

Abstract
Sleep apnea is characterized by increased sympathetic activity and is associated with systemic hypertension. Angiotensin (Ang) peptides have previously been shown to participate in the regulation of sympathetic tone and arterial pressure in the hypothalamic paraventricular nucleus (PVN) neurons. We investigated the role of endogenous Ang peptides within the PVN to control blood pressure in a rat model of sleep apnea-induced hypertension. Male Sprague Dawley rats (250g), instrumented with bilateral guide cannulae targeting the PVN, received chronic infusion of Ang antagonists (A-779, Ang-(1-7) antagonist; losartan and ZD7155, AT1 antagonists; PD123319, AT2 receptor antagonist, or saline vehicle). A separate group received an infusion of the GABAA receptor agonist (muscimol) to inhibit PVN neuronal activity independent of angiotensin receptors. After cannula placement, rats were exposed during their sleep period to eucapnic intermittent hypoxia (IH; nadir 5% O2; 5% CO2 to peak 21% O2; 0% CO2) 20 cycles/hour, 7 hours/day, for 14 days while mean arterial pressure (MAP) was measured by telemetry. In rats receiving saline, IH exposure significantly increased MAP (+12±2 mmHg vs Sham −2±1 mmHg P<0.01). Inhibition of PVN neurons with muscimol reversed the increase in MAP in IH rats (MUS: −9±4 mmHg vs vehicle +12±2 mmHg; P<0.01). Infusion of any of the Ang antagonists also prevented the rise in MAP induced by IH (A-779: −5±1 mmHg, losartan: −9±4 mmHg, ZD7155: −11±4 mmHg and PD123319: −4±3 mmHg; P<0.01). Our results suggest that endogenous Ang peptides acting in the PVN contribute to IH-induced increases in MAP observed in this rat model of sleep apnea-induced hypertension.
Classification terms: hypothalamic-pituitary-adrenal regulation, peptides: anatomy and physiology, cardiovascular regulation
Keywords: sympathetic activity, intermittent hypoxia, hypertension, renin-angiotensin system, angiotensin-(1-7)
Introduction
Chronic intermittent hypoxia (CIH), a characteristic of human sleep apnea, leads to sustained elevation of sympathetic nerve activity (SNA) and mean arterial pressure (AP). Although the mechanisms by which CIH augments AP are incompletely understood, activation of the sympathetic nervous system seems to be essential for the maintenance of hypertension (Sica et al., 2000; Fletcher, 2003; Prabhakar et al., 2005). Indeed, several methods of sympathetic blockade, including carotid body denervation, renal sympathectomy, adrenal demedullation and sympathetic nerve ablation prevent the rise in AP due to CIH exposure (Fletcher et al., 1999; Fletcher, 1992a).
The hypothalamic paraventricular nucleus (PVN) is an important site regulating arterial pressure and sympathetic activity (Dampney, 1994; Tagawa & Dampney, 1999;). Inhibition of PVN neurons with nitric oxide donor (Allen, 2002; Horn et al., 1994; Stern et al., 2003) or γ-aminobutyric acid (GABA) receptor agonist (Kannan et al., 1989; Allen, 2002; Silva et al., 2005), reduces renal SNA and systemic AP. Conversely, activation of PVN neurons by microinjection of an excitatory amino acid (Kannan et al., 1989) or GABAA-receptor antagonist, increases arterial pressure and sympathetic activity in anesthetized (Chen et al., 2003; Chen & Toney, 2003) and conscious (Kannan et al., 1989) rats. Several studies have suggested that augmented discharge of PVN neurons may be involved in the sustained sympathoexcitation in pathological conditions including congestive heart failure (Zucker et al., 2004), myocardial infarction (Zhang et al., 2001; 2002), salt-sensitive hypertension (Weiss et al., 2007), and genetic hypertension (Allen, 2002).
Angiotensin (Ang) peptides act as important neuromodulators in brain sites controlling sympathetic output and arterial pressure (Dampney et al., 2002; Veerasingham & Raizada, 2003). Specifically in the PVN, iontophoretic application of Ang II and Ang-(1-7), two peptides produced by the renin-angiotensin system (RAS) cascade, increase the firing rate of neurons by acting on specific receptors (Ambuhl et al., 1994; Cato & Toney, 2005). We have previously reported that both Ang II and Ang-(1-7) contribute to the maintenance of renal SNA and that microinjections of these peptides into the PVN increase sympathetic tone (Silva et al., 2005). In this study we investigated the contribution of Ang II and Ang-(1-7) receptors in the PVN to hypertension developed during fourteen days of IH exposure.
Results
IH exposure and GABAA inhibition
Animals were subdivided after histological analysis of cannulae placement: those with injection sites located within or on the borders of the PVN were the experimental group; animals with injection sites outside the PVN were analyzed as a "control" group. Table 1 shows baseline values of MAP and HR before IH or Sham exposure for all experimental groups.
Table 1.
