

Abstract
We present a cross-species chemogenomic screening platform using libraries of haploid deletion mutants from two yeast species, Saccharomyces cerevisiae and Schizosaccharomyces pombe. We screened a set of compounds of known and unknown mode of action (MoA) and derived quantitative drug scores (or D-scores), identifying mutants that are either sensitive or resistant to particular compounds. We found that compound–functional module relationships are more conserved than individual compound–gene interactions between these two species. Furthermore, we observed that combining data from both species allows for more accurate prediction of MoA. Finally, using this platform, we identified a novel small molecule that acts as a DNA damaging agent and demonstrate that its MoA is conserved in human cells.
Keywords: chemogenomics, evolution, modularity
Introduction
Understanding a compound's mode of action (MoA) is a requirement for rational therapeutics and for the understanding of drug resistance. Historically, this important endeavor has been primarily focused on case-by-case studies of antibacterial resistance, but technological developments have resulted in several genome-wide approaches to drug target discovery. Examples of these include studies of gene expression changes after treatment with a small molecule (Hughes et al, 2000; Lamb et al, 2006), physical isolation of a drug–target complex through affinity chromatography (Rix and Superti-Furga, 2009) and whole-genome sequencing of drug-resistant strains (Andries et al, 2005). In silico ligand docking provides a computational approach to identifying potential binding partners based on available crystallography- and NMR-derived protein structures (Kolb et al, 2009). Chemogenomics, or chemical genetic interaction profiling as a means for drug target identification, has been an area of active interest for more than a decade (Giaever, 2003; Wuster and Babu, 2008; Hoon et al, 2008b). A chemogenomic screen examines a drug's mode of action (MoA) by measuring the effect of a drug treatment on a collection of genetically distinct strains, typically a set with modified gene expression varying from multi-copy/overexpression to complete gene deletion. The development of mutant libraries in several model organisms, including Saccharomyces cerevisiae (Winzeler et al, 1999; Giaever et al, 2002), Schizosaccharomyces pombe (Kim et al, 2010; http://pombe.bioneer.co.kr/), Candida albicans (Rodriguez-Suarez et al, 2007) and Escherichia coli (Baba et al, 2006), has greatly accelerated chemogenomic screening. Combining individual drug–mutant relationships (i.e., resistance or sensitivity) into a profile provides a genome-wide view of a compound's effect on the cell. Comparing these drug fitness profiles to genetic interaction profiles composed of double mutant interactions can aid in the identification of drug targets (Parsons et al, 2004; Hoon et al, 2008a; Ho et al, 2009). Additionally, comparing drug profiles makes it possible to infer the MoA of a drug of interest by the similarity of its chemogenomic profile to profiles of drugs with known MoA (Hillenmeyer et al, 2008, 2010). Finally, fitness profile comparison has been used to identify pharmacophores in structurally related molecules (Giaever et al, 2004; Ericson et al, 2008).
In this study, we present a cross-species chemogenomic screening platform, involving two yeast species, S. cerevisiae and S. pombe, that we use to study the evolution of drug mechanism of action. To aid in this analysis, we have used genetic interaction data derived from double mutant analysis in both species (Schuldiner et al, 2005; Collins et al, 2007; Roguev et al, 2008; Wilmes et al, 2008; Fiedler et al, 2009) in conjunction with the chemogenomic profiles. We find that compound–functional module relationships are significantly more conserved than individual compound–gene interactions, suggesting that modularity is a key aspect of the conservation of drug response. Finally, we identify one small molecule predicted to be involved in inducing DNA damage and demonstrate that its MoA is conserved in S. cerevisiae, S. pombe and human cells.
Results
Screening of National Cancer Institute Diversity and Mechanistic Sets in S. cerevisiae and S. pombe
We screened the 2957-member National Cancer Institute (NCI) Diversity and Mechanistic Sets (http://dtp.nci.nih.gov/branches/dscb/repo_open.html) in both S. cerevisiae and S. pombe, using a high-throughput halo assay that measures inhibition of growth (Gassner et al, 2007; Woehrmann et al, 2010) and predicts EC50 values (see Materials and methods for description) for each compound (Figure 1A). The NCI Diversity Set is a collection of compounds selected for structural diversity, and the Mechanistic Set contains compounds that have been tested in the NCI human tumor 60 cell line screen. A total of 270 compounds were found to be bioactive in at least one species (Figure 1B), 132 of which had an effect in both fission and budding yeasts (Supplementary Table 1). We observed an overall ∼2:1 ratio between the predicted EC50 values in S. cerevisiae versus S. pombe, indicating that fission yeast may be globally more sensitive to drugs (Figure 1C). For each compound, nine chemical properties (ClogP, polar surface area (PSA), number of atoms, molecular weight, number of hydrogen-bond donors and acceptors, number of violations of Lipinski's Rules, number of rotatable bonds and molecular volume) were considered. We found no obvious difference between the properties of small molecules that were bioactive only in one species (see Supplementary information). However, when we compared the properties of compounds that were active in at least one species to those of the inactive compounds, we observed that the ClogP, the predicted octanol–water partition coefficient, was more than 80% higher among bioactive compounds (P<5.54 × 10−12) (see Supplementary information). Bioactive compounds also tended to have a lower PSA, a higher molecular weight, lower number of hydrogen bond acceptors, a lower number of hydrogen bond donors and a higher volume than the inactive compounds. Taken together, these data indicate that compact molecules that are non-polar or have intramolecular hydrogen bonding partners are the most bioactive in both yeast species.
Figure 1.

High-throughput yeast halo assay identified bioactive compounds and predicts EC50 values. (A) A total of 2957 compounds from the National Cancer Institute's Diversity Set were arrayed in 384-well plates (10 mM, DMSO). These small molecules were pin transferred to agar plates seeded with yeast cells and then incubated at 25°C. The plates were read with a plate reader to identify halos of death, and the EC50 value is predicted from the volume of the death halo. (B) Of the 2957 compounds screened, we found 271 to be active in at least one organism. Eighty-four compounds are active only in S. cerevisiae, 55 are active only in S. pombe and 132 are active in both species. (C) EC50 values are predicted for each small molecule based on halo volume. On average, we observed that S. pombe is approximately 2 × more sensitive to a small molecule than S. cerevisiae.
