2. On the other hand, if H2O2 inactivates the ability of TR to reduce Trx, the ΔA340 should not be affected upon addition of Trx to the assay. The initial reaction assay contained 10 mM H2O2, 50 nM WT TR, and 200 μM NADPH. The peroxidase activity was followed for 20 min (by monitoring ΔA340) after which time E. coli Trx was added to a final concentration of 90 μM. The background activities were determined through a series of control reactions looking at the amount of non-enzymatic peroxidase activity. These assays were done in a 0.5 mL reaction volume containing the standard assay buffer at room temperature.
In order to directly compare the Trx-reductase activity of mTR that had been treated with H2O2 to a control sample of TR in which H2O2 was omitted, we integrated a peroxide quenching step into the assay using catalase to remove excess H2O2 from the mTR sample. Removal of H2O2 would then allow us to directly compare the ΔA340 of the two samples. Enzyme (45 nM mTR in assay buffer) was incubated with either 1 or 50 mM H2O2 for 25 min. Catalase (14 units) was then added to the 0.5 mL assay and incubated for an additional 12 min. A fresh bolus of 200 μM NADPH was then added to the assay and the ΔA340 was monitored for an additional 2 min to make sure that the peroxidase activity had been quenched. This was followed by the addition of E. coli Trx to a final concentration of 90 μM. The control sample was treated in an identical manner except that H2O2 was not present in the assay mixture.
Attempt to Inhibit TR by Dimedone Treatment in the Presence of H2O2
The experiment described in this section was designed to help determine the redox state of the Se atom as it turns over H2O2. It has always been assumed that Sec-TR uses a selenenic acid (Sec-SeOH) intermediate during the redox cycle with H2O2 as the substrate. If this is so then TR should react with reagents that alkylate or “trap” cysteine-sulfenic acids (Cys-SOH), which would then inactivate TR. On the other hand, if another oxidation state of Sec is used to reduce H2O2, then the trapping reagent should not be expected to label and inactivate TR. For this experiment we added dimedone, a known sulfenic acid labeling reagent, to our assay containing H2O2 (33). For this experiment the assay mixture contained 200 μM NADPH and 50 mM H2O2 in 0.5 mL of assay buffer with the reaction initiated by the addition of 10 nM WT TR. The peroxidase activity was monitored by measuring the ΔA340. After 20 min of reaction time dimedone was titrated into the reaction assay incrementally (from a freshly prepared stock of 12.7 mM dimedone in ethanol) until a final concentration of 150 μM of dimedone in the assay was achieved. After 40 min of monitoring the reaction an additional bolus of NADPH (200 μM) was added to replenish the assay and the ΔA340 was monitored for an additional 5 min. After 45 min of total reaction time, E.coli Trx (90 μM) was added and the ΔA340 was monitored for an additional 15 min.
RESULTS AND DISCUSSION
Previously, it was reported that methaneseleninate (CH3SeO2−) was a substrate for TR1 (34). However, the previous report did not determine whether the reduction of CH3SeO2− was solely due to the Sec-containing C-terminal redox center, or whether the N-terminal redox center also contributed to the reduction of this substrate. In light of our recent work showing that the N-terminal redox center of mitochondrial mTR could reduce a number of substrates with high catalytic activity independent of the Sec-containing C-terminal redox center (10), we set out to determine whether CH3SeO2− could be reduced by our truncated mTR (mTRΔ8). The results show that at pH 7.4, the truncated enzyme has considerably less activity than the full-length TR containing the C-terminal Sec residue (Table 1). However, the results also show that the Sec→Cys mutant of mTR reduces CH3SeO2− with only a ~5-fold loss in activity indicating that Sec is not crucial for the reduction of this substrate. The Cys-mutant or mTRΔ8 could not be saturated with substrate at pH 7.4 and thus we cannot report kcat and Km values under these conditions.
Table 1.
Kinetic Constants of Various mTRs Towards Methaneseleninic Acid at pH 7.4
| Enzyme | Activity (mol NADPH/min/mol TR) |
kcat (min−1) | Km (μM) | |
|---|---|---|---|---|
| 100 μM CH3SeO2− | 200 μM CH3SeO2− | |||
| mTR-GCUG | 377 ± 16.4 | 593 ± 35.8 | 963 ± 49.0 | 134 ± 12.3 |
| mTR-GCCG | 77.7 ± 12.4 | 114 ± 10.5 | ND | ND |
| mTRΔ8 | 8.7 ± 1.2 | 12.4 ± 0.5 | ND | ND |
| TR-GCUGa | ND | ND | 840 | 26 ± 10 |
Cytosolic mouse TR. Data taken from (34).
ND is not determined.
Interestingly, an investigation of activity as a function of pH showed that the WT enzyme, the Cys-mutant, and the truncated enzyme each reduced CH3SeO2− with much higher activity at acidic pH (Table 2). Though we note that the Km of the Cys-mutant is ~3-fold larger than the Km for the WT enzyme. This may indicate that the on-rate for formation of the ES complex is slower in the mutant in comparison to the WT enzyme, although there are likely other possible reasons for this higher apparent Km value. A comparison of the activities of the truncated enzyme at its pH optimum (6.1) with the WT enzyme at its pH optimum (6.3) shows that the truncated enzyme only has ~4-fold less activity than the WT enzyme with CH3SeO2− as the substrate. All three enzymes could be saturated with substrate at their pH optima (unlike the situation at pH 7.4) so that kcat and Km values could be determined.
Table 2.