Basal values for MAP and HR before IH or sham xposure with drug infusion into the PVN.
| Experimental Group |
n | MAP (mmHg) |
HR (bpm) |
|---|---|---|---|
| Sham Saline | 4 | 111 ± 4 | 400 ± 11 |
| IH Saline | 5 | 113 ± 4 | 415 ± 4 |
| Sham A-779 | 6 | 116 ± 1 | 408 ± 10 |
| IH A-779 | 6 | 118 ± 2 | 419 ± 12 |
| Sham Losartan | 4 | 113 ± 4 | 431 ± 16 |
| IH Losartan | 5 | 115 ± 3 | 437 ± 8 |
| Sham ZD7155 | 5 | 112 ± 4 | 427 ± 7 |
| IH ZD7155 | 6 | 114 ± 3 | 423 ± 3 |
| Sham PD123319 | 6 | 106 ± 4 | 428 ± 6 |
| IH PD123319 | 5 | 113 ± 4 | 428 ± 9 |
| IH Muscimol | 5 | 113 ± 3 | 433 ± 7 |
Values are expressed as mean ± SEM.
IH exposure significantly increased MAP in vehicle-treated rats compared to vehicle-treated Sham rats (IH: +12±2 mmHg vs Sham: −2±1 mmHg; P<0.01; Figure 1).
Figure 1.
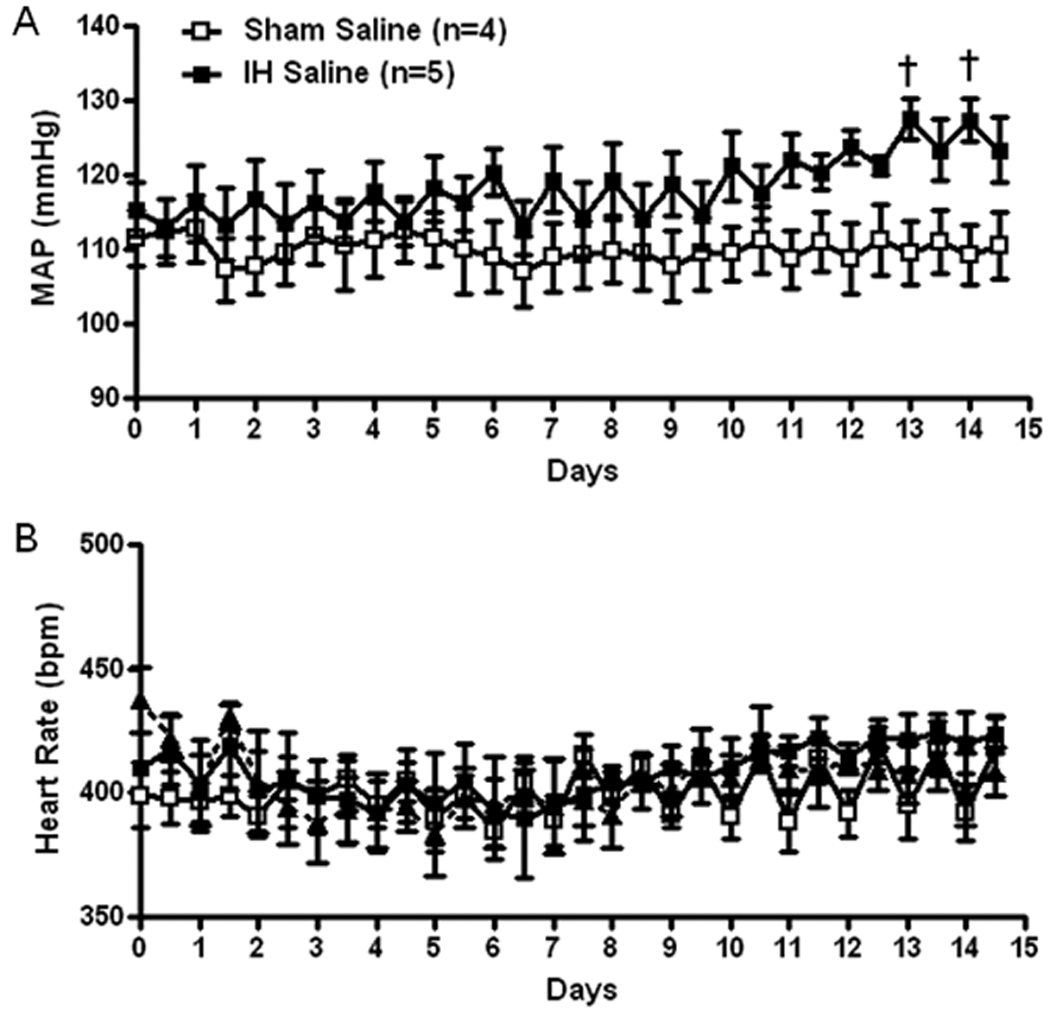
Grouped data showing 12 hour average recordings of mean arterial pressure (MAP, A) and heart rate (B) in Sham and IH exposed rats, chronically infused with saline (200 nL/hr) into the PVN. Daytime MAP and heart rate correspond to X-axis whole numbers while night time MAP and heart rate corresponds to values plotted between numbers.
