

Abstract
A major question in transcription factor (TF) biology is why a TF binds to only a small fraction of motif eligible binding sites in the genome. Using the estrogen receptor-α as a model system, we sought to explicitly define parameters that determine TF-binding site selection. By examining 12 genetic and epigenetic parameters, we find that an energetically favorable estrogen response element (ERE) motif sequence, co-occupancy by the TF FOXA1, the presence of the H3K4me1 mark and an open chromatin configuration in the pre-ligand state provide specificity for ER binding. These factors can model estrogen-induced ER binding with high accuracy (ROC-AUC=0.95 and 0.88 using different genomic backgrounds). Moreover, when assessed in another estrogen-responsive cell line, this model was highly predictive for ERα binding (ROC-AUC=0.86). Variance in binding site selection between MCF-7 and T47D resides in sites with suboptimal ERE motifs, but modulated by the chromatin configuration. These results suggest a definable interplay between sequence motifs and local chromatin in selecting TF binding.
Keywords: chromatin, DNA binding, modeling, recognition motifs, transcription factor
Introduction
Despite the primary importance of transcription factor (TF)–DNA interaction, little is known about how specificity and selection are determined during TF recruitment on a genomic scale (Farnham, 2009). In order to drive their transcriptional programs, TFs bind to specifically recognized DNA segments, commonly short and degenerative sequence-recognition motifs, which are often represented frequently in the genome and exert their action over variable distances to interact with the basal transcriptional machinery. However, TF proteins occupy only a very small fraction (typically <2%) of all their potential recognition motifs found in the genome. Moreover, this limited number of occupied sites might be significantly different between different cell types (Lupien et al, 2008). Access of regulatory proteins such as TF to DNA is regulated by chromatin and is an important aspect in controlling transcriptional regulation of specific gene loci and TF function (Gregory and Horz, 1998; Morse, 2003). DNA packaging into nucleosomes may also physically restrict the accessibility of the genome to regulatory proteins such as TFs (Cairns, 2007; Rando and Ahmad, 2007; Petesch and Lis, 2008). This restriction is dynamic and changes during development and in response to exogenous cues (Lee et al, 2007; Schones et al, 2008).
Taking estrogen receptor-α (ERα) as a model of an inducible TF (Ali and Coombes, 2000), we address how ER utilizes specific binding sites from its genomic repertoire. ERα acts by directly binding to a 13–19 bp canonical palindromic-recognition motif, the estrogen response element (ERE) or, less frequently, by indirectly binding DNA via interaction with another TF in a ‘tethered’ mode. Genome-wide positional analysis (Carroll et al, 2006; Lin et al, 2007; Hurtado et al, 2008; Welboren et al, 2009) showed that the vast majority of in silico predicted ERα-binding sites are not occupied in vivo in the MCF-7 human breast cancer cell line. These ERα studies also suggested the need for cooperating TFs such as the Forkhead protein, FOXA1 (Carroll et al, 2005), in facilitating ER binding to chromatin. Nuclear co-regulators, which often possess chromatin-modulating activities, appear to act cooperatively with ERα to establish patterns of gene expression and thus provide considerable functional flexibility in specifying transcriptional regulation (Strahl and Allis, 2000; McKenna and O’Malley, 2002; Cheng et al, 2006). However, these studies have not systematically addressed the predictive value of specific genomic features, either individually or in combination, in defining ligand-induced ER binding across the human genome.
To investigate this further on a genome-wide scale, we set out to map the epigenetic signatures that are important for ERα-binding site utilization. Here, using massive parallel sequencing, we analyzed the areas of open chromatin, and genome-wide occupancy of six histone methylation/acetylation marks in MCF-7 cells, and two factors likely to be co-localized with ER binding, FOXA1 and AP1 (FOS and JUN). RNA polymerase II (RNA Pol II) was also included in the analysis given the observation that ER-binding sites interact with the transcription start sites (TSSs) through a looping mechanism (Pan et al, 2008; Fullwood et al, 2009). We surmised that functionally active binding sites might be marked by RNA Pol II interaction. Overlapping these marks with an ERα-binding site map, we sought to define rules of ERE-binding site usage leading to downstream transcriptional regulatory control of ERα.
Results
Genome-wide mapping of ERα-binding sites
We first identified all ERα-binding sites by a chromatin immunoprecipitation (ChIP)-sequence strategy and generated more than 7 and 12 million uniquely mapped tags for estradiol (E2) and vehicle-treated samples, respectively, in MCF-7 cells and T47D cells (Supplementary Table I). As ChIP-seq is sensitive to biases engendered by gene amplification, we removed all sites residing in positions exhibiting significant copy number variation in MCF-7 cells (Shadeo and Lam, 2006). Using a global intensity threshold corresponding to a P-value of 0.001 and subsequent filtering by local normalization to a control input DNA library, we identified 16 043 peaks that were defined as binding sites in the non-amplified MCF-7 genome (De Santa et al, 2009) (see Supplementary Methods).
These ChIP-seq-derived-binding sites overlapped with up to 86% of all sites in previous genome-wide ER-binding mapping studies (Carroll et al, 2006; Lin et al, 2007; Hurtado et al, 2008; Welboren et al, 2009) (Supplementary Figure 1, Supplementary Tables II and III), and had a similar distribution relative to gene landmarks as previously reported: the majority of binding sites are located in the intragenic regions (40%) and distant 5′ and 3′ regions, with only 9% in promoters (within 5 kb upstream and 1 kb downstream from the TSS) (see Supplementary Figure 2A and B). We subjected 81 sites to validation by ChIP-qPCR and found good correlation between ChIP-seq tag count and qPCR quantification (Supplementary Figure 3, P=5.0E-8). Other measures of validity such as correlation of ER occupancy with the presence of an ERE-recognition motif were also observed confirming the quality of the library (data not shown). Adjusting the threshold settings used, we found that majority of the binding sites were already bound in the absence of E2, albeit often at low levels, but almost all sites showed significantly augmented binding after E2 stimulation (Supplementary Figure 4). These data suggest that ER occupancy is commonly present before ligand exposure and this occupancy is enhanced by ligand induction.