Kinetic Constants of Various TRs Towards Methaneseleninic Acid at the Optimal pH
| Enzyme | pH | kcat (min−1) | Km (μM) |
|---|---|---|---|
| mTR-GCUG | 6.3 | 1804 ± 118 | 87.7 ± 18.6 |
| mTR-GCCG | 6.5 | 644 ±75 | 293 ± 63 |
| mTRΔ8 | 6.1 | 417 ± 16 | 87.2 ± 7.8 |
The pH rate profile for the truncated enzyme shows a sharp optimum at pH 6.1, with the enzyme still retaining significant activity between pH 5 and 6 as shown in Figure 2A. The fast enzymatic reduction of CH3SeO2− at acidic pH by the truncated TR parallels the fast chemical reduction of a model alkylseleninic acid at acidic pH reported by Hilvert and coworkers (25). Reduction of the seleninic acid form of selenosubstilisin is also fast at acidic pH (25). We note that the truncated enzyme has an acidic pH optimum for a number of different substrates as shown in Figure 2B. These results show that the N-terminal redox center functions efficiently at acidic pH and implies that mitochondrial mammalian TR may have evolved to catalyze the reduction of mitochondrial Trx under acidic conditions. The intermembrane space of mitochondria is known to be acidic relative to the cytosol (35). To the best of our knowledge, the pH optimum for the reduction of mitochondrial Trx by mitochondrial mammalian TR (mTR3) is not known. However, with E. coli Trx as the substrate, mTR3 shows a broad pH rate profile maintaining a 50% level of activity between pH 6 to 8 (Figure S1 of the Supporting Information). We suggest that one possible reason for the acidic pH optimum of the N-terminal redox center is that it allows mTR to function at acidic pH without becoming inactivated by oxidation. If the C-terminal Sec residue were oxidized to Sec-SeO2− under acidic conditions, it would rapidly become reduced back to Sec-SeH. Indeed our experiments show that at pH 5.5–6, mTR does not lose significant activity when exposed to 50 mM H2O2 (see Figure S2 of the Supporting Information).
Figure 2.

(A) Activity vs. pH profile for mTRΔ8. As can be seen, the truncated enzyme has a sharp pH optimum near 6 for the reduction of CH3SeO2−. (B) The N-terminal redox center has an acidic pH optimum for a number of different substrates: selenite (open diamonds), lipoic acid (closed circles), DTNB (open squares).
We also investigated whether a sulfur analogue of CH3SeO2−, hydroxymethanesulfinate (HOCH2SO2−), could be a substrate for either the truncated enzyme or the full-length enzyme. We could not detect any reduction of this sulfur analogue (as measured by consumption of NADPH) in our assay with either enzyme. This result, combined with our previous study on the truncated mTR, shows that the truncated mTR can reduce a number of Se-containing substrates efficiently, while the S-analogues are either reduced very poorly or not at all. This fact, highlighted in Figure 3, helps to illuminate the role of Se in the enzymatic reaction mechanism of mTR. We posit that the reason Se-substrates are efficiently reduced by the truncated mTR is due to the superior electrophilic character of the Se atom (discussed extensively in our recent review – 20). We have also argued that the rate-limiting step in the mTR reaction mechanism is the thiol/selenosulfide exchange reaction that occurs between the N- and C-terminal redox centers (kex). The Se-containing substrates shown in Figure 3 are surrogate substrates for the known substrate of the N-terminal redox center – the selenosulfide bond of the oxidized Gly-Cys1-Sec2-Gly tetrapeptide motif of the C-terminal redox center. The results with these Se-surrogate substrates for the N-terminal redox center strongly imply that one way in which Se accelerates the enzymatic reaction in the holoenzyme is the superior ability of Se to accept electrons (electrophilicity) in the exchange reaction. Other chemical factors are likely to be important in the overall reaction mechanism also, though our point here is to highlight an unrecognized way in which the presence of Se in TR and other selenoenzymes can enhance the enzymatic rate.
Figure 3.
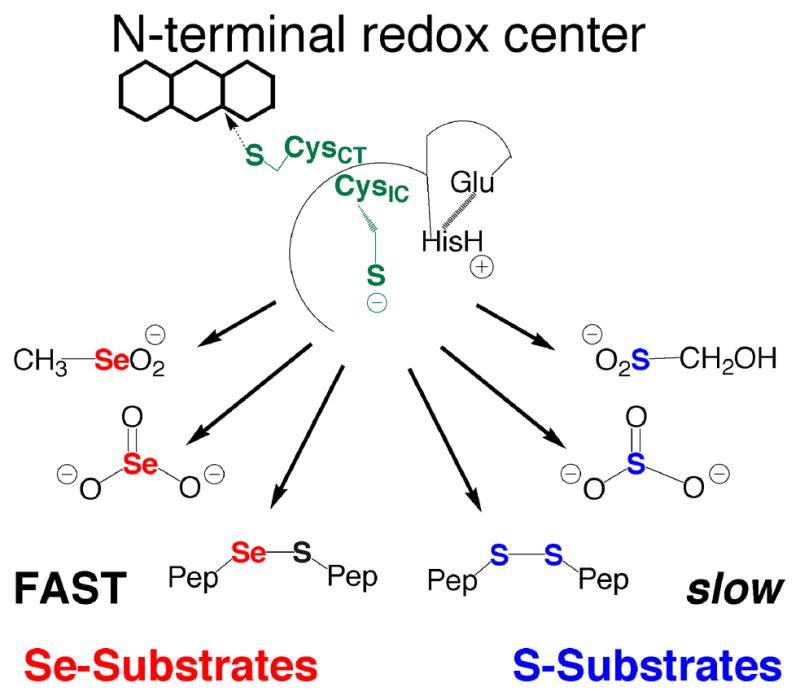
The importance of Se to substrate utilization by the N-terminal redox center. The N-terminal redox center will reduce small molecule Se-containing substrates such as SeO3−2, CH3SeO2−, and selenosulfide (Se–S) containing peptides. The S-analogs of these substrates are either not reduced at all such as HOCH2SO2− (reported here) and SO3−2 (10), or are reduced very slowly as in the case of cystine and other disulfide (S–S) containing peptides (12). We believe that the most likely reason that these Se-containing compounds are good substrates (reduced quickly) compared to the S-analogs is due to selenium’s strong ability to accept electrons (high electrophilicity). The explanation that high electrophilicity is the determining factor in substrate utilization by the N-terminal redox center is consistent with the fact that DTNB, lipoic acid, and quinones, all highly electrophilic compounds, are turned over by the N-terminal redox center as reported by us and others (20 and references therein).