Next, we used a growth assay (Figure 2A) to screen 21 bioactive compounds against libraries of S. pombe and S. cerevisiae deletion strains arrayed in agar plates (Table I). Of these 21 compounds, 12 are well-characterized compounds that were selected based on a wealth of previous information for benchmarking purposes, whereas the remaining were randomly selected from those found to be bioactive in both species in the halo assay (Table I). Using a previously described algorithm designed to quantitatively assign genetic interactions based on colony size (Collins et al, 2006), we generated drug scores (or D-scores) indicating the effects of compounds on individual mutations, either negatively (e.g., sensitivity) or positively (e.g., resistance) (Figure 2B). In this analysis, we assume a neutral model, in which the expected growth of a treated mutant would be the product of the growth rates of the untreated mutant and compound-treated wild-type cells. Growth less than this represents sensitivity (Figure 2B), and could arise when a drug negatively impacts on a component of a pathway acting in parallel to a pathway that has a component mutated (Figure 2C). Better than expected growth, or resistance, could arise from a situation where the drug target (or another protein functioning in the same pathway as the target) is deleted (Figures 2B and C). Another scenario where resistance could emerge would be one in which a compound binds to an enzyme and causes the protein to carry out a function that is harmful to cell viability. In this case, deletion of the protein target would relieve the detrimental effect, resulting in resistance (Figure 2C). An example of this latter case would be the binding of the DNA damaging agent camptothecin to the DNA/topoisomerase complex and inhibiting the dissociation of Top1, an event that triggers the DNA damage response (DDR) pathway. These examples are intended to represent possible interpretations of the chemogenomic data, but many other possibilities exist. Finally, collectively, the D-scores can be treated as a phenotypic signature and can be compared with genetic interaction profiles, generated from double mutations, to help identify potential drug targets, as a profile derived from a drug and a mutation of the target should, in principle, result in similar profiles (Figure 2D).
Figure 2.

Cross-species, chemogenomic screening platform. (A) Deletion strains arrayed in 1536-format on agar plates were pin transferred onto compound containing agar plates. These plates were then incubated and photographed. The colony sizes were measured, the raw colony sizes processed, and each strain was given a D-score that quantifies the deviation from an expected neutral growth model (see Materials and methods section). (B) The expected neutral value is equal to product of the growth rate of the deletion with the growth rate of the compound treated wild type. A negative D-score indicates that the deletion strain has a lower fitness than expected by the neutral combination of both perturbations. A positive one indicates that this deletion strain grows better under this condition than expected by the neutral model. (C) A neutral score could indicate that the compound target and the protein corresponding to the gene deletion are in unrelated pathways (Neutral). A negative score could indicate that the small molecule targets a cellular pathway that works in parallel to the pathway containing the genetic deletion (e.g., sensitivity). The positive phenotype can come about in a variety of ways; e.g., the query compound could target a gene that is in the same pathway as the deleted gene (resistance I). Additionally, the drug could alter the function of a protein (protein E in the toy example) creating a new function (function X) that is detrimental to the cell (resistance II(i)). In the latter case, deletion of the drug-altered protein (resistance II(ii)) would lead to resistance. (D) Chemogenomic (D-score) profiles and genetic (S-score) profiles can be compared via hierarchical clustering. This type of analysis can identify small molecules with similar mode of action (drug 1, drug 3), and can potentially pair drugs with their biological targets (drug 2, mutant 2).
Table 1. Compounds profiled in the chemogenomic screen with predicted EC50 value, screening concentration used, compound effect if known and structure.
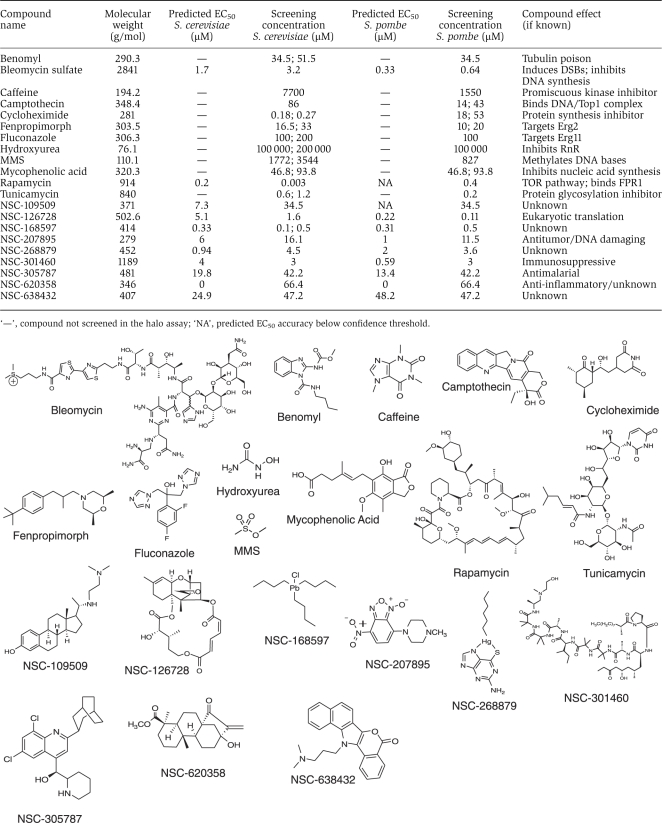
|
The set of 21 compounds were screened against a panel of 727 and 438 gene deletion mutants, representing a wide range of biological processes in S. cerevisiae and S. pombe, respectively, which contained 190 1:1 orthologs (Roguev et al, 2008; Figure 3A, see Supplementary Table 3). Two highly reproducible, independent screens were carried out in each species (rsc=0.72; rsp=0.76, P-value<10−100; Figure 3B) and the resulting data was averaged together to provide a final data set (see Supplementary Table 2). This data set recapitulates many of the previously known functional interactions for the well-studied compounds. For example, among the strongest negative interactions (D-scores <−10) with the DNA damaging agent MMS are deletions of genes belonging to the RAD52 epistasis group (RAD52, RAD55, RAD57), as well as the mutants of the Ubc13-Mms2 ubiquitin-conjugating enzyme, plus its interacting RING-finger gene Rad5, all known to be important for DNA damage repair. Also, as expected, the strongest positive (resistance) interaction with camptothecin was its target, topoisomerase I (TOP1), whereas the strongest negative interactions of benomyl (D-score <−15) are with factors involved in microtubule regulation (TUB3, CIN4, CIN1, PAC2, GIM4).
Figure 3.

Overview of chemogenomic data set. (A) A panel of 438 S. pombe deletion mutants and 727 S. cerevisiae mutants were screened. In total, 190 of these genes are orthologous between each species and are categorized into different functional processes. (B) The reproducibility of the final data sets. Before averaging, two independent data sets were correlated in each species. The data sets were highly correlated: r=0.72 for S. cerevisiae and 0.76 for S. pombe (P-value <10−100). (C) Scatterplot of correlation (r=0.44, P-value=2.2 × 10−16) of our data set with a previously published chemogenomics data set (Hillenmeyer et al, 2008).
To test the accuracy of the data set, we first checked if our quantitative score would correctly identify known sensitive strains. We used previously published S. cerevisiae chemical genetic data generated in a pooled competition liquid growth assay (Hillenmeyer et al, 2008) to define sensitive knockout strains (those with log2 ratio >1.5) for the 12 benchmark compounds. Additionally, compound sensitivities for MMS, mycophenolic acid and hydroxyurea were compared with results from previous non-competitive (i.e., not pooled in batch culture) studies in S. cerevisiae. In all cases, sensitive strains had much lower average D-scores than non-sensitive strains (Supplementary information, P-value <1.7 × 10−13 for small molecules screened at approximately the same concentration and P-value <0.003 for other compounds). We then compared the D-score with the log2 ratio score defined in the study by Hillenmeyer et al (2008). For compounds appearing in both data sets (at approximately the same concentration), we found these two measures to be significantly correlated (r=0.44, P-value=2.2 × 10−16; Figure 3C). Finally, we used an independent set defined by high-confidence compound–gene interactions obtained from the STITCH (Kuhn et al, 2010) (http://stitch.embl.de/) database to benchmark our data set and to compare it with previously published chemogenomic screens of S. cerevisiae. Using the area under the receiver operating characteristic (AROC) curve as measure of discriminatory power, we observed that the D-scores had a similar discriminatory power to identify these high-confidence compound–gene interactions as previous large scale chemogenomic experimental studies (Supplementary information). Together, these results argue that we have generated a robust, reproducible data set of high quality.