Characteristics of chromatin configuration of ER-binding sites
Using the 16 043 ERα-binding sites, we analyzed the population characteristics of the chromatin configuration of these ERα-binding sites. To this end, we performed ChIP-seq analysis for the occupancy configuration of each of the following marks before and after E2 exposure: RNA Pol II, the activation marks H3K4me1, H3K4me3, H3K9ac and H3K14ac, and the repression marks H3K9me3 and H3K27me3. As FOXA1 has been suggested to be a pioneering factor (Carroll et al, 2005, 2006) that potentially can direct ER binding, we also assessed whether the presence of FOXA1 binding based on ChIP-seq occupancy in the absence of estradiol exposure might be a predictor of ER binding after ligand exposure. Because of our finding of AP1 as a common co-motif at ER-binding sites, we included the two components of the AP1 complex, FOS and JUN in the ChIP-seq analysis. The details of the antibodies, the depth and coverage of sequencing for each of the libraries are shown in Supplementary Table I. In addition, we assessed the chromatin configuration of ERα-binding sites by deeply sequencing the fragments isolated by Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) (Giresi et al, 2007) which enriches for nucleosome-free genomic DNA in the aqueous phase of a phenol extraction. The tag count of FAIRE fragments reflects the nucleosome depletion at any given site. From the tag profile of FAIRE libraries we found an open chromatin conformation within 1 kb around ER-binding sites after as well as before E2 application (Figures 1 and 2B). We observed a gradient for this open chromatin conformation with each quartile of ER-binding sites, with quartile 1 (highest ER occupancy) binding sites showing the maximum openness, and quartile 4 (least occupancy) binding sites showing the least. In contrast, the EREs with no ER binding (unbound sites) lacked this open chromatin conformation, either in the E2 induced (Figure 1) or non-induced state (data not shown).
Figure 1.

Chromatin activation and repressive mark profiles of ERα-binding sites after E2 stimulation. The 16 043 ERα-binding sites defined by ChIP-seq sequence tag occupancy were arranged in descending order of their induction and subsequently divided into four quartiles (Q1–Q4). The 1st quartile group contains the strongest induced ERα-binding sites while the 4th quartile group contains the weakest induced binding sites (colored lines: Q1, Q2, Q3 and Q4). The tags from each ChIP-seq library (downsampled to 7 M tags) were mapped and then used to calculate the average count per bp in intervals ±2 kb relative to the center of ER-binding sites (ERBS) identified from ChIP-seq study. For comparison, the same average tag count profile is shown for 10 000 random sites unbound by ER (gray lines).
Figure 2.

Estradiol induces chromatin changes around the ERα-binding sites (ERBS). (A) Correlations between ER chromatin immunoprecipitation (ChIP)-seq peak height and tag counts of chromatin modification marks before (vehicle) and after (E2) stimulation (* indicates significance level at P<0.0001 for all groups). All ChIP-seq libraries were of equal size (N=7 million tags) to make the signal directly comparable. ChIP-seq signals for repressive histone marks H3K9me3 and H3K27me3 have no significant correlation with ER-binding intensity. (B) Effect of estrogen on the chromatin signals with respect to different marks. ER-binding sites from quartile 1 were analyzed for different chromatin marks.
H3K4 mono- and trimethylations, and H3K9 or H3K14 acetylations are generally associated with regions of transcriptionally active chromatin (Bernstein et al, 2004). The presence of these activation histone marks pointed precisely at the ERα-binding site and was correlated with ERα binding (Figure 1). Of note is that H3K4me1 signals in the absence of ligand were correlated with ERα occupancy (Figure 2A), which is consistent with the association of H3K4me1 with sites of enhancer function. Such an association has been noted in human HeLa cells for the p300-binding sites (Heintzman et al, 2007; Robertson et al, 2008), STAT1, predicted enhancers and FOXA2 sites from mouse adult liver cells (Robertson et al, 2008). Similarly, the H3K9 and H3K14 acetylation marks were also progressively enriched around the ERα-binding sites. The signal for activation histone marks correlated with ER occupancy at ER-binding sites (Figures 1 and 2A, Supplementary Figure 5). In contrast to the ERα-bound sites, none of these histone activation marks were enriched in non-bound EREs. Previous studies suggested that methylation of H3K27 correlated with gene repression (Lee et al, 2006; Roh et al, 2006) and methylation of H3K9 has been implicated in heterochromatin formation and gene silencing (Bannister et al, 2001). Our analyses showed that these signals were very low for both bound and non-bound sites with no significant difference between the two states. Thus, the activation marks are associated with ER binding whereas the repression marks are not.
As the ChIP-seq technology may possess unknown biases, we sought to validate the profiles of FAIRE and H3K4me1 signals around ER-bound sites using a custom made NimbleGen array containing 12 966 validated binding sites that had ERE-like sequences derived from two published genome-wide studies (Carroll et al, 2006; Lin et al, 2007) and 31 468 non-binding sites bearing computationally predicted high-affinity binding sites (Vega et al, 2006). These ChIP-chip results showed the same positive correlation between ER binding and FAIRE or H3K4me1 enrichment (Supplementary Figure 6).
After determining the distribution of chromatin marks in E2 and vehicle-treated samples, we then asked whether there was a gradient of association between each of the histone and chromatin marks at the pre-ligand state and ERα binding following ligand activation. Significantly, we observed strong correlation between ERα occupancy and the intensity of all marks associated with gene activation, H3K4me1, H3K4me3, H3K9ac, H3K14ac, FAIRE and RNA Pol II, either before or after ligand exposure, based on rank correlation statistics (Figure 2A). By contrast, the associations between ERα occupancy and the repression marks were not statistically significant. Taken together, we surmise that bona fide ER-binding sites have the general characteristics of exhibiting open chromatin, harboring activation marks on histone 3 and co-localizing with RNA Pol II.
We then asked whether the characteristics of the ERα-binding sites changed upon ligand induction of ER. We compared chromatin marks on ChIP-seq-defined ER-binding sites (quartile 1) before and after E2 treatment. The results (Figure 2B) show that the FAIRE tag profiles are, on average, the same before and after ligand induction, which suggests that there is no significant change in the open chromatin configuration after ligand induction. In contrast, we found H3K9ac, H3K14ac signals and especially RNA Pol II signals were increased after ligand exposure (Figure 2B). The dramatic increase with RNA Pol II after ligand exposure suggests that Pol II is progressively recruited to ER-binding sites upon ER activation. The repression marks of H3K9me3 and H3K27me3 did not change with E2 exposure. H3K4me3 showed only a subtle increasing change. H3K4me1 decreased as these sites were probably progressively methylated to H3K4me2 and/or H3K4me3. The trends of estrogen effect on chromatin signals were maintained for binding sites from other quartiles (Supplementary Figure 7). These results suggest that there is a dynamic interplay between ERα binding and specific characteristics of the local chromatin configuration with the ERα binding altering the chromatin state of the binding site. RNA Pol II binding after E2 induction was the most correlated with ER binding after ER activation. By contrast, H3K4me1 (histone lysine monomethylation) was the most significantly correlated of these initial factors in the absence of ligand (Figure 2A).