While the stronger nucleophilic character of Se relative to S is frequently cited as the reason for the occurrence of Se in enzymes (27), the observation that the electrophilic character of Se is an extremely important feature of the enzymatic reaction mechanism of mTR lead us to investigate why the strong electrophilicity of Se might be of biological importance to mTR and other selenoenzymes in general. The fact that CH3SeO2− was easily reduced by the truncated mTR was of particular interest to us, especially because the sulfur-analogue could not be reduced by the enzyme. It is well documented that sulfinic acids are chemically very resistant to reduction by thiols, with reduction only achieved under harsh conditions (21, 22). Biologically the only known example of a cysteine-sulfinic acid residue (Cys-SO2−) in a protein to be reduced back to a cysteine-thiol (Cys-SH) is the peroxidatic Cys residue of peroxiredoxin, which requires the action of another enzyme (sulfiredoxin) to reduce the Cys-SO2− residue to a Cys-SH residue (36). We reasoned that the reduction of CH3SeO2− by the truncated mTR could serve as a model for what happens to the holoenzyme when it is exposed to H2O2 and other oxidants as shown in Figure 4. Oxidation of a Se atom to the seleninic acid form (RSeO2−) will make the Se atom even more electrophilic and thus highly susceptible to attack by other thiols, either exogenous or enzymic. In mTR if the Sec residue is oxidized to Sec-SeO2−, it could be very quickly reduced back to Sec-SeH, due to the close proximity of Cys1 and CysIC (as shown in Figure 4). This led us to hypothesize that mTR should be highly resistant to inactivation by oxidation because oxidation of Sec-SeH to either Sec-SeOH or Sec-SeO2− should result in very fast reduction by enzymic thiols to the selenosulfide form of TR, which could then be reduced back to the selenolate via addition of NADPH.
Figure 4.
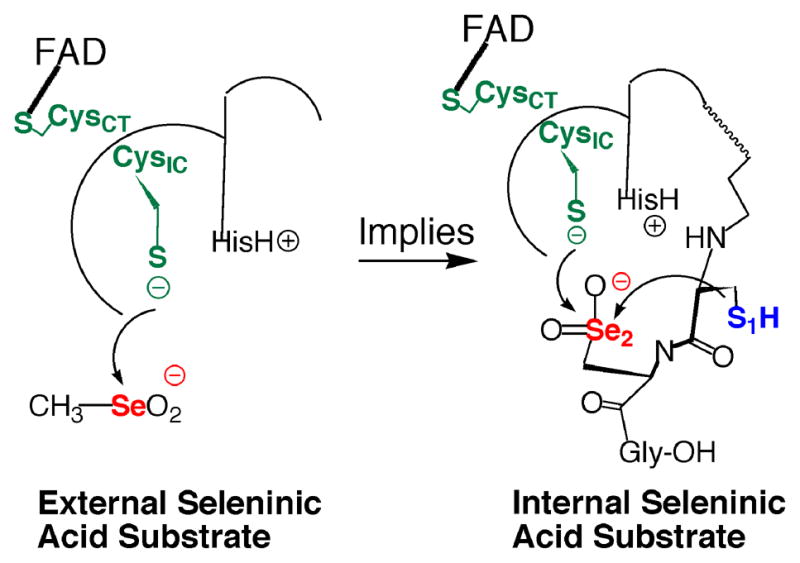
Our truncated mTR construct (mTRΔ8) will reduce CH3SeO2− as an external substrate (left structure). This implies (right structure) that if the Sec residue in the C-terminal tail, as part of the full-length enzyme, becomes oxidized to the Sec-SeO2− form, it can be easily reduced by either the N-terminal redox center or by the adjacent Cys residue (Cys1).
For this hypothesis to be correct, the N-terminal redox center must also resist oxidation so that it can reduce the C-terminal redox center. Conversion of CysIC-SH to CysIC-SOH or CysIC-SO2− by H2O2 would render the enzyme inactive. To test the N-terminal redox center’s ability to resist inactivation by oxidation, we incubated mTRΔ8 (truncated TR) with NADPH to reduce the enzyme followed by incubation with increasing concentrations of H2O2. The samples were then desalted and assayed for DTNB reductase activity. The results, shown in Table 3, demonstrate that the N-terminal redox center resisted very high concentrations of H2O2 and its ability to reduce DTNB was not affected, even when incubated with 50 mM H2O2. To further test the ability of the N-terminal redox center to resist inactivation by treatment with H2O2, we repeated the same experiment, but used racemic lipoic acid as the substrate instead. Previously we reported that mTRΔ8 could also reduce racemic lipoic acid due to the enhanced electrophilicity of its disulfide bond attributed to bond angle strain (10). The results of this experiment are summarized in Table 4. The results show that the activity of the truncated enzyme is very consistent over the range of 0.5 to 50 mM H2O2, and only loses ~20% of its activity over this range.
Table 3.
DTNB Reductase Activity of Truncated TR after H2O2 Incubation.a
| H2O2 (mM) | Activity (mol NADPH/min/mol of enzyme) |
|---|---|
| 0 | 640 ± 80 |
| 0.5 | 600 ± 40 |
| 1 | 580 ± 35 |
| 10 | 530 ± 10 |
| 50 | 670 ± 40 |
The concentration of DTNB in this assay was constant at 0.5 mM. For other assay conditions see text.
Table 4.
Lipoic Acid Reductase Activity of Truncated TR after H2O2 Incubation.a
| H2O2 (mM) | Activity (mol NADPH/min/mol of enzyme) |
|---|---|
| 0 | 60.6 ± 12.1 |
| 0.5 | 53.8 ± 2.4 |
| 1 | 47.4 ± 4.8 |
| 10 | 40.2 ± 2.3 |
| 50 | 48.7 ± 4.9 |
The concentration of lipoic acid in this assay was constant at 4.5 mM. For other assay conditions see text.