Compound–module interactions show higher cross-species conservation than compound–gene relationships
The availability of chemogenomic data for two different yeast species allows us to study how drug response has changed during evolution. D-scores for 190 orthologous genes were computed for both species and used to assess the evolutionary conservation of compound–gene interactions (Figure 4A, Supplementary Table 3). The low correlation between these scores (r=0.13), although statistically significant (P-value <4.4 × 10−11), indicates a large degree of evolutionary divergence of drug sensitivities among these genes. We have previously observed a similar conservation level (r=0.14, P-value < × 10−170) between a large set of genetic interactions from sets of orthologous genes in these two species (Roguev et al, 2008).
Figure 4.

Compound–module interactions are more conserved than compound–gene interactions. (A) A plot of the D-scores for the same compound–gene pairs in S. cerevisiae versus S. pombe. The level of observed conservation (r=0.13, P-value=4.4 × 10−11) is similar to the level of observed between gene–gene interaction partners (Roguev et al, 2008). (B) Use of the I-score to compare compound–gene versus compound–module interactions. We observe a higher conservation on the compound–module level by comparing the accuracy of using compound–gene interactions in S. pombe to predict compound–gene interactions in S. cerevisiae (AROC—area under the ROC curve=0.57) with using compound–module interactions for the same calculation (AROC=0.84). We observe that combining S. pombe compound–module interactions with S. cerevisiae compound–module interactions improves predictions over those made by either data set alone. Then we combine the S. cerevisiae data from Hillenmeyer et al (2008) with our S. cerevisiae data to carry out the same prediction, and we observe no significant improvement (compound–gene AROC=0.75, compound–module AROC=0.86). Finally, we use our cross-species data to predict small-molecule targets in human, and observe a predictive ability significantly higher than random (compound–gene AROC=0.64; compound–module AROC=0.72). (C) Comparison of compound–gene and gene–gene interactions. Compound–gene interactions are represented by dashed lines, gene–gene interactions are shown by solid lines. S. cerevisiae data are shown in red, and S. pombe data are shown in blue. Only highly significant interactions with P-value <0.005 are represented, and the line thickness is proportional to the significance of each interaction (−log(P-value)).
Although individual genetic interactions are generally poorly conserved between these fungi, conservation of genetic interactions among genes that code for physically interacting proteins tends to be significantly higher (Roguev et al, 2008). We reasoned that a similar principle could apply to compound–gene relationships. If a complex is important for the response to a small molecule in one yeast species then it might also be important in the other, even if the individual compound–gene interactions have diverged.
In order to address this question, we first obtained a set of previously reported compound–gene functional interactions in S. cerevisiae from the STITCH database (Kuhn et al, 2010; http://stitch.embl.de/). These interactions were then used to define a set of high-confidence compound–module interactions. Modules were defined as groups of proteins that are part of the same protein complex or share gene ontology terms (see Materials and methods section). Next, we developed the I-score, a metric that combines the D-score between a small molecule and a mutant with a measure of the similarity between the genetic and chemogenomic profiles derived from the same mutant and compound, respectively. The I-score was then used to quantify both compound–gene interactions and compound–module interactions (see Materials and methods section; Figure 4B, top).
We evaluated the ability of our data set to predict the high-confidence functional interactions from STITCH using the AROC curves, a standard metric that can be defined as the probability that the I-score will accurately discriminate a known compound–gene or module interactions from a random one. Using S. cerevisiae I-scores, an AROC value of 0.72 was observed for the discrimination of known compound–gene interactions from random (Figure 4B, S. cerevisiae to S. cerevisiae). These data also correctly identify known module interactions over random with an AROC of 0.85. In contrast to the above result, when S. pombe compound–gene I-scores are used to predict S. cerevisiae compound–gene associations, the prediction is only slightly better than random (57%) (Figure 4B, S. pombe to S. cerevisiae), supporting the idea that compound–gene interactions are poorly conserved. However, S. pombe data can predict S. cerevisiae compound–module interactions almost as well as S. cerevisiae data (84%) (Figure 4B, S. pombe to S. cerevisiae). These results suggest a greater conservation of compound–module interactions when compared with compound–gene interactions.
Combined cross-species chemogenomic data highlights relevant mode of action
To learn if using data from two different species would improve the prediction of a small molecule's MoA, we attempted to predict the high-confidence S. cerevisiae compound–gene and compound–module interactions by combining information from both species (see Materials and methods section). From all interactions found in STITCH, we identified 15 compound–gene and 36 compound–module interactions, for which we had experimental data in both yeasts. In accordance with the evolutionary patterns observed above, the combination of data from both species improves compound–module association predictions (from 85 to 94%), but does not increase the accuracy of compound–gene predictions (Figure 4B, both yeasts to S. cerevisiae). The improvement observed for prediction of module interactions is statistically significant (P-value <0.006) and does not depend on the STITCH cutoffs used (see Supplementary information for cutoff and statistics test). The difference observed could simply result from the combination of two imperfect predictors. To demonstrate that the improvement is related to the data coming from two different species, we combined our S. cerevisiae data with another larger chemogenomic S. cerevisiae data set (Hillenmeyer et al, 2008). Using the combination of both of these data sets, we observed no significant improvement in the ability to predict compound–gene or compound–module interactions from STITCH (Figure 4B, S. cerevisiae liquid+agar to S. cerevisiae).
In order to learn why combining cross-species chemogenomic data might improve MoA identification, we focused our analysis on the response to benomyl, a drug known to bind to and interfere with the function of microtubules. It is expected that the effects of benomyl should mimic the effects of knocking out microtubule components. For this reason, we used the genetic interaction data available for these species to search for modules that show strong genetic interactions with microtubule factors (see Materials and methods section). In Figure 4C, the benomyl–complex interactions (dashed lines) were compared with the complex–complex genetic interactions (solid lines) of microtubule-related factors for both species. As expected, there is a highly significant overlap between these two networks. Of 11 complexes that genetically interact with microtubules, 6 show significant interactions with benomyl. The strongest and most conserved benomyl association identified is in fact with the primary target (microtubules) (Davidse, 1986). In addition, we observed that the strongest benomyl–complex associations (with the prefoldin complex and kinetochore components) mimic the strong and conserved genetic interactions with microtubule factors, whereas weaker benomyl–complex associations are with components that have more divergent genetic interactions with microtubule factors.