It is known that certain histone marks are closely linked to specific gene boundaries such as promoter regions and, therefore, might be associated with ER binding by virtue of binding site location relative to gene boundaries. Therefore, we analyzed whether certain ERα-binding site-associated marks were specifically localized to such gene boundaries. We found that two histone modifications, H3K4me3 and H3K9ac, were present at much higher levels at promoter-associated ERα-binding sites (within 5 kb of TSS, Figure 2C). However, ChIP-seq enrichment of the remaining activation mark, H3K4me1, and the other binding site signatures (RNA Pol II and FAIRE) were significantly associated with ERα-binding sites regardless of their location relative to gene boundaries. Because of this positional bias to promoters, we posited that H3K4me3 and H3K9ac would not be the best predictive markers for ligand-induced ERα binding in genome scale, which was confirmed by prediction with cross-validation (Supplementary Tables VIII–XI).
Figure 2.

(C) The tag density profile for chromatin marks at the ERα-binding sites from different genomic locations relative to RefSeq genes: promoter (5 kb), intragenic, distal (5–100 kb upstream transcription start site; TSS) and gene desert. For both (B and C) the signal is average tag count in intervals ±2 kb of the center of the binding sites. Distance is shown in the x axis and the ChIP-seq tag count is presented in the y axis for each panel. Changes after E2 treatment are shown by arrows. For C, the symbol * indicates bias in ChIP-seq enrichment for promoter ERα-binding sites.
Co-localizing TFs
According to our motif analysis (see below), 52% of the 16K ER-binding sites were found to contain half-site ER-binding motif or no discernible motif. It has been suggested that co-localization of specific TFs with ER on DNA may contribute to the utilization of any site for ER binding. To this end, we examined the consensus TF-recognition sequences that are enriched in close proximity to bona fide ER binding half sites. Co-motif analysis was performed using MDscan (Liu et al, 2002) after masking out all ER half sites in the binding regions. Correlating the identified motifs with entries in TRANSFAC suggested that FOXA (presumably, FOXA1), AP1, AP2 and CACD (similar to SP1) factors were potentially associated with these sites (Supplementary Figure 8). When analyzed across all subcategories of ER-binding sites, the presence of the consensus motifs for these four TFs remain highly statistically significant as compared with random genomic segments except in the 2% of the sites that harbor no ERE motif. In this ERE-negative category, only FOXA1 and AP2 motifs were marginally enriched. Of interest is that the presence of AP1, AP2 and FOXA1 motifs were statistically more commonly adjacent to definite half sites than in full sites (P=0.0005 for AP1, P=0.0046 for AP2, and 0.025 for FOXA1 by Fisher's exact test) (Supplementary Figure 9, Supplementary Table IV).
The ER-binding sites were classified into four categories based on sequence motif (thermodynamic model scores, see below). Since, as the quality of the ER motif deteriorated from full sites to intermediate half sites, the likelihood of co-motif occurrence in the ER binding region increased uniformly for all four co-motifs (Supplementary Figure 9, Supplementary Table IV). Binding regions with no ERE showed no clear trend, perhaps because of the fact that they were too few in number to characterize statistically. The inverse relationship we observed between ER motif quality and co-motif occurrence is consistent with a model wherein low-affinity EREs are more likely to require assistance from other TFs in recruiting ER. This assistance could take the form of direct protein–protein interactions, as has been suggested for AP-1 and ER (Safe and Kim, 2008), or a cooperative or chromatin-modifying role as suggested for FOXA1 (Carroll et al, 2006).
Given the association of AP1 and FOXA1 with ER-binding sites described in the literature, we sought to assess the association of the conjoint Fos–Jun (components of the AP1 complex) and FOXA1 with ER-binding sites. We observed that the components of the AP1 complex, cFos and cJun, or the intersect of the two factors (representing the intact AP1) at vehicle treatment were poorly correlated with ER binding whereas the presence of FOXA1 was highly correlated. When taken together, at the pre-ligand state, FOXA1, H3K4me1, RNA Pol II and FAIRE had the highest correlation with subsequent E2-induced ER binding (Figure 2A).
Owing to technical variation, ChIP-seq libraries had different number of mappable binding sites (Supplementary Table I), which may alter the assessment of individual factors associated with ERα-bound sites. To account for this variance, we downsampled all histone modification ChIP-seq libraries to a consistent minimal available size (N=7 million) by random removing of reads and verified the obtained correlations and ROC-AUC scores (Supplementary Table VII).
Energetics in sequence selectivity
ERα binds preferentially to the consensus estrogen receptor element, a 13–19 bp palindromic sequence consisting of two oppositely oriented 5–8 bp ‘half sites’ separated by a 3-bp spacer. ERα can also bind to isolated half sites, though with less specificity. In order to systematically assess the binding affinity of half and full sites of the ∼16,000 ERα binding regions, we fitted a widely used thermodynamic model of TF–DNA binding to the ChIP-seq data (Foat et al, 2006; Zhao et al, 2009).
Our model was constructed by applying a new algorithm called Thermodynamic Modeling of chip-Seq (TherMoS) (Sun et al, manuscript in preparation; MATLAB code available upon request). The algorithm employs nonlinear regression to fit the observed distribution of sequence tags in a ChIP-seq library, and produces an estimate of the position-specific energy matrix (PSEM), i.e., the matrix of binding free energy changes induced by all possible single mutations of the consensus-binding sequence. Once trained, the model provides an estimate of the G-score, i.e., the binding free energy, of ERα to any sequence. The G-score can then be converted to an estimate of TF occupancy, or binding probability, using elementary statistical mechanics (see Materials and methods). In this manner, the appropriateness of any sequence to act as a substrate for ERα binding can be quantified and compared across sites.