In order to determine how the activity of the truncated mTR might be affected in the presence of H2O2, we repeated our activity assays with DTNB and lipoic acid in the presence of increasing H2O2 concentrations. In these experiments NADPH is always present so that the N-terminal redox center is always maintained in the reduced state. The SH group of CysIC should therefore be directly exposed to oxidant in this experiment. In the case of the DTNB assay under these conditions, the activity begins to decline at concentrations of 10 mM H2O2 and above (Table 5). We note that 10 mM H2O2 is a far higher concentration of H2O2 than what occurs in vivo. The decrease in activity seen in this experiment at high concentrations of H2O2 is most likely due to re-oxidation of the TNB anion to DTNB in the presence of H2O2. Re-oxidation of product to substrate would cause a corresponding decrease in absorbance at 412 nm, which is the wavelength where the TNB anion (product) absorbs. This would result in a decrease in the calculated activity. To substantiate this possibility we carried out a non-enzymatic control reaction involving the addition of 10 μM dithiothreitol (DTT) to 0.5 mM DTNB followed by the addition of 10 mM H2O2 (Figure S3). Upon addition of DTT to the cuvette containing DTNB an immediate color change was observed indicative of the rapid reduction of DTNB to the TNB anion. This observation was confirmed through spectral monitoring at A412 where a large increase in absorbance was observed. After the sample had been exposed to DTT for 2 min, 10 mM H2O2 was added to the cuvette, which resulted in a sharp decrease in A412. After 7 min the A412 had almost returned to its original level where only DTNB was present. Additionally, the color of the reaction mixture had reverted back from yellow to a transparent solution, further evidence that the TNB anion had re-oxidized back to the original DTNB form upon treatment with H2O2. These results signify that the observed decrease in A412 and corresponding loss in calculated activity with 10 mM H2O2 in the reaction is due to the non-enzymatic re-oxidation of TNB anion back to DTNB by peroxide.
Table 5.
DTNB Reductase Activity of truncated TR in the presence of H2O2.a
| H2O2 (mM) | Activity (mol NADPH/min/mol of enzyme) |
|---|---|
| 0 | 610 ± 50 |
| 0.5 | 540 ± 10 |
| 1 | 600 ± 10 |
| 10b | 470 ± 20 |
The concentration of DTNB in this assay was constant at 0.5 mM. For other assay conditions see text.
As discussed in the text and shown in Figure S3 of the Supporting Information, the decrease in activity at 10 mM H2O2 is most likely due to reoxidation of the TNB anion back to DTNB.
To further assess the activity of the N-terminal redox center under oxidative stress we repeated this experiment with lipoic acid as the substrate. This assay does not monitor product formation by measuring the absorbance of the newly formed product, but instead determines enzyme activity by measuring NADPH consumption. The results show that NADPH consumption is not affected by increasing the H2O2 concentration from 0 to 10 mM in the reduction of lipoic acid by the truncated mTR (Table 6). However, one noticeable trend was the increase in background (no mTR added) activity as a function of increased H2O2 concentration, most notably with 10 mM H2O2 in the reaction. Upon further investigation we found that when 50 mM H2O2 was reacted with 4.5 mM lipoic acid, in a reaction also containing 200 μM NADPH, significant non-enzymatic activity was observed (data not shown). Chemically, this can most likely be explained by the H2O2-mediated oxidation of the disulfide bond of lipoic acid to the thiosulfinate form of lipoic acid (β-lipoic acid). β-lipoic acid is a very good electrophile that can easily accept a hydride from NADPH, which could facilitate its chemical reduction.
Table 6.
Lipoic Acid Reductase Activity of truncated TR in the presence of H2O2.a
| H2O2 (mM) | Activity (mol NADPH/min/mol of enzyme) |
|---|---|
| 0 | 59.4 ± 5.2 |
| 0.5 | 56.6 ± 0.6 |
| 1 | 54.3 ± 3.1 |
| 10 | 53.0 ± 4.1 |
The concentration of lipoic acid in this assay was constant at 4.5 mM. For other assay conditions see text.
The overall results above show that the truncated mTR is remarkably resistant to high concentrations of H2O2. We would like to note that this is especially remarkable since the pKa of CysIC-SH was determined to be 5.8 (12). At pH 7.0, this thiol group is mostly ionized and it might be expected to oxidize to the sulfenic acid form when it comes into contact with H2O2. The CysIC-SOH group could be reduced by the resolving CysCT residue of the CICVNVGCCT motif, which would imply that the truncated TR has peroxidase activity. However, we could not detect peroxidase activity with the truncated TR (data not shown). We hypothesize that the way in which CysIC-S− avoids being oxidized is by being a weak nucleophile, which would preclude attack onto the -O–O- bond of peroxides. If CysIC-S− is in an ion-pair relationship with a nearby HisH+ residue (serving as a general acid/base catalyst) as has been proposed (37), then the electrons of the thiolate would not be readily available for attack onto electrophiles. In order for electrons to be transferred from CysIC-S− to a small molecule substrate in the truncated enzyme, the small molecule would have to be a good acceptor of electrons (strong electrophile). This point is exactly illustrated in Figure 4 and discussed by us elsewhere (20). It is interesting to contrast the inability of CysIC-S− (pKa = 5.8) to be oxidized with the ability of the selenolate of the C-terminal Sec residue (pKa ~ 5.2) to attack a peroxy-bond as demonstrated by the peroxidase activity of Sec-TR, while the Cys-mutant has no such activity (30).
We next tested the ability of the full-length enzyme (containing Sec) to resist inactivation by H2O2 by measuring its ability to reduce Trx after its pretreatment with H2O2. For this experiment we incubated 50 nM mTR with 10 mM H2O2 in the presence of NADPH. Under these conditions mTR will reduce H2O2 and release water as the product (30). The results of this experiment, depicted in Figure 5A, show that NADPH is consumed by mTR when H2O2 is added to the reaction mixture. This consumption of NADPH is a result of the Sec residue being oxidized by H2O2 and subsequently reduced by the N-terminal redox center. The peroxidase activity of mTR is inferior to that of Gpx-1, but the steady consumption of NADPH by mTR in the presence of H2O2 shows that the Se atom must be exposed to H2O2 in order for substrate turnover to occur. The rate of NADPH consumption (as measured by the change in slope) only changed 15% from the beginning to the end during the 20 min that the reaction was monitored, most likely resulting from ever decreasing concentrations of H2O2 in the assay. This shows that at high concentrations of H2O2, the enzyme is not deactivated (unlike Gpx-1) with respect to its peroxidase activity. After this 20 min reaction, the lid to the spectrometer was opened and 90 μM Trx was added to the cuvette. As can be seen in Figure 5A, there is a sharp decrease in the slope signifying that there is rapid consumption of NADPH. This consumption of NADPH is due to the rapid reduction of Trx by mTR. This latter result suggested to us that the ability of mTR to reduce Trx is not impaired by a high concentration of H2O2.