Angiotensin receptor blockade
Chronic blockade of Ang(1-7) receptors with A-779 prevented an increase in MAP in the IH exposed rats so that MAP was not different from Sham exposed rats on any day (Figure 2A). Heart rate was also not different between groups (Figure 2B). The AT2 receptor antagonist also prevented an increase in MAP in the IH exposed group but MAP still tended to be higher in the compared to Sham rats receiving PD123319 (Figure 2C). The AT2 receptor blocker also tended to decrease HR and HR was significantly lower than during the control period on day 14 in both groups (Figure 2D). Figure 3 shows MAP and HR in Sham and IH rats receiving chronic infusion of the AT1 receptor blockers, losartan (Figure 3A and 3B) or ZD7155 (Figure 3C and 3D). Both antagonists reduced MAP in the IH and Sham exposed rats compared to vehicle treated rats but did not change HR. In the rats receiving the AT1 receptor blockers, MAP was not different between Sham and IH rats.
Figure 2.
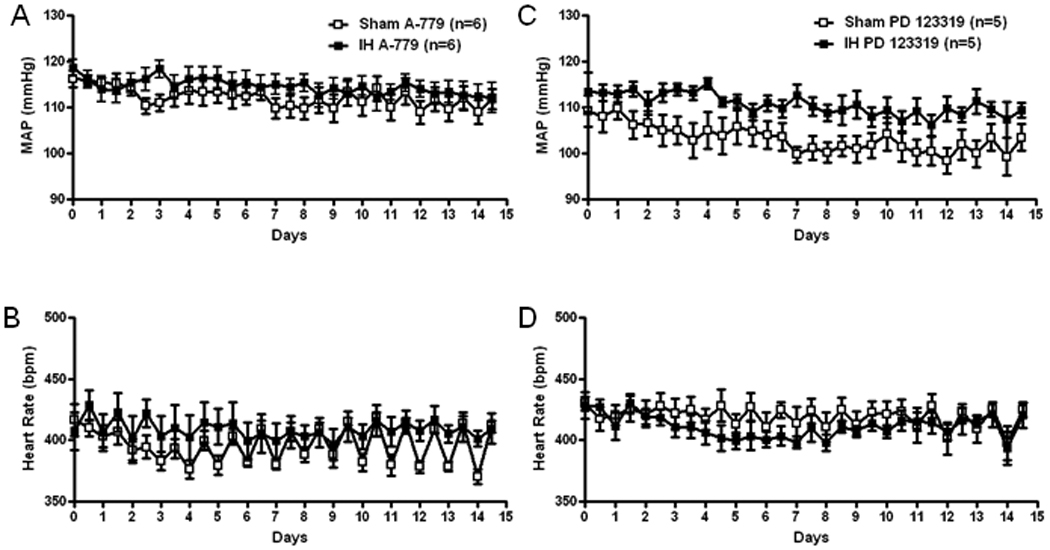
Grouped data showing 12 hour average recordings of mean arterial pressure (MAP) and heart rate (HR) in Sham and IH exposed rats chronically infused with A-779 (A and B, 2 nmol/h) or PD 123319 (C and D, 2 nmol/h) into the PVN. Daytime MAP (A and C) and heart rate (B and D) correspond to X-axis whole numbers while nighttime MAP and heart rate correspond to values plotted between numbers.
Figure 3.
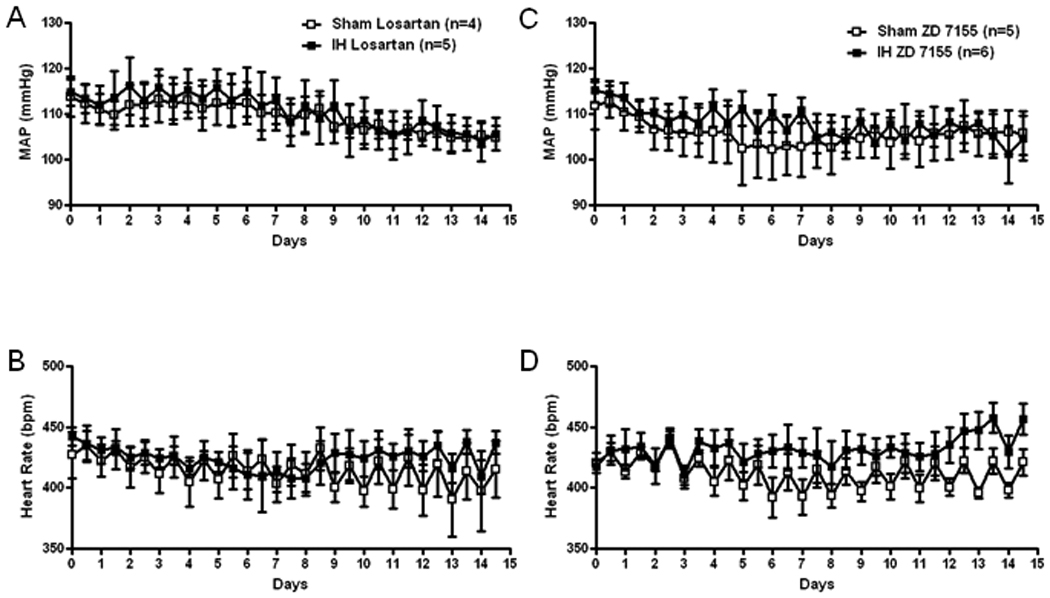
Grouped data showing 12 hour average recordings of mean arterial pressure (MAP, A and C) and heart rate (B and D) in Sham and IH exposed rats chronically infused with losartan (A and B, 2 nmol/h) or ZD 7155 (C and D.0.2 nmol/h) into the PVN. Daytime MAP and heart rate correspond to X-axis whole numbers while nighttime MAP and heart rate corresponds to values plotted between numbers.