As ERα has been known to bind palindromic EREs and also isolated half sites (Krishnan et al, 1994; Vega et al, 2006; Safe and Kim, 2008), we based our motif search on a two-dimensional score composed of the G-scores of the left and right 7-mers within a 17-mer ERα motif (Figure 3A). Enrichment of 17-mers within ±50 bp of ChIP-seq peaks, relative to randomly chosen non-coding regions, was evaluated in this two-dimensional score space (Figure 3B). The resulting enrichment plot shows that perfect or near-perfect matches to the palindromic ERE consensus (Figure 3B, bottom left corner) are highly enriched in ERα binding regions. These ‘full sites’ presumably bind both subunits of an ERα dimer. We also observe enrichment of half-site motifs, defined as 17-mers with a low G-score in only one half of the palindrome (Figure 3B, yellow streaks along the x and y axes), suggesting that there exists a significant population of binding sites at which only one ERα molecule is in direct contact with DNA. We found that the average asymmetry between left and right half site G-scores (GL and GR) decreased smoothly as the estimated 17-mer occupancy increased, indicating that ERα-binding sites exist on a continuum between half sites and palindromic full sites (Figure 3C; see Supplementary Methods). Based on the inflection point of this half site asymmetry plot, we defined ‘definite full sites’ as 17-mers with predicted occupancy >0.05. Intermediate full sites were defined as 17-mers with lower predicted occupancy than definite full sites, and more than twofold enrichment along the dotted diagonal line, i.e., 0.02<occupancy⩽0.05 (Figure 3D). A similar enrichment-based criterion gave GL/R <1.65 as the threshold for definite half sites (Figure 3E). Intermediate half sites were defined with a looser G-score threshold, and ‘no ERE’ ChIP-seq peaks were defined as binding regions in which not a single 17-mer above the threshold could be found.
Figure 3.

De novo ER motif discovery and definition of five subcategories of ERα-binding site. (A) Sequence logo of palindromic ER motif deduced by Thermodynamic Modeling of chip-Seq (TherMoS) from MCF-7 ChIP-seq data. (B) Binding free energy of 17-mers relative to that of the consensus-binding sequence is decomposed into contributions (G-scores) from the left (GL; x axis) and right (GR; y axis) half sites. The plot shows log10-scale enrichment of G-score pairs in 100 bp regions centered on ChIP-seq peaks, relative to randomly chosen non-coding regions in the genome. Predicted probability of binding, i.e., occupancy  τ=2.3396. Area I: definite full site (occupancy>0.05); area II: intermediate full site (0.02<occupancy⩽0.05); area III: definite half site; area IV: intermediate half site; area V: no ERE. (C) Occupancy threshold for definite full (palindromic) sites was defined as the value below which GL and GR become anti-correlated, i.e., asymmetric. (D) Occupancy threshold for intermediate sites corresponds to the point on the dotted diagonal line in B wherein the enrichment is twofold. The same point on the diagonal line is used to define the G-score threshold for intermediate half sites (dashdot line in B). (E) G-score threshold for definite half sites is the point on the dashed vertical line in B where the enrichment is twofold.
τ=2.3396. Area I: definite full site (occupancy>0.05); area II: intermediate full site (0.02<occupancy⩽0.05); area III: definite half site; area IV: intermediate half site; area V: no ERE. (C) Occupancy threshold for definite full (palindromic) sites was defined as the value below which GL and GR become anti-correlated, i.e., asymmetric. (D) Occupancy threshold for intermediate sites corresponds to the point on the dotted diagonal line in B wherein the enrichment is twofold. The same point on the diagonal line is used to define the G-score threshold for intermediate half sites (dashdot line in B). (E) G-score threshold for definite half sites is the point on the dashed vertical line in B where the enrichment is twofold.
We thus classified the 16 043 binding regions into definite full sites (31%), intermediate or possible full sites (17%), definite half sites (38%), intermediate half sites (12%) and binding sites that show no evidence of an ERE (2%) (Supplementary Figure 10A). Thus, most binding regions harbor sequences that are energetically recognizable as an ERE (86%, if one excludes intermediate half sites and no-ERE regions). We observed a linear relationship between the quartile assignment of the ERα-binding site and the probability of harboring a definite full site, suggesting that as the binding motif becomes progressively weaker, the strength of ER binding decreases (Supplementary Figure 10B). The appropriateness of the full site, half site and no-ERE categories was independently confirmed by de novo motif analysis using MEME (Bailey et al, 2009) (Supplementary Figure 10C). ERE-like sequences were discovered in all but the ‘no-ERE’ category, though the signal in the intermediate half site group was variable and considerably weaker requiring initialization from the consensus sequence AGGTCA. Thus, the binding site categories defined on the basis of free energy scores were independently confirmed using MEME. Overall, these results suggest that a significant gradient of progressive degeneracy exists in the bona fide binding sites for ERα.
Factors predicting ER-binding site utilization
It has been estimated that there are over 1 million potential ER-binding sites in the human genome as computationally identified by motif scans (Vega et al, 2006), but we noted only 16 043 true binding sites in the MCF-7 genome. Using a stringent TherMoS-based predicted occupancy threshold of 0.05 which represents only optimal or near-optimal full ERE motifs, we calculated that there are 229 663 sequence-optimal binding sites in the reference human genome. As the MCF-7 genome was found to contain only 5105 of these full-ERE regions that were bound by ERα, we estimate that only ∼2% of ‘full EREs’ in the genome are detectably bound in that cell type. Given that we had genome-wide data on epigenetic signatures of ER-binding sites, and a robust module for assessing DNA sequence characteristics for optimal ERα binding, we sought to identify specific characteristics of a DNA segment before E2 exposure that determine whether the site would be subsequently bound by ER after ligand addition. We constructed logistic regression models for characteristics best associated with ligand-induced ER binding using the ChIP-seq tag densities from the pre-ligand state in 500 bp windows for all chromatin marks (H3K4me1, H3K4me3, H3K9ac, H3K9me3, H3K14ac and H3K27me3), for FOXA1, c-Fos, c-Jun, FAIRE and RNA Pol II, and the occupancy score for the TherMoS-derived ER PSEM.
We assessed the classification performance of our predictive models using receiver-operator characteristic (ROC) curves (Figure 4). We fitted logistic regression models for all possible combinations of the 12 (see Supplementary Tables VIII–XI) features and recorded the best area under the ROC curve (ROC-AUC score) for each N-feature combination. The problem of modeling ER site selectivity can be approached from different angles depending on the specific question one wants to ask. First, we asked whether regions that are bound by ER in MCF-7 cells on E2 treatment could be distinguished from random genomic background. To eliminate any possible bias related to proximity to promoters, we used 14 338 non-TSS-proximal ER ChIP-seq determined binding sites (whether they contained an ERE or not) and used as control, 820 000 regions that neither overlapped with ER ChIP-seq peaks nor were near TSSs.