Figure 5.

(A) The plot monitors the consumption of NADPH by following the decrease in A340. When 10 mM H2O2 is added to a reaction containing 50 nM mTR-GCUG and NADPH, mTR reduces H2O2 to water and becomes oxidized. The oxidized mTR is reduced by NADPH resulting in a decrease in A340. After prolonged exposure (20 min) to excess H2O2, 90 μM Trx is added to the cuvette and the sharp increase in NADPH consumption (shown by a larger negative slope) shows that Trx is rapidly reduced and that a large excess of H2O2 does not inhibit the enzyme. (B) Comparison of activity progress curves for mTR treated with H2O2 (blue = 50 mM, green = 1 mM) and control mTR (red = no H2O2). After approximately 25 min, all of the NADPH in the reaction is consumed (for the enzyme treated with 50 mM H2O2) shown by a plateau in the slope. The samples were then treated with 14 units of catalase to remove excess H2O2 for 12 min. During this quenching step the A340 was not monitored, but we have added a line to the plot for continuity. Since all of the NADPH was consumed in the sample treated with 50 mM H2O2, an additional bolus of NADPH was then added to the reaction to achieve a final concentration of 200 μM. The reaction was then monitored for 2 additional min at 340 nm to ensure all of the H2O2 was removed and then 90 μM Trx was added to each sample. The activity progress curves are extremely similar, even for the sample treated with 50 mM H2O2. The overall results show that mTR is very resistant to inactivation, even though the ability of the enzyme to turnover H2O2 shows that the Se-atom must be exposed to the oxidant.
We next designed an assay in which we could compare the Trx-reductase activity of Sec-TR that had been exposed to a very large excess of H2O2 to that of a control sample not exposed to H2O2. Because Sec-TR has peroxidase activity, we integrated a quench step using catalase to remove the very large excess of H2O2 remaining in the sample after a 25 min incubation step with H2O2. The removal of H2O2 by catalase allowed us to directly compare the Trx-reductase activity of the control enzyme to the enzyme treated with H2O2 because there would be no background peroxidase activity as measured by the decrease in A340. This experiment served as a further, more stringent test of the ability of Sec-TR to resist inactivation by oxidation.
The results of this experiment is shown in Figure 5B. The results show that when TR is exposed to 50 mM H2O2 a decrease in A340 is observed for 20 min indicating all of the NADPH has been consumed and that the Se atom in TR has been exposed to H2O2. The plateau in A340 was observed for another 5 min after which catalase was added to the cuvette and the sample was incubated for 12 additional min. The formation of bubbles was observed during this interval indicating the reduction of H2O2 by catalase was occurring. Once bubble formation had stopped, we added a fresh bolus of 200 μM NADPH to the cuvette and monitored the absorbance at 340 nm for 2 min. No change was observed during this interval. We then added E. coli Trx to the cuvette (final concentration was 90 μM) and again monitored A340. The results show that the resulting progress curve of the control sample and the H2O2 treated sample were nearly identical. Figure 5B shows the results for both 1 and 50 mM H2O2 in the sample. These results show that TR is remarkably resistant to inactivation by oxidation with H2O2.
Previously, it has been supposed that the oxidation state in the peroxidase reaction of mTR is the selenenic acid (Sec-SeOH) form (38). There is no direct evidence for this supposition. In order to address this question, we attempted to trap the Sec-SeOH form of TR by treating it with the sulfenic acid (R-SOH) trapping reagent dimedone in the presence of H2O2 and NADPH. Dimedone will react with Cys-SOH to form a stable covalent adduct. Dimedone thus inhibits thiol peroxidases that form transient sulfenic acid residues during their catalytic cycles. Presumably, dimedone will also react with a selenenic acid to form a selenoether adduct. We note that when Gpx-1 is treated with dimedone the enzyme is not inhibited (39). When dimedone was added to our reaction mixture containing WT mTR, NADPH and H2O2, we could not detect any inhibition even when dimedone was in great excess of mTR. This experiment is shown in Figure 6. The result of this experiment does not exclude the possibility that mTR does in fact use the Sec-SeOH form to catalyze the reduction of H2O2. One possibility is that the Sec-SeOH form of TR is reduced very quickly to the selenosulfide form by the adjacent thiol group of the Gly-Cys1-Sec2-Gly motif. This would prevent the trapping of the selenenic acid form of TR by dimedone. The other possibility, suggested by our results with the surrogate substrate CH3SeO2−, is that the C-terminal Sec-SeH residue is oxidized to a Sec-SeO2− residue which is quickly reduced back to Sec-SeH by the possible pathways shown in Figure 4.
Figure 6.