Similar to the AT1 receptor antagonists, the GABAA receptor agonist, muscimol, prevented the increase in MAP and decreased HR in IH rats and tended to lower MAP in the Sham exposed rats so that MAP was not different between SHAM and IH rats treated with muscimol (Figure 4).
Figure 4.
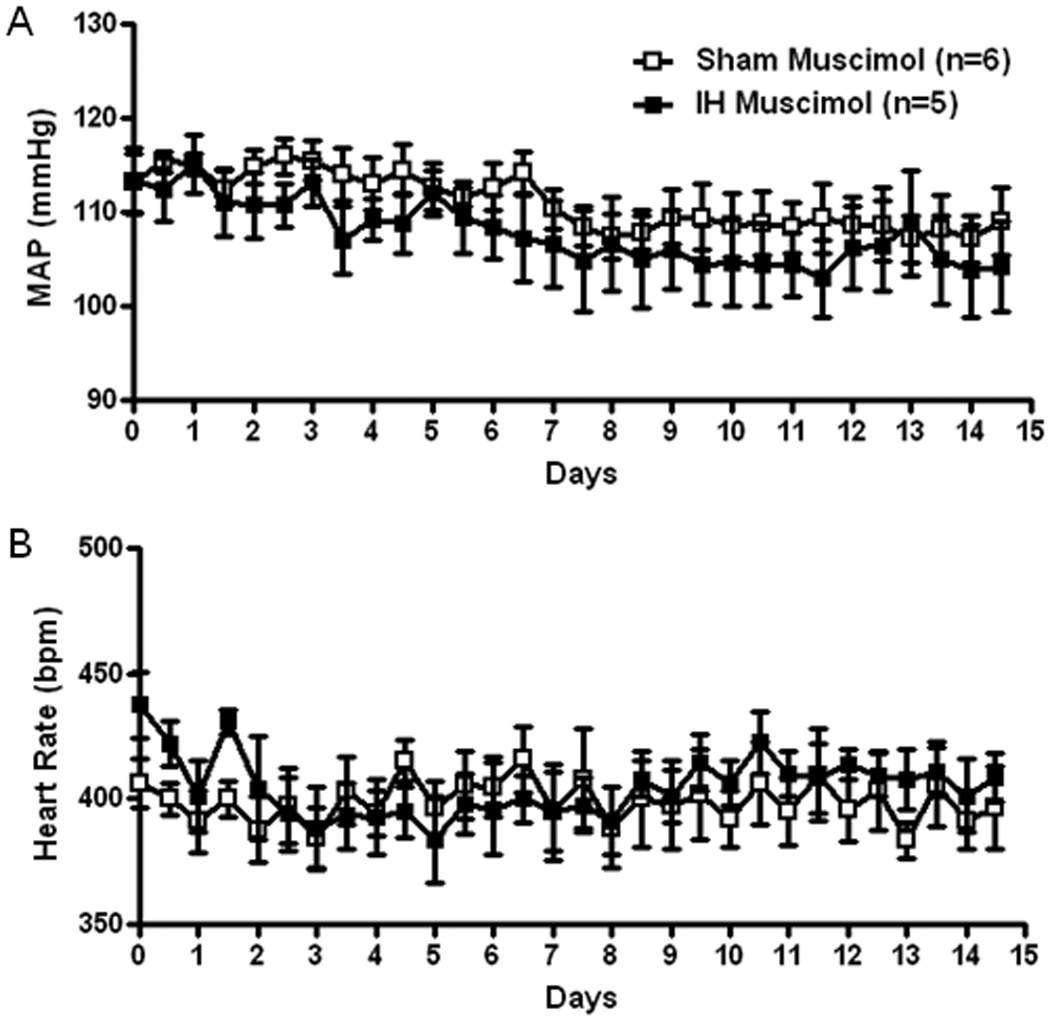
Grouped data showing 12 hour average recordings of mean arterial pressure (MAP, A) and heart rate (B) in Sham and IH exposed rats chronically infused with muscimol (20 pmol/h) into the PVN. Daytime MAP and heart rate correspond to X-axis whole numbers while nighttime MAP and heart rate corresponds to values plotted between numbers.