Figure 4.

Predictive factors of ERα binding. The values are averages of five runs and bars show standard errors. The curves are ROC curves for (A) logistic regression models (the best-performing one-, two-, three-, four-, and six-feature models; see Supplementary Table VIII) that discriminate between distal ER-bound (N=14 338) and random genomic (N=820 000) sites in MCF-7, (B) the performance of the best models (Supplementary Tables X and XI) versus three-parameter models using FOXA1, H3K4me1 and Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) in discriminating between bound and non-bound Thermodynamic Modeling of chip-Seq (TherMoS)-predicted EREs (transcription start site; TSS-proximal and distal EREs are plotted separately) in MCF-7, and (C) a validation of the logistic regression model derived from MCF-7 data tested on T47D data.
We assessed all possible combinations of all factors and found that a four-parameter model that included the TherMoS ER affinity score, FOXA1, H3K4me1, and FAIRE represented the most efficient in terms of identification of ligand-induced ER-binding sites resulting in an ROC-AUC of 0.95, as judged by testing on an independent test set (Supplementary Table VIII). This four-parameter combination had essentially the same area under the curve (AUC) for the ROC as the model using six features while the addition of more than six features caused the ROC-AUC to deteriorate because of over-fitting to the training set (Figure 4A). In addition, these four selected features always (five out of five validations) represented the top-scoring four-parameter model, while the best 5-, 6-, 7-feature (etc.) models varied between the five runs (Supplementary Table VIII). Thus, the four-parameter model is the most stable and parsimonious predictive model for ER-binding site selection. After having established that it is possible to model with a considerable degree of accuracy which distal regions will be occupied by ER after E2 treatment, we asked whether ER-bound proximal promoters could be distinguished from non-ER-bound proximal promoters. In this scenario, the best resulting logistic regression model (which used all the features) had a ROC-AUC in excess of 0.92 (Supplementary Figure 13). A model using only TherMoS score, H3K4me1 and FOXA1, though, reached the almost equivalent ROC-AUC score of 0.915 owing to the open chromatin nature of promoter proximal regions (Supplementary Table IX).
Next, we asked the following question: Given the presence of optimal thermodynamically predicted EREs, can we distinguish, based on chromatin marks, which will actually be ER bound after E2 treatment? To answer this, we took the 229 663 thermodynamically assigned full EREs as the ‘universe’ of EREs and attempted to predict the 5105 sites that would be experimentally bound. As the TherMoS affinity score had been used to define the training and test data sets, this parameter was omitted in the predictor. This restricted analysis revealed that the ROC-AUC values reached 0.88 for distal sites (see Supplementary Table X for the best N-parameter models) and 0.80 for TSS-proximal sites (Figure 4b and Supplementary Table XI). Thus, using the best ERE motifs, the presence of FOXA1 co-occupancy, H3K4me1 histone modification and open chromatin could identify the majority of the sites that would be selected by activated ER. Interestingly, FOXA1 was the most informative feature for discriminating TSS-proximal sites, while H3K4me1 was the most informative feature for the distal sites.
To validate this predictive model, we performed ChIP-seq analysis of ERα and FOXA1 binding, H3K4me1 and FAIRE status before and after ligand induction in a different ER positive breast cancer cell line, T47D. We then tested the four-parameter-binding site predictive model derived from MCF-7 on T47D cells using all T47D ChIP-seq ER peaks and found an ROC-AUC of 0.86 (Figure 4C). When the model was tested on only the TherMoS-predicted EREs in T47D and assessed for the ability to discriminate bound versus not bound ERE sites in T47D (using only the three features H3K4me1, TherMoS score and FOXA1), the resulting ROC-AUC for this task was 0.93. Thus, our ER binding model is predictive in two independent cell lines.
Interplay of ERE and chromatin state in determining ERα-binding sites in different cell lines
We asked the question whether differences in the binding sites selected in ER positive breast cancer cell lines could uncover any systematic hierarchies that control binding site selection. Of particular interest is whether ER-binding sites common in the two cell lines are systematically different from those sites found uniquely in either MCF-7 or T47D. Our results showed that of the 5421 binding sites in T47D, 3597 or 66% were in common with MCF-7-binding sites, while 12 446 sites were MCF-7 unique, with only 1824 sites being T47D unique. The specificity of these binding sites as common or cell line unique was confirmed using qPCR in 73 selected sites (Supplementary Figure 14). The ER-binding sites common to both cell lines had the highest levels of each of the four predictive parameters: TherMoS score, H3K4me1, FOXA1 occupancy and FAIRE. In the cell line unique sites, the thermodynamically modeled ER affinity scores, which reflect the fixed parameter dictated by sequence, were discernibly lower than in the common sites (Figure 5G). Consistent with this, we found that the common sites harbor the highest proportion of full and intermediate full EREs (62%) whereas the cell line unique sites have a lower proportion (from 25% in the case of T47D unique sites to 42% for MCF-7 unique sites) (Supplementary Figure 15). The distinction between the two cell line unique categories, however, is that despite similar low ER affinity scores, all other parameters (H3K4me1, FAIRE, and FOXA1 occupancy) were significantly enriched in the MCF-7 unique sites as compared with the T47D unique sites in MCF-7 cells (Figure 5A–C; see also Supplementary Figure 16 and 17). Our parallel ChIP-seq experiments in T47D cells confirmed higher enrichment of FAIRE and FOXA1 marks in the T47D unique ER-binding sites as compared with the values at the same sites in MCF-7 cells (Figure 5D–F, as compared with MCF-7 unique sites). Thus, these two factors clearly determine cell line specificity of ER binding. H3K4me1 has in average, lower enrichment in T47D unique ER-binding sites, although statistically higher than unbound ERE sites or background genomic segments in T47D cells. Taken together, this suggests that sites with optimum ERE motifs will be preferentially used across cell lines of the same lineage (breast cancer), and that sites with weaker recognition motifs will depend more greatly on the chromatin ‘milieu’ for site selection which may vary from cell line to cell line.
Figure 5.