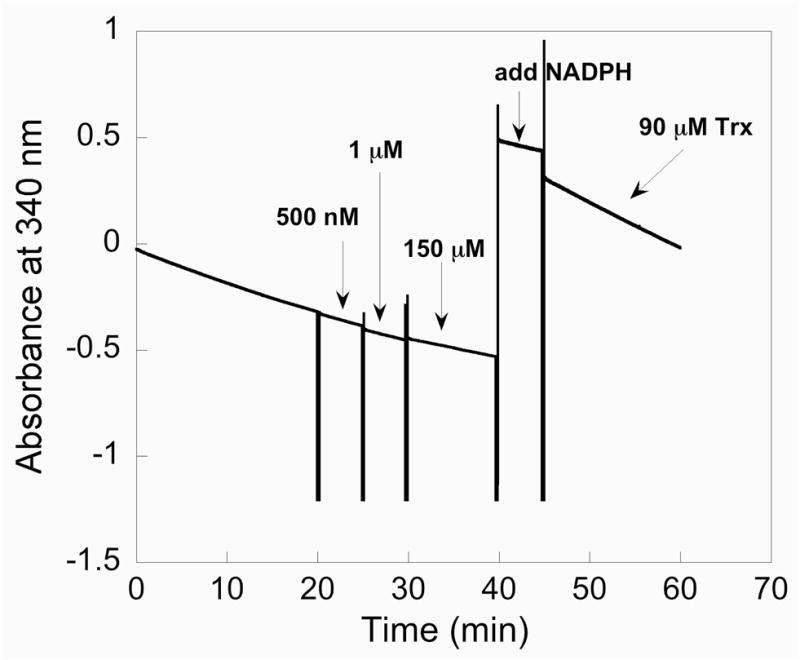
Dimedone/WT mTR trapping experiment. In this experiment we are attempting to trap a selenenic acid (Enz-SeOH) intermediate by adding the sulfenic acid trapping reagent dimedone to the WT enzyme in the presence of H2O2. Here 10 nM mTR is added to a cuvette containing 50 mM H2O2 and 200 μM NADPH. As shown by the decrease in A340, the enzyme is consuming NADPH due to the enzyme’s peroxidase activity. After 20 min we began a series of dimedone additions, starting with 500 nM dimedone and gradually increasing the concentration of dimedone to 150 μM. No change in the slope is apparent even after adding a large excess of dimedone to the reaction, showing the enzyme continues to turnover H2O2. After 40 min, a fresh bolus of NADPH is added and the peroxidase activity is monitored for several more minutes after which 90 μM Trx is added to the reaction. Upon addition of Trx, the consumption of NADPH is sharply increased as shown by the decrease in A340.
Irrespective of the oxidation state of the Se atom in mTR after it is exposed to H2O2, our overall results show that unlike Gpx-1, mTR is not deactivated with respect to its peroxidase and Trx-reductase activities even when exposed to very high concentrations of H2O2. There are two potential fates of a Sec-SeO2− residue in a selenoenzyme. The first possibility is β-elimination to produce a DHA residue with release of H2SeO2, as recent evidence shows is the case for Gpx-1 (19). The second fate is rapid reduction back to a selenol by several thiol equivalents. Direct experimental support for the reduction of an Enz-Sec-SeO2− form to an active Enz-Sec-SeH form has been provided by Hilvert and coworkers, who showed that the Enz-Sec-SeO2− form of selenosubtilisin (a synthetic selenoenzyme) could be reduced under mild conditions (25).
The results of the study on the inactivation of Gpx-1 by H2O2 from Rhee and coworkers (19) are not necessarily at odds with our results and our hypothesis that selenoenzymes are resistant to inactivation by oxidation. An important point that was not mentioned in their study was that the Cys-mutant of Gpx-1 was also found to be sensitive to inactivation by H2O2 (40). Rocher hypothesized that the reason for the sensitivity of the Cys-mutant of Gpx-1 to inactivation by H2O2 was that the active-site Cys-SH residue was over-oxidized to Cys-SO2−, which then underwent β-elimination to form DHA (40). Rocher and coworkers further hypothesized that the use for Sec in Gpx-1 was to enable the enzyme to be resistant to inactivation by oxidation. In their words:
“In conclusion, the most significant results of this study are…a weak activity of the Cys-mutant Gpx and a marked tendency to inactivation in the presence of hydroperoxides, which underlines the selective advantage of Sec-Gpx, which may have evolved to catalyze the fast reduction of hydroperoxides without undergoing significant self-inactivation.”
Thus while Rhee and coworkers may have indeed found that Sec-Gpx-1 was partially inactivated by H2O2, it is not presently known how much more resistant Sec-Gpx is to inactivation by H2O2 than is the Cys-mutant Gpx. The results of our present study lead us to predict that the Sec-Gpx is considerably more resistant to inactivation by oxidation than is the Cys-mutant. Support for this idea comes from a study of the class 4 Gpx from S. mansoni (41). Dimastrogiovanni and coworkers constructed the Cys-mutant of this phospholipid hydroperoxidase (termed SmGpx-1) and found that it was inactive despite the presence of reducing agent at all stages of purification. The mutant was crystallized and the structure revealed that the active-site Cys-SH residue was oxidized to the sulfonic acid (Cys-SO3−), revealing the reason for its loss in activity.
CONCLUSIONS
Mechanistically, we believe that the data with the truncated mTR and small molecule substrates shows that strong substrate electrophilicity is an important feature for transferring electrons from the N-terminal redox center of mTR to the C-terminal redox center as can be seen from the pattern of substrate usage in Figure 3. From a mechano-enzymatic perspective (or chemico-enzymatic as we say in our recent review - 20), the low nucleophilicity of CysIC can explain the requirement for a strong electrophilic center in the C-terminal redox center of mammalian TR. The superior electrophilicity of Se relative to S can at least in part explain the manner in which Se accelerates the reduction of oxidized Trx and other substrates; selenium is needed in mTR because it accelerates the transfer of electrons from the N-terminal redox center to the C-terminal redox center. We note that this position is based upon our studies with the mitochondrial enzyme. The dependence on the presence of Se for reduction of substrates is very different in cytosolic mTR (30) and the mechanistic similarities/differences between the two types of TRs are as yet unresolved.
We posit that the electrophilic character of the Se atom in Sec-SeOH and Sec-SeO2− residues in the C-terminal redox center of mitochondrial Sec-TR is critical for mechanistic recovery from oxidative stress. This chemical property confers the enzyme with an overall ability to resist irreversible inactivation by oxidation due to the much faster rate of reduction of Se-oxides in a selenoenzyme relative to S-oxides in a sulfur-enzyme, with the difference in reduction of Sec-SeO2− being exceptionally fast relative to Cys-SO2−. While at the moment the oxidation state of the Sec residue in mTR is not known upon encountering H2O2, our finding that CH3SeO2− can be reduced by the enzyme’s N-terminal redox center strongly suggests that an oxidized Sec-SeO2− residue in mTR can be rapidly reduced by enzymic thiols, restoring the activity of the enzyme under conditions of oxidative stress. We have also shown that not only does the C-terminal redox center resist irreversible oxidation, but the sulfhydryl of CysIC in the N-terminal redox center also resists oxidation, most likely through a weak ability to attack the peroxy bond in H2O2. The N-terminal redox center’s ability to resist oxidation is critical for rescuing the activity of the enzyme under conditions when the C-terminal Sec-residue becomes oxidized to various oxidation states. Thus the entire modular architecture of the enzyme is constructed to resist inactivation by very high levels of H2O2. Our results imply that the redox signaling function of the thioredoxin system can remain operating (as long as NADPH is present) during conditions of oxidant stress since Sec-TR is very resistant to inactivation (3).