As noted above, the benomyl–complex interactions tend to mirror the genetic interactions of benomyl's primary target. The generality of this finding was assessed by comparing the conservation of compound–gene interactions with genetic interactions for orthologous pairs in both species. For a given pair of orthologs, we used the similarity of their compound–gene score vectors and their genetic interaction scores as a proxy for the conservation of both types of interactions. We analyzed 89 pairs of orthologs, for which we had at least 100 genetic interaction scores in common to calculate a similarity score. We observed that these two metrics show a modest but significant positive correlation (r=0.28, N=89, P-value=0.0078), suggesting that the divergence of compound–gene interactions can be explained to some extent by the divergence of genetic interactions. Some of the small-molecule interactions identified with chemogenomic screens are with components that genetically interact with the primary targets. We believe that, given that the most important functional interactions of the primary drug targets are more likely to be conserved across species, the availability of chemogenomic data for multiple species allows us to identify these biologically relevant drug associations.
Finally, we wanted to test if these data from both species could be used to make informed predictions of drug MoA in human cells. We compiled a high-confidence list of compound–gene and compound–module interactions for Homo sapiens derived from the STITCH database (see materials and methods). We identified 56 compound–gene and 33 compound–module interactions, for which we have experimental data from either fungi by orthology. These predictions can discriminate a known compound–gene and a compound–complex interactions from random with an AROC of 0.64 and 0.71, respectively (Figure 4B, both yeasts to human). These results clearly show the potential of our platform as a model system to study drugs’ MoAs with application to human cells.
Prediction of MoA for uncharacterized compounds
Having shown that combining chemogenomic data from both species improves the capacity to predict known compound–complex associations, we set out to make predictions for the MoA of all the small molecules screened (Table I). We used two approaches: correlation of the small-molecule profiles and high-confidence predictions for compound–complex associations. First, we calculated all pairwise correlations for all compounds in each species (Figure 5A). Correlations for compounds that are known to have a similar effect (e.g. MMS, hydroxyurea) are higher than average (median correlation coefficient of 0.34 versus 0.20, P-value=0.026 with a Kolmogorov–Smirnov test; Figure 5A, red nodes). We therefore reasoned that strong correlations between a well-studied small molecule and one of unknown MoA could be used to predict MoAs. Using this approach, we identified 17 of these highly correlated compound pairs between an NSC compound and one of the benchmark small molecules (see Supplementary Table 4). The most highly correlated compound profiles are those of MMS/NSC-207895 and camptothecin/NSC-207895, suggesting that this nitrobenzofuroxan is triggering the DDR pathway (Figure 5A). Additionally, when one correlates the S. cerevisiae and S. pombe profiles for the same compound, only using D-scores for the 190 orthologs, NSC-207895 appears as the most highly correlated pair (Figure 5B, r=0.48). That DNA damaging agents have the most highly correlated profiles in both species could be related to the fact that DNA damage response is a highly conserved biological function. It should be noted that the genes used to determine these profile correlations were weighted toward DNA damage response functions (∼20%) (Figure 3A).
Figure 5.

Correlation analysis of small-molecule profiles in S. cerevisiae and S. pombe. (A) A plot comparing pairs of small-molecule profiles from both fission and budding yeasts derived from the 190 orthologs. In red are profile pairs for which the compounds have a similar MoA. (B) Correlation coefficients between profile pairs, comparing the profile for each compound in S. cerevisiae versus the profile for that same compound in S. pombe, using only the scores for the ortholog pairs. The NSC-207895 profiles are most conserved between the two species. (C) Network diagram showing contributions from each species to predicted compound–module interactions (red=S. cerevisiae, blue=S. pombe, purple=equal contribution; see text for details). Line thickness is proportional to the significance of each interaction (−log(P-value)).
We used the combined data from both species to predict compound–complex interactions (Figure 5C, see Materials and methods for details). Expected compound–complex associations are observed: e.g., benomyl has strong predicted association scores with microtubules, the kinetochore and the prefoldin complex, while the DNA damaging agents all show strong interactions with the DNA repair and/or replication fork complexes. Some of these results highlight the complexity in extrapolating MoA from chemogenomic data. For example, we predict strong associations between tunicamycin with both the RPD3-C(L) complex and ribosomes, while it has been shown that tunicamycin inhibits the synthesis of N-linked glycoproteins (Figure 5C). These seemingly unlikely functional interactions are justified based on previous literature; the compound-induced impairment of the secretory pathways triggers the unfolded protein response, leading to a decrease in ribosome production (Warner and Nierras, 1999) that is regulated by the RPD3-C(L) complex (Sandmeier et al, 2002). This compound–complex network also allows us to study previously uncharacterized compounds. For example, the small-molecule NSC-109509 shows strong associations with chromatin modifiers/remodelers COMPASS, RSC and SET3-C (Figure 5C), suggesting that it might have an effect on chromatin regulation. The compound NSC-207895 shows strong functional interactions with both DNA repair and replication fork complexes, much like MMS and camptothecin, suggesting a role for this compound in some aspect of DNA metabolism.
Chemogenomic screen identifies NSC-207895 as a DNA damaging agent
Several pieces of evidence point toward NSC-207895, a 4-nitrobenzofuroxan derivative, being a DNA damaging agent. Previously, compounds in this family were shown to inhibit DNA and RNA synthesis in mammalian cells (Ghosh and Whitehouse, 1968; Whitehouse and Ghosh, 1968). Furthermore, the profiles for NSC-207895 are highly correlated with those of known DNA damaging agents, MMS and camptothecin, in both yeast species (Figure 5B). Finally, Figure 5C illustrates the similarity between compound–complex interactions of those three compounds. Based on these findings we further investigated the MoA of NSC-207895.
To determine if NSC-207895 had an effect on DNA integrity in vivo, we measured phosphorylation of the DNA checkpoint factor, Rad53, a hallmark of the DDR pathway. After treatment with this uncharacterized compound, we found that the phosphorylation levels of Rad53 had increased significantly (Figure 6A), suggesting that it is involved in some aspect of DNA damage in vivo. This response is specific to NSC-207895, as eight other randomly selected compounds do not result in a similar activation of Rad53 (Supplementary information). We then asked whether the compound would cause physical damage to DNA in vitro. Damaged DNA is known to have decreased transformation efficiency. We developed a plasmid-based transformation assay for DNA damage (see Materials and methods section) and used it to assess the effects of MMS, hydroxyurea or NSC-207895 as DNA damaging agents. In the case of hydroxyurea we observed little effect, consistent with its role as a ribonucleotide reductase inhibitor (Figure 6B). MMS, on the other hand, showed the expected dose-dependent decrease in transformation efficiency due to the known DNA-methylating action of MMS. NSC-207895 showed no effect on transformation efficiency (Figure 6B). In order to gain a better understanding of the MoA of the NSC-207895 compound, we tested its effect on cell cycle progression. We released synchronized untreated cells or cells treated with MMS, hydroxyurea or NSC-207895 and used flow cytometry (FACS) to follow the effect of the different conditions (Figure 6C). We observed that NSC-207895 caused a very significant replication delay, one that was similar to the one observed in the presence of MMS.