Binding site selection between ER positive breast cancer cell lines. Average ChIP-seq tag count of H3K4me1, Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE), FOXA1 in MCF-7 (A–C) and T47D cells (D–F) correspondingly at ERα-binding sites (±250 bp proximity of ER site center) for common and specific sites for MCF-7 and T47D cell lines. ChIP-seq data before E2 activation and normalized to the ChIP library size. ERα-binding sites common to the cell line (common) are significantly highly enriched by chromatin marks from cell line specific binding sites in both cell types, as well as have higher sequence affinity (panel G, free energy-based ER affinity score). Common and MCF-7-specific sites are enriched by (A) H3K4me1 (B) FOXA1 and (C) FAIRE marks in MCF-7 cells, while T47D-specific ERα-binding sites have significantly lower ChIP-seq counts than MCF-7-specific sites (see P-values in Supplementary Table VI). (D–F) show ChIP-seq tag enrichment for H3K4me1, FOXA1 and FAIRE correspondingly, in T47D cells, where it is evident that FAIRE and FOXA1 in T47D unique ER-binding sites show significantly higher signal as compared with MCF-7 unique sites.
Discussion
A central problem in TF biology is how binding sites are selected given the near ubiquity of short and degenerate recognition motifs and the small fraction of high-affinity sites that are actually bound. Investigations in different TFs and their individual binding sites point to co-factors, heterodimerization partners and enhanceosome organization as potential contributors. Herein, we assess, on a genome-wide basis, the potential rules for binding sites selection using the nuclear hormone receptor, ERα, as the model system. We used logistic regression modeling on 12 features to characterize the binding site selection by ERα: the ChIP-seq tag densities from the pre-ligand state in 500 bp windows for all chromatin marks (H3K4me1, H3K4me3, H3K9ac, H3K9me3, H3K14ac and H3K27me3), for FOXA1, c-Fos, c-Jun, FAIRE and RNA Pol II, and the occupancy score for the thermodynamically derived ER PSEM which is based on the primary genomic sequence. The advantage of this system is the natural biochemical inducibility of ERα by its ligand, estradiol, which allows for assessment of potential binding sites in the uninduced state of the TF. This avoids the possibility that certain chromatin characteristics may have been recruited by the TF binding itself. This possibility was seen in our data set wherein RNA Pol II was dramatically recruited to ER-binding sites after ligand induction and would have been considered a major predictive factor. However, RNA Pol II binding in the pre-ligand state in genome scale was significantly more modest and its importance disappeared in the combined analysis.
Taken together, our observations reveal that the fixed presence of an optimal ERE, the pre-ligand state presence of the enhancer mark, H3K4me1, pre-ligand occupancy by the pioneering factor FOXA1, and an open chromatin configuration provide optimum prediction of ER binding at any specific site. Though the open chromatin configuration as determined by FAIRE had a role in the model building, its impact was modest compared with the other parameters. Previous work has shown that H3K4me1 is associated with enhancer function (Heintzman et al, 2007) and, therefore, is consistent with the action of ERα. Though there is an association with activation marks H3K9ac, H3K14ac and ERα binding, these marks do not add further predictive power to the four-parameter model. This predictive model was validated on another estrogen-responsive cell line, T47D with a similarly high performance characteristic (ROC-AUC=0.86).
The comparisons between experimental binding sites that are shared between the two ER positive breast cancer cell lines (common to MCF-7 and T47D), and those sites that are present only in the individual cell lines (cell line unique binding sites) provide evidence for a differential impact of chromatin configuration depending on the strength of the binding site motif. This suggests a hierarchy of site usage such that those sites with energetically optimal binding motifs will be preferentially used across different cell types assuming the presence of the appropriate TFs. Instead of a clear binding/non-binding demarcation of sequences for the ERE, there appears to be a continuous probability gradient for ERE-like sequences of considerable degeneracy to bind with ERα. We observed that the less optimal motifs, which still represent a significant number of sites, are subject to greater influence by chromatin effects for ER-binding site utilization. This observation was underscored by the fact that binding sites with full ERE motifs were marginally aided by the other chromatin characteristics in the predictive model. By contrast, the binding sites with lower motif scores (e.g., half-ERE sites) needed the addition of chromatin information to achieve optimal prediction. Primary sequence is the structurally immutable component of the four parameters that define our binding site model. Of note is that even FOXA1 binding appears to be guided by the common presence of a FOXA1 response element in close vicinity to the ER-binding site. This suggests a model wherein the sites that have the best ERα binding by sequence may be destined to be the preferred binding sites across different cell lines. The variations across cell lines would then reside in those binding sites with suboptimal motifs (e.g., definite and intermediate half-ERE sites). If we assume that the union of ERα-binding sites in T47D and MCF-7 represent the universe of detectable ER-binding sites using our conditions, then up to 80% of these sites harbor less ‘optimal’ binding motifs. The ERα binding at this set would be significantly modulated by the chromatin configuration and the presence of activation and enhancer histone marks.
A few studies have previously reported integration of chromatin structure, epigenetic marks, TF binding and sequence motifs into predictive models of binding site selection. Cheng et al (2009) performed a study focused on the erythroid TF GATA1 based on two histone marks and restricted the cofactor ChIP experiments to chromosome 7. In addition, their analysis was correlative rather than discriminative. Ernst et al (2010) introduced the concept of ‘General Binding Preference’ (GBP)—a TF-independent measure of genomic properties that can be combined with a specific motif to more sensitively detect binding sites. In contrast to our ER- and MCF-7-centric study, this is a general approach to TF binding. In order to compare our method to that of Ernst et al, we defined more than 280 000 high-scoring putative ER-binding sites using the ER_Q6 PWM from TRANSFAC, and attempted to predict which of those that would actually be bound in MCF-7 genome (after E2) using either the GBP or our MCF-7-specific data (Supplementary Table XII). The comparison showed that while the GBP is a good predictor of binding (ROC-AUC=0.788) despite its cell type independence, it is not good at H3K4me1 occupancy before E2 (ROC-AUC=0.848) or the combination of the signals we have identified as the most predictive (H3K4me1 combined with FOXA1 occupancy; ROC-AUC=0.861). It is clear that the GBP score captures some aspects of chromatin structure which are partly predictive of TF binding, and this score may be a helpful tool for binding prediction when combined not only with motif information (as in Ernst et al, 2010) but also with cell-type-specific information.
When taken together, we have developed a highly predictive model for ERα-binding site selection that takes into account binding site motif degeneracy and specific chromatin characteristics. The use of a ligand inducible TF such as ERα permits the ascertainment of the relative impact of factors that determine the binding of the TF under pre-ligand and inactivated conditions. Our observations point to an interplay between the strength of recognition motifs and the intensity and combinations of chromatin characteristics at putative binding sites especially at the majority of sites not bearing optimal recognition motifs. Integrating these data defines a system that uses definable rules to achieve the common outcome of ERα binding to specific genomic sites.