Supplementary Material
Acknowledgments
We would like to thank Mr. Adam Lothrop for reading this manuscript and providing critical comments. We would also like to thank Mr. Adam Mirando for assistance with some experiments.
Footnotes
These studies were supported by National Institutes of Health Grant GM094172 to RJH
A340, absorbance at 340 nm; A412, absorbance at 412 nm; βME, beta-mercaptoethanol; CH3SeO2H, methaneseleninic acid; CH3SeO2K, potassium methaneseleninate; Cys, cysteine; Cys-SO2−, sulfinic acid form of Cys; CysCT, charge-transfer cysteine; CysIC, interchange cysteine; DHA, dehydroalanine; dimedone, 5,5-dimethyl-1,3-cyclohexanedione; DTT, dithiothreitol; DTNB, 5,5′-dithio-bis(2-nitrobenzoic acid); E.coli, Escherichia coli; EDTA, ethylenediaminetetraacetic acid; Gly, glycine; H2O2, hydrogen peroxide; HOCH2SO2Na, sodium hydroxymethanesulfinate; Mr, molecular ratio; mTR, mouse thioredoxin reductase; NADPH, β-nicotinamide adenine dinucleotide phosphate- reduced; Sec, selenocysteine; Sec-SeO2−, seleninic acid form of Sec; Ser, serine; TNB, thionitrobenzoic acid; Tris, tris-(hydroxymethyl)aminomethane; Trx, thioredoxin; TR, thioredoxin reductase; U, the one letter code for Sec; WT, wild type.
We refer to the mouse WT TR as mTR-GCUG. The abbreviation mTR refers to mouse thioredoxin reductase, while the letters GCUG refer to the sequence of the last four amino acids in the C-terminus of TR, glycine-cysteine-selenocysteine-glycine (GCUG). As the Cys and Sec residues are in an adjacent redox dyad, we often use the subscripts 1 and 2 to further describe their positions relative to each other as in mTR-GlyCys1Sec2Gly-OH. We find this useful when describing the redox dyads of other high Mr TRs such as the one from D. melanogaster, which uses Cys in place of Sec. We therefore describe the redox dyad from this TR as Cys1Cys2 as in DmTR-Ser-Cys1Cys2Ser-OH. All of our work with TR has been with the mitochondrial enzyme and we often abbreviate the WT enzyme as mTR3, where the number 3 denotes the cellular compartment (mitochondria). In this nomenclature system, mTR1 is the cytosolic enzyme and mTR2 is the testes specific TR (3). The two mutant enzymes in this study are the truncated TR, missing the last 8 C-terminal amino acids (abbreviated mTRΔ8) and the Sec → Cys mutant of TR (abbreviated as mTR-GCCG).
SUPPORTING INFORMATION AVAILABLE
There are three Figures in the Supporting Information. Figure S1 shows the pH rate profile for the reduction of Trx by mTR3. Figure S2 shows activity progress curves of TR (control and H2O2 treated samples) at acidic pH. Figure S3 shows the rapid oxidation of the TNB anion in the presence of 10 mM H2O2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bock A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Selenocysteine: the 21st amino acid. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 2.Atkins JF, Gesteland RF. The twenty-first amino acid. Nature. 2000;407:463–465. doi: 10.1038/35035189. [DOI] [PubMed] [Google Scholar]
- 3.Sun QA, Wu Y, Zappacosta F, Jeang KT, Lee BJ, Hatfield DL, Gladyshev VN. Redox regulation of cell signaling by selenocysteine in mammalian thioredoxin reductases. J Biol Chem. 1999;274:24522–24530. doi: 10.1074/jbc.274.35.24522. [DOI] [PubMed] [Google Scholar]
- 4.Carugo O, Cemazar M, Zahariev S, Hudáky I, Gáspári Z, Perczel A, Pongor S. Vicinal disulfide turns. Protein Eng. 2003;16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- 5.Hudaky I, Gaspari Z, Carugo O, Cemazar M, Pongor S, Perczel A. Vicinal disulfide bridge conformers by experimental methods and by ab initio and DFT molecular computations. Proteins. 2004;55:152–68. doi: 10.1002/prot.10581. [DOI] [PubMed] [Google Scholar]
- 6.Turanov AA, Kehr S, Marino SM, Yoo MH, Carslon BA, Hatfield D, Gladyshev VN. Mammalian thioredoxin reductase1: roles in redox homoestasis and characterization of cellular targets. Biochem J. 2010 doi: 10.1042/BJ20091378. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SR, Bar-Noy S, Kwon J, Levine RL, Stadtman TC, Rhee SG. Mammalian thioredoxin reductase: oxidation of the C-terminal cysteine/selenocysteine active site forms a thioselenide, and replacement of selenium with sulfur markedly reduces catalytic activity. Proc Natl Acad Sci USA. 2000;97:2521–2526. doi: 10.1073/pnas.050579797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorlatov SN, Stadtman TC. Human thioredoxin reductase from HeLa cells: Selective alkylation of selenocysteine in the protein inhibits enzyme activity and reduction with NADPH influences affinity to heparin. Proc Natl Acad Sci U S A. 1998;95:8520–8525. doi: 10.1073/pnas.95.15.8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bar-Noy S, Gorlatov SN, Stadtman TC. Overexpression of wild type and SeCys/Cys mutant of human thioredoxin reductase in E. coli: the role of selenocysteine in the catalytic activity. Free Radicals in Biology and Medicine. 2001;30:51–61. doi: 10.1016/s0891-5849(00)00448-2. [DOI] [PubMed] [Google Scholar]