Figure 5 shows MAP and HR at day 7 and 14 in Sham and IH exposed rats receiving chronic infusion of saline, A-779, losartan, ZD7155 or PD123319 bilaterally into the PVN. IH exposure induced a gradual increase in MAP peaking at day 14, as shown in Figure 1. Chronic treatment with A-779 and ZD7155 lowered MAP compared to vehicle treatment at day 7 (A-779: −3±1 mmHg and ZD7155: −6±1 mmHg vs vehicle: +3±1 mmHg; P<0.01) and all angiotensin antagonists lowered MAP even more by day 14 completely preventing the increase in MAP observed in the vehicle group at day 14 (A-779: −5±1 mmHg, losartan: −9±4 mmHg, ZD7155: −11±4 mmHg and PD123319: −4±3 mmHg vs vehicle: +12±2 mmHg; P<0.01). HR did not change in any group, but ZD7155 infusion induced a significant increase in HR compared to the other angiotensin antagonists (ZD7155: +21±9 bpm vs A-779: −14±6 bpm, losartan: −12±8 bpm and PD123319: −18±4 bpm; P<0.01; at day 14).
Figure 5.
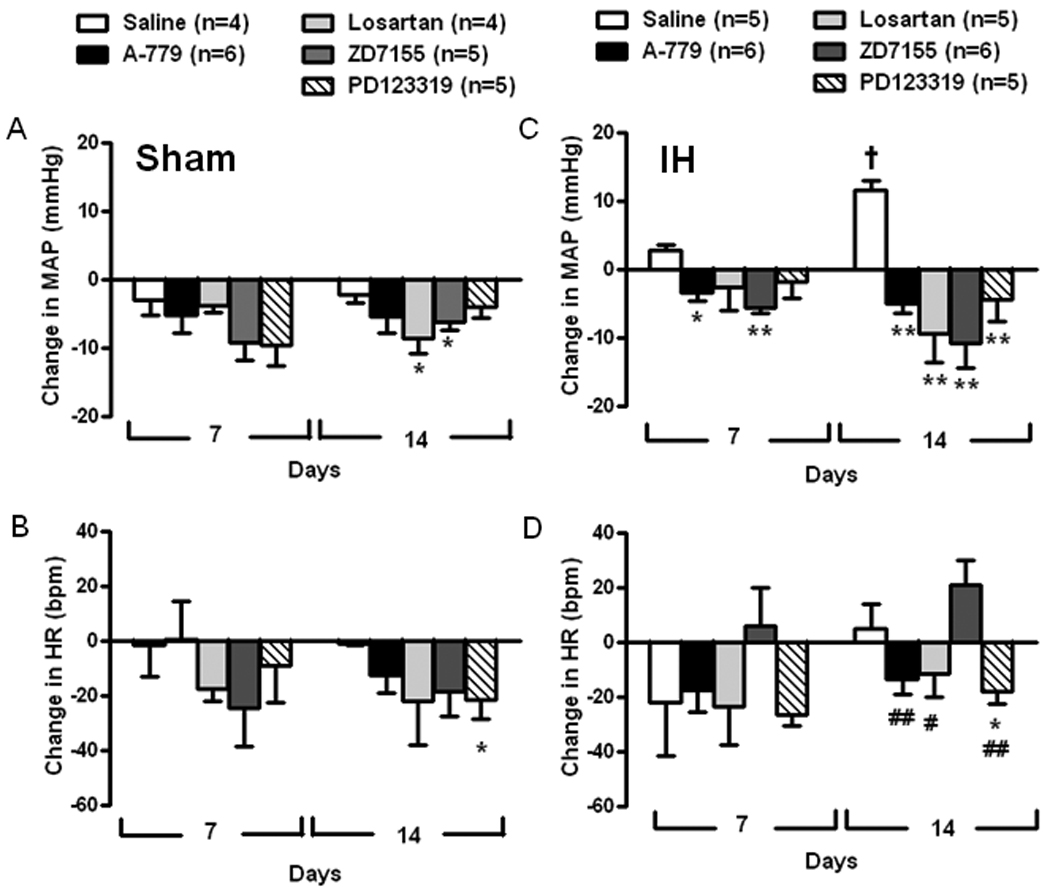
Grouped data showing the change in mean arterial pressure (MAP) and heart rate (HR) on days 7 and 14 of Sham exposed rats (A and B) and IH exposed rats (C and D) chronically infused with saline (200 nL/h), A-779 (2 nmol/h), losartan (2 nmol/h), ZD7155 (0.2 nmol/h) or PD123319 (2 nmol/h) into the PVN. Values are expressed as the averages of the change from starting MAP for each rat. *P<0.05 and **P<0.01 compared to saline; #P<0.05 and ##P<0.01 compared to IH ZD7155; †P<0.01 for IH saline on day 14 compared to day 7.