Materials and methods
Cell culture, estradiol treatment, and preparation of FAIRE and ChIP DNA samples
MCF-7/T47D cells were grown to 70–80% confluence in D-MEM/F-12 (Invitrogen/Gibco) (for MCF-7) or RPMI medium 1640 (for T47D) (Invitrogen /Gibco) supplemented with 10% FBS (Hyclone) in 150 mm dishes. In preparation for the 17 beta-estradiol (‘estrogen/E2’, Sigma) treatment, cells were then split into 1:3 into serum starving medium (phenol red-free D-MEM/F-12 medium (Invitrogen/Gibco) or RPMI medium 1640 supplemented with 5% charcoal-dextran stripped FBS; Hyclone) and cultured for 72 h to 70–80% confluence. Hormone-depleted cells were treated with E2 to a final concentration of 10 nM for 3 h before the ChIP/FAIRE procedure. The control cells were treated with an equal volume and concentration of vehicle, DMSO (Merck), for 3 h. For a ChIP/FAIRE experiment, we routinely used ∼1 × 108 cells from 5 to 6, 150 mm diameter cell culture plates. For isolating the open chromatin regions in the genome we performed FAIRE as described previously (Giresi et al, 2007). All chromatin immune precipitation (ChIP) experiments were carried out as described earlier (Lin et al, 2007). Details of the antibodies used for ChIP preparations are given in Supplementary Table I.
Sequencing of ChIP/FAIRE enriched DNA samples and data analysis
ChIP or FAIRE enriched DNA was further processed before subjecting to ChIP-seq library construction as per the Illumina Solexa ChIP-seq sample processing methods. The processed ChIP or FAIRE-enriched DNA fragments were then used for Illumina single read sequencing analysis. The sequence tag data was mapped to hg18 genome, and enrichment peaks for corresponding libraries were identified using ChIP-seq peak calling algorithm as previously described (Chen et al, 2008). For the ERα libraries, the identified peaks were filtered in three steps (see Supplementary Methods) and resulted in 16 043 ERα-binding sites (under E2 stimulation) from non-amplified regions, for the downstream analysis.
ChIP-seq data are available from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) under accession numbers GSE23701 and GSE23893.
Chromatin profiles
The tag density profile for each chromatin mark was constructed in intervals of ±2 kb of the centre of ER-binding sites at the same scale. The signal for each chromatin mark represents the average number of ChIP-seq tags for each group of binding sites. To compare profiles across ChIP-seq libraries for each library data were normalized (sampling to minimal available library size by removing excess sequence reads from the count).
Motif search
We initially analyzed the presence of the ERE motif in the ERα-binding sites identified in this study, according to a previously described method (Lin et al, 2007). We then sought to leverage our ChIP-seq data to construct the ERE motif in greater detail using the TherMoS algorithm (Sun et al, manuscript in preparation). Instead of the traditional position-specific scoring matrix, the algorithm fits an explicit thermodynamic model of TF–DNA binding (PSEM) to ChIP-seq data. The PSEM ΔΔGij represents the free energy contribution of each possible nucleotide i at position j in the binding site. The total binding energy (G-score) of any particular n-mer is simply obtained by summing over the free energy contributions from each nucleotide in the n-mer. In the case of palindromic motifs for homodimer binding, it is convenient to split the G-score into the contributions from the left (GL) and right (GR) half sites. The probability that a given DNA sequence is bound, i.e., the ‘occupancy’ of the sequence, is given by 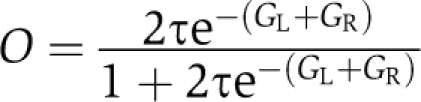 , where τ is a scale factor proportional to the intranuclear TF concentration (Zhao et al, 2009). We used this thermodynamic model to quantify ER-binding affinity at 16 043 binding regions identified by ChIP-seq in the non-amplified MCF-7 genome (see Supplementary Methods). We also systematically analyzed the set of TFs that modulate estrogen receptor function, by examining co-occupant proteins that might be enriched at the ERE half sites or no-ERE sites defined by TherMoS analysis, using MDscan (Bailey et al, 2009) (see Supplementary Methods). Genome coordinates of ER-binding sites in MCF-7 and T47D cells and corresponding background ERE sets together with binding affinity scores are available at the website http://www.gis.a-star.edu.sg/~liue/sup/ and as Supplementary Dataset 1.
, where τ is a scale factor proportional to the intranuclear TF concentration (Zhao et al, 2009). We used this thermodynamic model to quantify ER-binding affinity at 16 043 binding regions identified by ChIP-seq in the non-amplified MCF-7 genome (see Supplementary Methods). We also systematically analyzed the set of TFs that modulate estrogen receptor function, by examining co-occupant proteins that might be enriched at the ERE half sites or no-ERE sites defined by TherMoS analysis, using MDscan (Bailey et al, 2009) (see Supplementary Methods). Genome coordinates of ER-binding sites in MCF-7 and T47D cells and corresponding background ERE sets together with binding affinity scores are available at the website http://www.gis.a-star.edu.sg/~liue/sup/ and as Supplementary Dataset 1.
Predicting sites that will be bound by ER after ligand induction
We used logistic regression to assess how well various chromatin features were able to discriminate between ER bound and non-bound regions in three different scenarios or ‘classification tasks’. The features were either an ER affinity score (see main text) or tag count from a ChIP-seq library downsampled to minimal size 7 million tags (for MCF-7 libraries) or 12.5 million tags (for T47D libraries) for unbiased comparison of predictive chromatin marks. Logistic regression was performed using the ‘lrm’ command in R. The predictive performance of the resulting models was summarized using precision/recall and receiver operating characteristic (ROC) curves generated using the ROCR package for R (Sing et al, 2005). In classification task 1, 70% of the data was used for model construction and 30% was then used as the test set. For the TherMoS ER affinity scores, we needed to make sure that data that had been used to fit the thermodynamic model was not in any way involved in the fitting or evaluation of the predictive models. Therefore, we fitted a PSEM five times, each time using 80% of the data set, and used the resulting PSEM to score the remaining 20%. These five non-overlapping sets of 20% each were then divided into a training set (70%) for fitting the logistic regression model and a test set (30%) for evaluating the accuracy of the model. The resulting ROC and precision-recall curves are thus averages of five rounds of this procedure.