- 10.Lothrop AP, Ruggles EL, Hondal RJ. No selenium required: Reactions catalyzed by mammalian thioredoxin reductase that are independent of a selenocysteine residue. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonilla M, Denicola A, Novoselov SV, Turanov AA, Protasio A, Izmendi D, Gladyshev VN, Salinas G. Platyhelminth mitochondrial and cytosolic redox homeostasis is controlled by a single thioredoxin glutathione reductase and dependent on selenium and glutathione. J Biol Chem. 2008;283:17898–907. doi: 10.1074/jbc.M710609200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacey BM, Eckenroth BE, Flemer S, Hondal RJ. Selenium in thioredoxin reductase: a mechanistic perspective. Biochemistry. 2008;47:12810–12821. doi: 10.1021/bi800951f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forchhammer K, Leinfelder W, Bock A. Identification of a novel translation factor necessary for the incorporation of selenocysteine into protein. Nature. 1989;342:453–456. doi: 10.1038/342453a0. [DOI] [PubMed] [Google Scholar]
- 14.Leinfelder W, Zehelein E, Mandrand-Berthelot MA, Bock A. Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature. 1988;331:723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- 15.Heider J, Baron C, Bock A. Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein. Embo J. 1992;11:3759–3766. doi: 10.1002/j.1460-2075.1992.tb05461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Squires JE, Berry MJ. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life. 2008;60:232–235. doi: 10.1002/iub.38. [DOI] [PubMed] [Google Scholar]
- 17.Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- 18.Jukes TH. Genetic code 1990. Outlook Experientia. 1990;46:1149–1157. doi: 10.1007/BF01936925. [DOI] [PubMed] [Google Scholar]
- 19.Cho CS, Lee S, Lee GT, Woo HA, Choi EJ, Rhee SG. Irreversible inactivation of glutathione peroxidase-1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2009.2701. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hondal RJ, Ruggles EL. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2010 doi: 10.1007/s00726-010-0494-6. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finlayson AJ, MacKenzie SL, Finley JW. Reaction of alanine-3-sulfinic acid with 2-mercaptoethanol. Can J Chem. 1979;57:2073–2077. [Google Scholar]
- 22.Oae S, Togo H, Numata T, Fujimori K. Facile reduction of sulfinic acid to disulfide with thiol and chlorotrimethylsilane. Chem Lett. 1980;9:1193–1196. [Google Scholar]
- 23.Kice JL, Lee TWS. Oxidation-reduction reactions of organoselenium compounds. 1 Mechanism of the reaction between seleninic acids and thiols. J Am Chem Soc. 1978;100:5094–5102. [Google Scholar]
- 24.House KL, Garber AR, Dunlap RB, Odom JD, Hilvert D. 1H-NMR spectroscopic studies of selenosubtilisin. Biochemistry. 1993;32:3468–3473. doi: 10.1021/bi00064a034. [DOI] [PubMed] [Google Scholar]
- 25.Bell IM, Fisher ML, Wu ZP, Hilvert D. Kinetic studies on the peroxidase activity of selenosubtilisin. Biochemistry. 1993;32:3754–3762. doi: 10.1021/bi00065a030. [DOI] [PubMed] [Google Scholar]
- 26.Reich HJ, Gudmundsson BO, Green DP, Bevan MJ, Reich IL. The role of ate complexes in the lithium-sulfur, lithium-selenium and lithium-tellurium exchange reactions. Helv Chim Acta. 2002;85:3748–3772. [Google Scholar]
- 27.Arnér ES. Selenoproteins - what unique properties can arise with selenocysteine in place of cysteine? Exp Cell Res. 2010 doi: 10.1016/j.yexcr.2010.02.032. In Press. [DOI] [PubMed] [Google Scholar]
- 28.Bauer H, Massey V, Arscott LD, Schirmer RH, Ballou DP, Williams CH., Jr The mechanism of high Mr thioredoxin reductase from Drosophila melanogaster. J Biol Chem. 2003;278:33020–33028. doi: 10.1074/jbc.M303762200. [DOI] [PubMed] [Google Scholar]
- 29.Eckenroth BE, Rould MA, Hondal RJ, Everse SJ. Structural and biochemical studies reveal differences in the catalytic mechanisms of mammalian and Drosophila melanogaster thioredoxin reductases. Biochemistry. 2007;46:4694–4705. doi: 10.1021/bi602394p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong L, Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J Biol Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 31.Arnér ESJ, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 32.Sekharam M, Cunnick JM, Wu J. Involvement of lipoxygenase in lysophosphatidic acid-stimulated hydrogen peroxide release in human HaCaT keratinocytes. Biochem J. 2000;346:751–758. [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gromer S, Gross JH. Methylseleninate is a substrate rather than an inhibitor of mammalian thioredoxin reductase. Implications for the antitumor effects of selenium J Biol Chem. 2002;277:9701–9706. doi: 10.1074/jbc.M109234200. [DOI] [PubMed] [Google Scholar]
- 35.Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem Biophys Res Commun. 2005;326:799–804. doi: 10.1016/j.bbrc.2004.11.105. [DOI] [PubMed] [Google Scholar]
- 36.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 37.Huang HH, Arscott LD, Ballou DP, Williams CH., Jr Acid-base catalysis in the mechanism of thioredoxin reductase from Drosophila melanogaster. Biochemistry. 2008;47:1721–1731. doi: 10.1021/bi702040u. [DOI] [PubMed] [Google Scholar]
- 38.Björnstedt M, Hamberg M, Kumar S, Xue J, Holmgren A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J Biol Chem. 1995;270:11761–11764. doi: 10.1074/jbc.270.20.11761. [DOI] [PubMed] [Google Scholar]
- 39.Toppo S, Flohé L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Rocher C, Lalanne JL, Chaudière J. Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur J Biochem. 1992;205:955–960. doi: 10.1111/j.1432-1033.1992.tb16862.x. [DOI] [PubMed] [Google Scholar]
- 41.Dimastrogiovanni D, Anselmi M, Miele AE, Boumis G, Petersson L, Angelucci F, Nola AD, Brunori M, Bellelli A. Combining crystallography and molecular dynamics: the case of Schistosoma mansoni phospholipid glutathione peroxidase. Proteins. 2010;78:259–270. doi: 10.1002/prot.22536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.