Figure 6.

NSC-207895 is a DNA damaging agent. (A) We treated S. cerevisiae with known DNA damaging agents MMS and hydroxyurea, and with NSC-207895. Rad53-phosphorylation is triggered under all three conditions, indicating that NSC-207895 does trigger the DDR pathway in S. cerevisiae (B) A LEU plasmid was treated with DNA-methylating agent MMS, hydroxyurea or NSC-207895, and then transformed into BY4741. Decreased transformation efficiency compared with the control indicates that DNA has been physically damaged, as demonstrated with MMS. NSC-207895, like hydroxyurea, does not show decreased transformation efficiency, and therefore is not likely to be causing physical damage to the DNA in the plasmid environment. (C) S. cerevisiae synchronized cells were released into media containing either no compound, MMS, hydroxyurea or NSC-207895, and flow cytometry was used to track cell cycle progression. NSC-207895 caused a significant replication delay that was similar to the action of MMS. (D, E) To explore the conservation of compound MoA for NSC-207895, we treated S. pombe and H. sapiens U2OS cells with MMS, hydroxyurea and NSC-207895. As in S. cerevisiae, we observed phosphorylation of cds1 in S. pombe and CHK1 and CHK2 in H. sapiens, indicating that treatment with both control small molecules and the unknown compound all trigger the DNA damage response pathway in all three species. (D) Lane 1 is an untreated control, while lanes 2–5 contain NSC-207895 dissolved in DMSO at final concentration of 2.0, 4.0, 6.0, 8.0 μM, respectively. For pCHK1 S317, the upper band is the phosphorylated form, while the lower is likely a degraded species. For pCHK2 T68, the lower band is the phosphorylated form, whereas the upper band is likely pCHK2 T68 with other, as of yet, unidentified post-translational modifications.
To determine if this response was evolutionarily conserved, as suggested by the cross-species profiling, we monitored the phosphorylation levels of the S. pombe ortholog of Rad53, Cds1, and found a similar increase in its phosphorylated form (Figure 6D). Finally, we tested the effect of this compound in human cells and again found that the phosphorylation levels of Chk2, the functional counterpart of budding yeast Rad53, was increased (Figure 6E), indicating that the NSC-207895-induced DDR is evolutionarily conserved. Whereas in budding yeast, the Mec1 (human ATR homolog)–Rad53 (human CHK2 homolog) pathway is the major DDR pathway activated in response to a wide range of DNA damage and DNA replication stress (Zhou and Elledge, 2000), there are two main DDR pathways in mammalian cells (ATM-CHK2 and ATR-CHK1). Moreover, ATM and ATR activation is preferentially triggered by different genomic stresses. For example, while DNA replication stress mainly activates the ATR kinase (CHK1), the ATM kinase (CHK2) is activated by DNA double-strand break repair. When we continuously tested the DDR effect of this compound in human cells, we found that the phosphorylation levels of Chk2, the functional counterpart of budding yeast Rad53, was increased in a dose-and time-dependent manner in human U2OS cells (Figure 6E). More strikingly, a close examination of these phosphorylation profiles revealed that NSC-207895 triggers an immediate and early activation of ATR kinase at 1 h, which was significantly decreased at 3 h. In contrast, the ATM kinase activation is weak at 1 h, but is greater at the 3 h point (Figure 6E).
Taken together, these results show that NSC-207895 activates the DDR leading to a delay in cell cycle progression, but does not covalently damage DNA in vitro. It is likely to trigger the DDR pathway through an indirect mechanism, such as intercalation, as it is an electron-deficient, planar aromatic compound or requires a protein target to mediate its effect in vivo.
Discussion
In the late 90s, Alberts (1998) and Hartwell et al (1999) both suggested that the cell's components are organized into functional groups or modules, and that the reductionist approach of studying each element of a group in isolation was limiting. They argued that cell biologists should strive to understand how these elements work together to perform critical cellular functions. Previous evolutionary studies have underscored the importance of studying these functional groups' interactions. For example, we have observed that positive genetic interactions between genes coding for co-complex members are more conserved than average in S. cerevisiae and S. pombe (Roguev et al, 2008). Furthermore, in concordance with this finding, Jensen et al (2006) studied the evolution of cell cycle control of gene expression and noted that, although the timing of cell cycle regulation of different complexes is conserved across species, the specific genes that are regulated in each complex have significantly diverged. Finally, cross-species studies of phosphorylation demonstrated that the average levels of phosphorylation of functional modules are conserved, although the proteins that are phosphorylated can diverge quickly (Beltrao et al, 2009; Holt et al, 2009; Tan et al, 2009).
The evolutionary analysis of compound interactions presented here reveals a very similar pattern, whereby the compound–module interactions show higher conservation than the underlying compound–gene interactions. These different observations point to a similar and intuitive trend that could be explained by a faster change of the relative importance of functional module subunits relative to the slowly evolving function of the whole module. It should be noted that this apparent difference could, at least in part, be due the higher statistical power associated with group analysis relative to individual scores. However, beyond the evolutionary interpretations, these observations have immediate practical implications for the study of drugs' MoAs. The availability of similar data for a larger number of species will further elucidate the evolutionary dynamics of these stress/drug response functions.
Recent work has shown genetic interactions to be quite divergent (Roguev et al, 2008; Tischler et al, 2008). With this in mind, one should be critical when applying genetic discoveries made in fungi to animal systems (divergence ∼1.4 billion years ago; Hedges, 2002). However, fungal model systems are very convenient for preliminary studies, and so devising a method to add value to yeast-based predictions is highly desirable. To improve yeast-based predictions of drug MoA, we combine data from two model systems. We show that using cross-species chemogenomic data result in a significant improvement in the prediction of drug targets over those predicted from either species alone. Collecting compound–gene interactions data in S. cerevisiae and S. pombe allowed us to quantitatively compare drug behavior in two yeasts. This information lead to the observation that compound NSC-207895 behaves like known DNA damaging agents in both species, and allowed us to accurately predict that it would also behave as a DNA damaging agent in human cells. It should be noted that if two drug profiles are highly correlated in both species, not only do they share a MoA, but they are also probably affecting a highly conserved biological process. Identification of compound pairs, which are highly correlated in only one organism, provides information about biological pathways that are not conserved. This information could, in principle, be used to identify pathogen-specific drugs.
We believe that the strategy presented here has relevance for the study of drug–drug interactions. Lehár et al (2009) have shown that drug combinations tend to be synergistic only in specific cellular contexts (i.e., species or cell types). The availability of chemogenomic and genetic interaction data for different species also opens the door for the study of drug combinations and their evolutionary dynamics. Genetic interaction data should allow for a rationalization of the changes in drug synergy under different conditions observed by Lehár et al (2009). This knowledge could be quite useful; e.g., in the quest for combinatorial therapeutics, which specifically target pathogenic microbes or diseased tissues, leaving the host or symbiotic species unharmed.