Supplementary Material
Supplementary information, Supplementary Figures S1–17, Supplementary Tables SI–XIII
Acknowledgments
We thank Kartiki V Desai, Vinsensius B Vega., Sebastian Pott, Vipin Narang, Haixia Li, Han Xu, Yew Kok Lee, Tze Howe Charn, and RKM Karuturi for technical assistance, advice and science discussion. This research was supported by A*STAR of Singapore funds and also in part by the 6th European Community Framework Program grant CRESCENDO (FP6-018652) to ETL. Author contributions: RJ and ETL designed the experimental strategy. RJ, prepared histone ChIP samples, FAIRE DNA (MCF-7 cells), and performed ChIP-chip microarray validation. YFP, and EC prepared ERα (MCF-7 and T47D) and histone ChIP samples (T47D cells). SLK prepared ChIP samples for Fos, Jun, FOXA1 libraries (MCF-7) and FAIRE (T47D). YLO, MH and NDC performed major computational and statistical analyses. YLO, MH, RJ, SP, and ETL analyzed experimental data and interpreted the results. LU and JST performed validation experiments in MCF-7 and T47D cells. ML, WS and SP performed the TherMoS analysis of ER binding affinity and co-localizing motifs. YR provided support for Solexa sequencing of samples. ETL, RJ, YLO, MH and SP wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Ali S, Coombes RC (2000) Estrogen receptor alpha in human breast cancer: Occurrence and significance. J Mammary Gland Biol Neoplasia 5: 271–281 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL (2004) Global nucleosome occupancy in yeast. Genome Biol 5: R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR (2007) Chromatin remodeling: insights and intrigue from single-molecule studies. Nat Struct Mol Biol 14: 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M (2005) Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122: 33–43 [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M (2006) Genome-wide analysis of estrogen receptor binding sites. Nature Genetics 38: 1289–1297 [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK et al. (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117 [DOI] [PubMed] [Google Scholar]
- Cheng AS, Jin VX, Fan M, Smith LT, Liyanarachchi S, Yan PS, Leu YW, Chan MW, Plass C, Nephew KP, Davuluri RV, Huang TH (2006) Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol Cell 21: 393–404 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wu W, Kumar SA, Yu D, Deng W, Tripic T, King DC, Chen KB, Zhang Y, Drautz D, Giardine B, Schuster SC, Miller W, Chiaromonte F, Zhang Y, Blobel GA, Weiss MJ, Hardison RC (2009) Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res 19: 2172–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Narang V, Yap ZH, , Tusi BK, , Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola S, Testa G, Sung WK, Wei CL, Natoli G (2009) Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. The EMBO J 28: 3341–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Plasterer HL, Simon I, Bar-Joseph Z (2010) Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome Res 20: 526–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham PJ (2009) Insights from genomic profiling of transcription factors. Nat Rev Genet 10: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foat BC, Morozov AV, Bussemaker HJ (2006) Statistical mechanical modeling of genome-wide transcription factor occupancy data by Matrix REDUCE. Bioinformatics 22: e141–e149 [DOI] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EGY, Huang PYH, Welboren WJ, Han Y, Ooi HS, Ariyaratne PN, Vega VB, Luo Y, Tan PY, Choy PY et al. (2009) An oestrogen-receptor-α-bound human chromatin interactome. Nature 462: 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD (2007) FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PD, Horz W (1998) Chromatin and transcription—how transcription factors battle with a repressive chromatin environment. Eur J Biochem 251: 9–18 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (2008) Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 456: 663–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Wang X, Safe S (1994) Estrogen receptor-Spl complexes mediate estrogen-induced Cathepsin D gene expression in MCF-7 human breast cancer cells. J Biol Chem 269: 15912–15917 [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K et al. (2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125: 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C (2007) A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39: 1235–1244 [DOI] [PubMed] [Google Scholar]
- Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, Yeo A, George J, Kuznetsov VA, Lee YK, Charn TH, Palanisamy N, Miller LD, Cheung E, Katzenellenbogen BS, Ruan Y et al. (2007) Whole genome cartography of estrogen receptor α binding sites. PLOS Genet 3: e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Brutlag DL, Liu JS (2002) An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 20: 835–839 [DOI] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M (2008) FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132: 958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna NJ, O’Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108: 465–474 [DOI] [PubMed] [Google Scholar]
- Morse RH (2003) Getting into chromatin: how do transcription factors get past the histones? Biochem Cell Biol 81: 101–112 [DOI] [PubMed] [Google Scholar]
- Pan YF, Wansa KD, Liu MH, Zhao B, Hong SZ, Tan PY, Lim KS, Bourque G, Liu ET, Cheung E (2008) Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements. J Biol Chem 283: 32977–32988 [DOI] [PubMed] [Google Scholar]
- Petesch SJ, Lis JT (2008) Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell 134: 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Ahmad K (2007) Rules and regulation in the primary structure of chromatin. Curr Opin Cell Biol 19: 250–256 [DOI] [PubMed] [Google Scholar]
- Robertson AG, Bilenky M, Tam A, Zhao Y, Zeng T, Thiessen N, Cezard T, Fejes AP, Wederell ED, Cullum R, Euskirchen G, Krzywinski M, Birol I, Snyder M, Hoodless PA, Hirst M, Marra MA, Jones SJ (2008) Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding. Genome Res 18: 1906–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Cui K, Zhao K (2006) The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci USA 103: 15782–15787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S, Kim K (2008) Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol 41: 263–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132: 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadeo A, Lam WL (2006) Comprehensive copy number profiles of breast cancer cell model genomes. Breast Cancer Res 8: R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T (2005) ROCR: visualizing classifier performance in R. Bioinformatics 21: 3940–3941 [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- Vega VB, Lin CY, Lai KS, Kong SL, Xie M, Su X, Teh HF, Thomsen JS, Yeo AL, Sung WK, Bourque G, Liu ET (2006) Multiplatform genome-wide identification and modeling of functional human estrogen receptor binding sites. Genome Biol 7: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG (2009) ChIP-Seq of ERα and RNA polymerase II defines genes differentially responding to ligands. The EMBO J 28: 1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Granas D, Stormo GD (2009) Inferring binding energies from selected binding sites. PLOS Comput Biol 5: e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information, Supplementary Figures S1–17, Supplementary Tables SI–XIII