Angiotensin antagonist infusion into sites completely outside PVN borders had no effect on the IH exposure-induced rise in MAP or in HR demonstrating that the effects of Ang blockade are due to antagonist-receptor interactions within the PVN (Supplemental Figure 1A). Examples of the injection sites considered within and out of the PVN are illustrated in Supplemental Figure 1B
The activity data for the two groups indicated a tendency for all of the IH exposed rats independent of antagonist treatment to move less during the dark hours (awake) and more during the light hours (sleep/exposure time) but due to the between animal variability, there were no significant differences in activity between groups or within groups over time (data not shown). Therefore the differences in MAP and HR between groups do not appear to be due to different levels of activity.
Discussion
The main findings in this study were 1) PVN neurons appear to modulate IH-induced increases in AP in rats; 2) angiotensin-(1-7) receptors along with both AT1 and AT2 angiotensin II receptors are necessary for this PVN-dependent increase in AP; 3) both AT1 and AT2 receptors appear to regulate AP in Sham rats while Ang(1-7) receptors only contribute to AP control after IH.
Furthermore, the effect of muscimol to lower AP and HR in Sham as well as in IH rats reflects that PVN neurons are normally active and control AP (Decavel & Van den Pol, 1990; Allen, 2002; Silva et al., 2005). This also indicates that GABAA mediated inhibition is functional following IH exposure. Thus the increased AP following IH is more likely due to exaggerated excitatory mechanisms in the PVN rather than to loss of GABAA receptor function.
Sleep apnea is characterized by recurrent brief cessations in breathing during sleep which repeatedly reduces arterial blood oxygen saturation (hypoxia) accompanied by small increases in circulating CO2 (Wolk et al., 2003; Prabhakar et al., 2007). Although hypoxia is thought to be the primary stimulus for inducing hypertension in sleep apnea patients and IH models, combining hypoxia with hypercapnia more profoundly stimulates the sympathetic nervous system (Morgan et al., 1995; Fletcher, 2001; Xie et al., 2001; Cutler et al., 2004). Cutler et al showed that sympathetic activation stimulated by hypoxia combined with either hypercapnia or eucapnia increased sympathetic activity more than hypocapnic hypoxia (Cutler et al., 2004). Our model of IH similarly combined increased inhaled CO2 during hypoxia periods to maintain eucapnia in the face of the hypoxia-induced hyperventilation (Snow et al., 2008) to more closely mimic the changes in arterial gases and sympathetic activation experienced in sleep apnea.
The mechanisms by which IH exposure leads to hypertension are not completely understood, but activation of the sympathetic nervous system appears necessary (Fletcher et al., 1992a; Fletcher et al., 1992b; Fletcher, 2003). Several studies have suggested hypoxia stimulation of carotid body chemoreceptors (Smith et al., 1996; Kara et al., 2003; Narkiewicz & Somers, 2003) leads to sustained reflex sympathetic activation (Narkiewicz & Somers, 2003; Prabhakar et al., 2007) with eventual resetting of sympathetic tone and arterial pressure to higher set points (Narkiewicz et al., 1998). This is supported by the observation that IH leads to sensitization and long-term facilitation of the carotid body responses to hypoxia (Peng et al., 2003) and enhanced sympatho-excitatory responses to chemoreflex activation after IH exposure (Greenberg et al., 1999; Braga et al., 2006; Dick et al., 2007).
The hypothalamic PVN is one of the major premotor neuron cell groups contributing to sympathetic control of the cardiovascular system and arterial pressure (Dampney, 1994). The PVN modulates SNA and AP responses to chemoreflex activation through both direct projections to the sympathetic preganglionic neurons in the spinal cord and via indirect synaptic relays with the rostral ventrolateral medulla (RVLM) and connections with the nucleus tractus solitarii (NTS) (Badoer, 2001; Olivan et al., 2001). Olivan et al demonstrated that bilateral electrolytic lesion of the PVN attenuated pressor responses to chemoreflex activation in awake rats (Olivan et al., 2001). In addition, blockade of neurotransmission in the PVN in anesthetized rats attenuated renal and phrenic nerve responses to peripheral chemoreflex activation (Reddy et al., 2005) while peripheral chemoreflex activation increased Fos expression in PVN neurons (Cruz et al., 2008). Thus the PVN appears to be an integral part of the central neuronal circuitry processing the sympathoexcitatory component of the peripheral chemoreflex (Olivan et al., 2001; Reddy et al., 2005; Cruz et al., 2008).