Materials and methods
Halo assay
Compounds were taken up in DMSO at 10 mM final concentration in 384-well plates. Agar plates containing YPD or YES were seeded with an overnight culture of either wild-type S. cerevisiae or S. pombe, and the compounds were then pin transferred to the seeded agar plates. The compound-treated plates were incubated and the OD of the agar was measured using a plate reader. The presence of a ‘halo of death’ indicates an active compound, and the size of this halo can be correlated to an inhibitory growth concentration (predicted EC50) for each compound. The compounds were defined as bioactive if they had a measurable halo, indicative of growth inhibition. The cut-off was chosen to maximize accuracy and still allow for prediction of EC50s on the order of ∼200 μM. The statistical analysis, cut-offs and prediction of EC50 were performed as described previously (Woehrmann et al, 2010).
Chemogenomic screen
In order to perform a comparative analysis of compound–gene interactions, we selected a set of 21 for chemogenomic screening (Table I). Of these 21 compounds, 9 were a random subset of the bioactive compounds identified in the halo assay and the remaining were selected based on the availability of previous information for benchmarking purposes. The strains screened were selected from the commercially available S. cerevisiae and S. pombe haploid deletion libraries, and were chosen to represent a wide range of biological functions. As compound availability was a concern, we attempted to minimize the number of strains screened while maximizing the number of overlapping 1:1 genetic orthologs. Strains were also selected in order to maximize complementarity with previously collected genetic interaction data for subsequent analysis. In total, we selected 727 S. cerevisiae strains and 438 S. pombe strains, with a 190-strain overlap of orthologs, arrayed into 1536-format. In order to guarantee that the chemogenomic data sets for the different yeasts are comparable, profiling in both species was performed with approximately the same concentrations. Different screening concentrations around known or predicted EC50 were tested and we kept those that would produce strong and reproducible results. Specifically, the final concentrations were picked based on the following criteria: (a) the resulting distribution of D-scores for a profile had to have at least 2 × the standard deviation of the D-scores from an untreated profile, indicating that there is a signal due to the compound and (b) the replicate profiles needed to correlate with each other with a Pearson's correlation r>0.4, indicating that the data are reproducible.
Analysis of molecular properties of NCI library compounds
Two data sets measuring the bioactivity of small molecules in yeast were used. The first data set comes from the halo assay described in Figure 1A (data set A). It describes the predicted EC50 values of 2957 NCI compounds in both wild-type S. cerevisiae and wild-type S. pombe. The second data set (data set B) describes the bioactivity of 87 264 NCI compounds on S. cerevisiae strains. In the initial stage of the anticancer drug screen that produced this data set, 87 264 small molecules were tested against six different strains of S. cerevisiae. Of these, 12 068 had a strong effect on yeast growth and 75 196 did not (http://dtp.nci.nih.gov/yacds/index.html). We downloaded the molecular structures of each of the small molecules (ftp://ftp.ncbi.nih.gov/pubchem/) and computed nine properties for each of them in order to assess whether one of these properties was significantly distinct for the small molecules that affect growth. Small-molecule properties were computed using Molinspiration's mib tool (http://www.molinspiration.com/). A local version of the mib tool was kindly provided by John J Irwin of the University of California, San Francisco.
Transformation efficiency assay
We treated plasmid pRS315 (carrying a LEU2 marker, 449 ng/μl stock solution) with MMS (450, 112 mM), hydroxyurea (200, 50 mM) and NSC-207895 (10, 2.5 mM) in an equal volume of AB buffer (Stokes and Michael, 2003) for 30 min at 30°C (reaction volumes ∼20 μl). The plasmids were then purified using a Qiagen clean-up kit; reaction mixtures were diluted with 100 μl PB buffer, agitated and filtered in a Qiagen spin column. The precipitate was washed with 0.75 ml PE buffer, spun 1 × . The filtrate was discarded, and then the column was re-spun to remove residual buffer. Columns were transferred to clean, dry 1.5-ml Eppendorf tubes. Fifty μl of EB was gently pipetted onto the filters, and allowed to soak 1 min, then spun down. The same filtrate was used to re-soak the filter, and was re-spun down. Average yields for these reactions were ∼25 ng/μl. These drug-treated plasmids were transformed into S. cerevisiae (BY4741) according to the LiAc/SS carrier DNA method (Gietz and Schiestl, 2007), and the resulting colonies were tallied manually.
Phosphorylation of Rad53/cds1
Yeast Strains PGY1834: S. cerevisiae W303 lys2Δ ade2 leu2 his3 trp1 ura3 RAD53-HA∷TRP1; NB2118: S. pombe h-leu1-32 ura4-D18 cds1-2HA6his:ura4+. All experiments were performed in rich medium: YM-1 with 2% dextrose for S. cerevisiae and YE5S for S. pombe, at 30°C. PGY1834 cells in log phase were treated with no addition, DMSO, nocodazole (0.25, 1.25, 2.5 or 7.5 μg/ml), MMS (0.05, 0.25, 0.5 or 1.5 mM), HU (3.75, 18.75, 37.5 or 112.5 mM) or NSC-207895 (0.5, 2.5, 5 or 15 μM) for 2 h. NB2118 cells in log phase were treated with medium alone, DMSO, nocodazole (0.25, 2.5 or 25 μg/ml), MMS (0.05, 0.5 or 5 mM), HU (3.75, 37.5 or 375 mM) or NSC-207895 (0.5, 5 or 50 μM) for 4 h at 30°C. Cell pellets from both species were rinsed and frozen at −80°C, and then lysed in hot SDS sample buffer. Cell extracts from S. cerevisiae were run on a 4–20% gradient polyacrylamide gel (BioRad), whereas S. pombe cell extracts were run on 8% polyacrylamide gels polymerized with 25 μM Phos-tag Acrylamide reagent (NARD Institute Ltd). Gels containing Phos tag were washed in transfer buffer with 5 mM EDTA, and then transfer buffer alone. Both types of gels were transferred to PVDF and blotted with anti-HA (16B12, Covance). To control for loading, the membrane from the S. cerevisiae experiment was stained with Ponceau S, whereas the gel from the S. pombe experiment was stained with Coommassie.
Phosphorylation of CHK1 and CHK2
U2OS cells were plated into six-well plates and cultured to 70–80% of confluency. Compound NSC-207895 was dissolved in DMSO (1000 × ), added to each well with final concentration at 2.0, 4.0, 6.0, 8.0 μM, respectively. Control wells were treated with equal amount of DMSO. Cells lysates were prepared at 1.0, 3.0 h treatment point by direct addition of 1 × SDS–PAGE loading buffer. About 20 μg of each lysate were separated on 4–12% SDS–PAGE gradient gels. Proteins were transferred to PVDF membrane, followed by probing with antibodies against Chk1 S317 (Bethyl), Chk2 T68 (Cell signaling), r-H2AX (Millipore) and GAPDH (Santa Cruz).
Synchronization, drug treatment and FACS
For synchronized experiments, PGY1834 cells in log phase were arrested in 10 μg/ml α-factor for 3 h (with an additional 10 μg/ml α-factor added after 2 h), then released into medium alone, 5.3 mM MMS, 48 μM NSC-207895 or 300 mM hydroxyurea. Aliquots were removed at the indicated times and fixed in 70% ethanol. Samples were subsequently treated with RNase A and proteinase K, stained with Sytox Green, and analyzed by FACS.
High-confidence set of compound–gene and compound–module interactions derived from the STITCH database
The STITCH database maintains a list of compound–gene association scores that are derived from the weighted combination of different data source. These include information on direct protein physical interactions and functional interactions obtained by literature mining. In STITCH, each compound–gene association has a score ranging from 0 to 1 that relates to the strength of the functional association. We defined high-confidence associations as having a STITCH score >0.65. The results obtained do not vary with the threshold selected, and we provide in Supplementary information the analysis performed with cut-offs of different stringency. For each small molecule, we derived a list of module interactions using the high-confidence gene interactions from STITCH and a set of manually curated complexes (Güldener et al, 2006; Collins et al, 2007) and Gene Ontology annotations (Ashburner et al, 2000). We used these to search for ‘modules’ (defined here as a complex or a Gene Ontology group) with a statistically significant enrichment of subunits among the STITCH compound–gene interactions (P-value <0.01 based on random sampling). Compound–gene pairs defined as high-confidence interactions can be found in Supplementary Table 5.
We obtained known compound–gene associations for both S. cerevisiae and human; however, similar associations were not available in S. pombe. Orthology assignment between S. pombe and S. cerevisiae was obtained from the Fungal Orthogroups Repository (Wapinski et al, 2007); orthology assignment between the two fungi and human was obtained from the Inparanoid database (O’Brien et al, 2005).
Combining chemogenomic data with genetic interaction data (I-score)
To combine all available information into a single score useful for comparisons and prediction, we developed the two-variable I-score. The first variable is the D-score, which is the scored interaction for a specific small molecule/gene pair, provided by the chemogenomic screen (see Figure 2B–D). The second variable incorporates data from previous work in genetic–interaction screening and compares chemogenomic profiles with genetic interaction profiles for each small molecule/gene pair. We have empirically observed that both tails of the D-score distribution and the positive side of the correlation coefficient distributions are indicative of known compound–gene interactions. We use Pearson's correlation to quantify the similarity between a compound-based D-score vector and a genetic S-score vector for all compound/gene profile pairs in both S. cerevisiae and S. pombe. In order to obtain a final score, we have z-score normalized both the D-score (Z-scoreD) and correlation coefficients (Z-scoreCCs). We have thus calculated a combined score as:
I-scores were used as the measure of compound–gene association and I-scores for both species can be found in Supplementary information (see Supplementary Table 5).
In order to predict known S. cerevisiae compound–gene associations based on the combined data from both fungi, the I-scores for S. pombe are first conferred to S. cerevisiae genes based on orthology assignments and summed with the corresponding I-score in S. cerevisiae. The same was done when predicting known human compound–gene associations. Human compound–gene pairs were annotated with data from both yeast species by orthology and the two scores summed.
Compound–module and module–module association scores
We used the above-defined I-score as a measure of compound–gene association, as determined by our screening approach. In order to predict module interactions, we calculated the average I-score of each module (defined here as a complex or a gene ontology group) and the probability of observing a similar or higher average score based on random sampling of an equal number of proteins (i.e., compound–module P-value). The strength of each compound–module association was then defined as −log(P-value). We excluded all modules with less than three members with a calculated I-score, as well very unspecific modules composed of more than 200 members. When using the experimental data from both fungi to predict compound–module associations, the P-value for a module interaction was calculated interdependently for each species and the final score was defined as:
Compound–module interaction scores for both species are provided in Supplementary information (see Supplementary Table 5).
Figure 4C shows complexes with significant genetic interactions with microtubules. For both species, we used previously available genetic interaction data to obtain the average of the absolute S-score between microtubules and other protein complexes. We then used random sampling to calculate the likelihood of observing a similar of higher value by chance. A cut-off of P-value <0.005 was used and the line thickness was set to be proportional to −log(P-value).
Supplementary Material
Halo assay data derived from S. cerevisiae and S. pombe
Complete D-score datasets for both species
List of orthologs used for cross-species compound profiling and their respective D-scores
Pearson's correlations for compound profile pairs in each species
Compound-gene I-scores, compound-module p-value scores and compound-module interaction scores
Supplementary Information, Supplementary Figures S1–3, Supplementary Tables S1–12
Acknowledgments
We thank Sean Collins, Sourav Bandyopadhyay, Assen Roguev, Hannes Braberg, Harlizawati Jahari, Trey Ideker, Regis Kelly and Douglas Crawford for valuable discussion, and Paul Russell for strains. We also thank Mike Shales for graphical work. PB was supported by a Long-Term Postdoctoral Fellowship from the Human Frontier Science Program. We acknowledge funding from NIH, Searle and Keck Foundations, as well as from Vincent Low. Author contributions:LK, SJE, DT, MB, RSL, NJK designed experiments; LK, TJB, NG, CZ performed experiments; LK, PB, AW, JW, NJK analyzed data; and LK, PB and NJK wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Alberts B (1998) The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92: 291–294 [DOI] [PubMed] [Google Scholar]
- Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307: 223–227 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, Harris M, Hill D, Issel-Tarver L, Kasarskis A, Lewis S, Matese J, Richardson J, Ringwald M, Rubin G, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko K, Tomita M, Wanner B, Mori H (2006) Construction of Escherichia coli K-12 in-frane, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2, 2006 0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Trinidad J, Fiedler D, Roguev A, Lim WA, Shokat KM, Burlingame A, Krogan N (2009) Evolution of phosphoregulation: comparison of phosphorylation patterns across yeast species. PLoS Biol 7: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, Schuldiner M, Krogan N, Weissman J (2006) A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol 7: R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, Ding H, Xu H, Han J, Ingvarsdottir K, Cheng B, Andrews B, Boone C, Berger SL, Hieter P, Zhang Z et al. (2007) Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446: 806–810 [DOI] [PubMed] [Google Scholar]
- Davidse L (1986) Benzimidazole fungicides: mechanism of action and biological impact. Ann Rev Phytopathology 24: 43–65 [Google Scholar]
- Ericson E, Gebbia M, Heisler LE, Wildenhain J, Tyers M, Giaever G, Nislow C (2008) Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genetics 4: e1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler D, Braberg H, Mehta M, Chechik G, Cagney G, Mukherjee P, Silva A, Shales M, Collins SR, van Wageningen S, Kemmeren P, Holstege F, Weissman JS, Keogh M, Koller D, Shokat K, Krogan N (2009) Functional organization of the S. cerevisiae phosphorylation network. Cell 136: 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassner NC, Tamble CM, Bock JE, Cotton N, White KN, Tenney K, Onge RPS, Proctor MJ, Giaever G, Nislow C, Davis RW, Crews P, Holman TR, Lokey RS (2007) Accelerating the discovery of biologically active small molecules using a high-throughput yeast halo assay. J Nat Prod 70: 383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Whitehouse M (1968) Potential antileukemic and immunosuppressive drugs. Preparation and in vitro pharmacological acitivity of some benzo-2,1,3-oxadiazoles (benzofurazans) and their N-oxides (benzofuroxans). J Med Chem 11: 305–311 [DOI] [PubMed] [Google Scholar]