Previous studies indicate RAS peptides importantly modulate sympathetic output and that a dysfunctional brain RAS participates in the pathogenesis of some forms of hypertension (Dampney et al., 2002; Veerasingham & Raizada, 2003). Immunocytochemical studies have identified both Ang II (Egli et al., 2000) and Ang-(1-7) (Block et al., 1988), as well as AT1, AT2 (Wright & Harding, 1994; Allen et al., 1998; Shelat et al., 1998; McKinley et al., 2003) and Mas receptors (Becker et al., 2007) in the PVN. Iontophoretic application of Ang II or Ang-(1-7) has also been shown to increase firing in PVN neurons (Ambuhl et al., 1994; Becker et al., 2007) and we reported previously that Ang II and Ang-(1-7) administered into the PVN increase RSNA and AP (Silva et al., 2005). Others have shown that AT1 receptors in the PVN regulate both baseline AP (Zhu et al., 2002; Silva et al., 2005) and the augmented AP and RSNA observed during central hyperosmolar stimulation (Chen & Toney, 2001) or when PVN neurons are disinhibited (Chen & Toney, 2003). Thus angiotensin receptors appear to modulate PVN neuronal activity under basal and excited conditions.
In pathophysiological states with elevated sympathetic discharge from the PVN such as congestive heart failure (Shelat et al., 1998; Zhang et al., 2001; 2002; Zucker et al., 2004) and hypertension (Weiss et al., 2007; Allen, 2002), both overexpression of AT1 receptors (Zucker, 2002; Zucker et al., 2004) and downregulation of inhibitory nNOS (Zhang et al., 2001) and GABAA (Zhang et al., 2002) pathways have been reported. Very recently, IH exposure was shown to be associated with reduced nNOS expression in the PVN and augmented AT1 expression in circumventricular organ (CVO) neurons projecting to the PVN (Weiss et al., 2007). The incomplete blood-brain barrier of the CVO (Wright & Harding, 1994) would allow PVN neurons to be stimulated during IH by Ang peptides produced locally or delivered by neuronal projections from the CVO which are stimulated by the cyclic hypoxia (Weiss et al., 2007).
In this study, we observed that the Ang-(1-7) receptor blocker did not affect AP in Sham rats while AT1 receptor antagonists in the PVN significantly reduced and the AT2 receptor antagonist tended to reduce AP in Sham rats. These findings corroborate previous demonstrations that AT1 receptors in the PVN contribute to AP control under normal conditions through tonic activation (Zhu et al., 2002; Silva et al., 2005) and further suggesting that AT2 receptors in the PVN may also play a role in tonic AP control.
AT1 and AT2 receptors are 34% homologous and have high affinity for Ang II (Stoll & Unger, 2001). Although most brain areas express only one subtype, a few regions, including the PVN, express both (McKinley et al., 2003). The PVN has a predominance of AT1 receptors, with especially high densities in the cardiovascular and sympathetic regulatory parvicellular area (Blair et al., 1996; Allen et al., 1998; Badoer, 2001; McKinley et al., 2003). Thus cardiovascular actions exerted by Ang II in this region may be mostly mediated by AT1 receptors but this study and our previous work (Silva et al., 2005) suggest AT2 receptors in the PVN can also mediate a significant portion of Ang II effects slightly different from those exerted by AT1 receptors as shown by the greater fall in AP at day 14 in the Sham rats receiving the AT1 receptor antagonist.
Strong staining for Mas in both parvi and magnocellular portions of the PVN (Becker et al., 2007), are consistent with Ang-(1-7) receptors also regulating sympathetic tone originating in the PVN. Additionally, direct activation of Ang-(1-7) receptors in the PVN increases the firing rate of PVN neurons (Santos et al., 2003). Thus the effects on arterial pressure of the different antagonists may have occurred through blockade of unique receptors which each separately contribute to AP regulation. Alternatively there may be functional interactions between the receptors as shown in previous demonstrations of interactions between these receptor subtypes. For example, it has been demonstrated that the excitatory effects produced by Ang II, Ang III and Ang-(1-7) in the PVN can be blocked by an AT2 receptor antagonist (Felix et al., 1991). In addition, the excitatory effects produced by Ang-(1-7) in PVN neurons can be blocked by AT1 or AT2 receptor antagonists (Ambuhl et al., 1992) and a functional interaction between Ang-(1-7) and AT1 receptors has been demonstrated in other brain regions (Von Bohlen und Halbach et al., 2000). Thus the simplest explanation for the current findings seems to be that following 14 days of IH exposure, multiple angiotensin receptors on PVN neurons contribute to the vasomotor response in conscious rats.