- Giaever G (2003) A chemical genomics approach to understanding drug action. Trends Pharmacol Sci 24: 444–446 [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, Bakkoury ME, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW (2004) Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. PNAS 101: 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH (2007) Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protocols 2: 35–37 [DOI] [PubMed] [Google Scholar]
- Güldener U, Münsterkötter M, Oesterheld M, Pagel P, Ruepp A, Mewes H, Stümpflen V (2006) MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res 34: D436–D441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell L, Hopfield J, Leibler S, Murray A (1999) From molecular to modular cell biology. Nature 402: C47–C51 [DOI] [PubMed] [Google Scholar]
- Hedges SB (2002) The origin and evolution of model organisms. Nat Rev 3: 838–849 [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Ericson E, Davis RW, Nislow C, Koller D, Giaever G (2010) Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol 11: R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G (2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320: 362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CH, Magtanong L, Barker SL, Gresham D, Nishimura S, Natarajan P, Koh JLY, Porter J, Gray CA, Andersen RJ, Giaever G, Nislow C, Andrews B, Botstein D, Graham TR, Yoshida M, Boone C (2009) A molecular barcoded yeast ORF library enables mode of action analysis of bioactive compounds. Nat Biotechnol 27: 369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt L, Tuch B, Villén J, Johnson A, Gygi S, Morgan D (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, Fung E, Proctor M, Shokat KM, Zhang C, Davis RW, Giaever G, Onge RPS, Nislow C (2008a) An integrated platform of genomic assays reveals small molecule bioactivities. Nat Chem Biol 4: 498–506 [DOI] [PubMed] [Google Scholar]
- Hoon S, St Onge RP, Giaever G, Nislow C (2008b) Yeast chemical genomics and drug discovery: an update. Trends Pharmacol Sci 29: 499–504 [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J et al. (2000) Functional discovery via a compendium of expression profiles. Cell 102: 109–126 [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Jensen TS, de Lichtenberg U, Brunak S, Bork P (2006) Co-evolution of transcriptional and post-translational cell-cycle regulation. Nature 443: 594–597 [DOI] [PubMed] [Google Scholar]
- Kim DU, Hayles J, Kim D, Wood V, Park HO, Won M, Yoo HS, Duhig T, Nam M, Palmer G, Han S, Jeffery L, Baek ST, Lee H, Shim YS, Lee M, Kim L, Heo KS, Noh EJ, Lee AR et al. (2010) Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 28: 617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P, Ferreira RS, Irwin JJ, Shoichet BK (2009) Docking and chemoinformatic screens for new ligands and targets. Curr Opin Biotechnol 20: 429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Szklarczyk D, Franceschini A, Campillos M, von Mering C, Jensen L, Beyer A, Bork P (2010) STITCH 2: an interaction network database for small molecules and proteins. Nucleic Acids Res 38: D552–D556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR (2006) The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313: 1929–1935 [DOI] [PubMed] [Google Scholar]
- Lehár J, Krueger A, Avery W, Heilbut A, Johansen L, Price E, Rickles R, Short Gr, Staunton J, Jin X, Lee M, Zimmermann G, Borisy A (2009) Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 27: 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien K, Remm M, Sonnhammer E (2005) Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 33: D476–D480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C (2004) Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol 22: 62–69 [DOI] [PubMed] [Google Scholar]
- Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B et al. (2006) Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126: 611–625 [DOI] [PubMed] [Google Scholar]
- Rix U, Superti-Furga G (2009) Target profiling of small molecules by chemical proteomics. Nat Chem Biol 5: 616–624 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Suarez R, Xu D, Veillette K, Davison J, Sillaots S, Kauffman S, Hu W, Bowman J, Martel N, Trosok S, Wang H, Zhang L, Huang L-Y, Li Y, Rahkhoodaee F, Ransom T, Gauvin D, Douglas C, Youngman P, Becker J et al. (2007) Mechanism-of-action determination of GMP synthase inhibitors and target validation in candida albicans and aspergillus fumigatus. Chem Biol 14: 1163–1175 [DOI] [PubMed] [Google Scholar]
- Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, Qu H, Shales M, Park H-O, Hayles J, Hoe K-L, Kim D-U, Ideker T, Grewal SI, Weissman JS, Krogan NJ (2008) Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science 322: 405–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeier J, French S, Osheim Y, Cheung W, Gallo C, Beyer A, Smith J (2002) RPD3 is required for the inactivation of yeast ribosomal DNA genes in stationary phase. EMBO J 21: 4959–4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, Weissman JS, Krogan NJ (2005) Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123: 507–519 [DOI] [PubMed] [Google Scholar]
- Stokes MP, Michael WM (2003) DNA damage-induced replication arrest in Xenopus egg extracts. J Cell Bio 163: 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner M, Jørgensen C, Bader G, Aebersold R, Pawson T, Linding R (2009) Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci Signal 2: ra39. [DOI] [PubMed] [Google Scholar]
- Tischler J, Lehner B, Fraser A (2008) Evolutionary plasticity of genetic interaction networks. Nat Genet 40: 390–391 [DOI] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A (2007) Automatic genome-wide reconstruction of phylogenetic gene trees. Bioinformatics 23: i549–i558 [DOI] [PubMed] [Google Scholar]
- Warner J, Nierras C (1999) Protein kinase C enables the regulatory circuit that connects membrane synthesis to ribosome synthesis in Saccharomyces cerevisiae. J Biol Chem 274: 13235–13241 [DOI] [PubMed] [Google Scholar]
- Whitehouse M, Ghosh P (1968) 4-nitrobenzofurazans and 4-nitrobenzofuroxans: a new class of thiol-neutralising agents and potent inhibitors of nucleic acid synthesis in leucocytes. Biochem Pharmacol 17: 158–161 [DOI] [PubMed] [Google Scholar]
- Wilmes G, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins S, Whitworth G, Kress T, Weissman J, Ideker T, Guthrie C, Krogan N (2008) A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell 32: 735–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, Bakkoury ME, Foury Fo, Friend SH, Gentalen E, Giaever G et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906 [DOI] [PubMed] [Google Scholar]
- Woehrmann MH, Gassner NC, Bray WM, Stuart JM, Lokey RS (2010) HALO384: a halo-based potency prediction algorithm for high-throughput detection of antimicrobial agents. J Biomol Screen 15: 196–205 [DOI] [PubMed] [Google Scholar]
- Wuster A, Babu MM (2008) Chemogenomics and biotechnology. Trends Biotechnol 26: 252–258 [DOI] [PubMed] [Google Scholar]
- Zhou B, Elledge S (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Halo assay data derived from S. cerevisiae and S. pombe
Complete D-score datasets for both species
List of orthologs used for cross-species compound profiling and their respective D-scores
Pearson's correlations for compound profile pairs in each species
Compound-gene I-scores, compound-module p-value scores and compound-module interaction scores
Supplementary Information, Supplementary Figures S1–3, Supplementary Tables S1–12