Previous studies have demonstrated that the Ang peptides play an excitatory role in the PVN (Ambuhl et al., 1994; Cato & Toney, 2005). Although acute blockade of Ang-(1-7) and AT2 receptors in normotensive anesthetized rats dramatically reduced renal SNA in a previous study, it did not affect AP (Silva et al., 2005). In the present study, AP did not change significantly in Sham animals receiving A-779 or PD123319, but both antagonists prevented AP increases in the IH rats and the AT2 antagonist tended to decrease AP in the Sham rats below that in the IH rats throughout the exposure period suggesting that acute and chronic effects of Mas and AT2 receptor activation may differ. In a previous study in anesthetized rats, blockade of AT1 receptors in the RVLM increased sympathetic activity under hypoxic conditions (Sheriff et al., 2006) and Ang-(1-7) attenuated the pressor effect of Ang II in the anterior hypothalamus (Hocht et al., 2006). Our results in conscious rats suggesting an excitatory role for both Ang II and Ang-(1-7) in the PVN following IH suggest the effect of central angiotensins on sympathetic activity might depend upon the balance between tonic excitatory and inhibitory effects on sympathetic premotor neurons (Dampney et al., 2007). This balance may be altered in pathophysiological conditions such as chronic IH since only blockade of AT1 receptors decreased AP in Sham animals and IH rats below baseline. Together, these observations suggest that under normal conditions, AT1 and perhaps AT2 receptors mediate tonic excitation in PVN neurons to modulate AP. However, after IH exposure, both Ang-(1-7) and Ang II receptors in the PVN participate in generating the increased AP. Although only direct measures of sympathetic activity can confirm the suggested changes in Ang regulation of sympathetic discharges following exposure to IH, the differential effects of the antagonists combined with previous studies (Zhu et al., 2002; Silva et al., 2005) suggest that the different angiotensin receptor types all regulate AP but through different mechanisms and under different conditions.
Perspectives
Our results suggest sympathetic premotor neurons in the PVN importantly mediate increases in AP induced by IH in rats. Although other mechanisms may be recruited to participate in the systemic pressor response, activation of angiotensin receptors in the PVN appears necessary for increased AP in this rat model of simulated sleep apnea. Clinical studies also suggest the RAS is activated in sleep apnea. Therefore, the ability of the Ang-(1-7) antagonist to lower arterial pressure in the IH rats and not in the Sham rats suggests this might be an important target for targeted pharmacological interventions.
Experimental Procedures
Experiments were performed in male Sprague-Dawley rats (250 to 300 g, Harlan Sprague Dawley). All animal protocols were reviewed and approved by the institutional animal care and use committee of the University of New Mexico Health Sciences Center and conform to National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Telemeter implantation and PVN cannula placement
Rats were instrumented with blood pressure telemetry devices (DSI model PA-C40, Arden Hills, MN) under isofluorane (2%) anesthesia. The catheter portion was inserted into the abdominal aorta through the femoral artery and the transmitter fixed in the peritoneal cavity, allowing 24 hour recordings of mean arterial pressure (MAP) and heart rate (HR).
After 5 days of recovery, two 30-gauge microinjection needles connected to a primed osmotic minipump (Alzet model 2002) were inserted bilaterally into the paraventricular nucleus (PVN) using a stereotaxic apparatus and cemented into place. The coordinates used were 1.8 mm posterior and 0.5 mm lateral to the bregma, at a depth of 7.8 mm below dura (Silva et al., 2005). Rats were allowed to recover for 2 more days, before the initiation of exposure protocols.
Eucapnic Intermittent Hypoxia Protocol
Rats were housed in Plexiglas boxes with free access to food and water and exposed to either intermittent hypoxia with CO2 supplementation to maintain eucapnia (IH; nadir 5% O2; 5% CO2 to peak 21% O2; 0% CO2 every 90 seconds) or room air (Sham; same environmental conditions as IH, however rats were constantly maintained at 21% O2; 0% CO2). The gas composition inside the chambers was controlled by timed solenoid switches operating 7 hr/day during the rats’ sleep period for 14 days as described previously (Allahdadi et al., 2005). The chosen frequency of apneas was used to mimic moderate sleep apnea, and the reduction in breathing oxygen levels was sufficient to induce hypoxia without promoting arousal after acclimatization. During the 14 days of IH exposure, MAP and HR were recorded continuously and osmotic minipumps delivered either vehicle or drugs bilaterally into the PVN (0.5 µl/hr). The drugs administered were: A-779 (Ang-(1-7) antagonist, 2 nmol/hr); losartan (AT1 receptor antagonist, 2 nmol/hr, a kind gift from Dr. Robson Santos); ZD7155 (AT1 receptor antagonist, 0.2 nmol/hr); PD123319 (AT2 receptor antagonist, 2 nmol/hr).The doses of drugs administered were based on the ability of the same doses to acutely lower either blood pressure or renal sympathetic nerve activity in similarly prepared anaesthetized rats (Silva et al., 2005). Additionally, we compared the effects of Ang antagonists with those evoked by muscimol (20 pmol/hr), a powerful GABAA agonist known to reduce MAP when injected acutely into the PVN. NaCl 0.9 % was used as vehicle since all administered drugs were prepared in saline solution. At the end of all protocols, rats were deeply anesthetized with sodium pentobarbital (150 mg/kg i.p.) and brains fixed (paraformaldeheyde 4% and sucrose 30%) to stain for histological identification of injection sites, as described previously (Silva et al., 2005) (Supplemental Figure 1).
Statistical Analysis
All data are expressed as mean ± SEM. MAP and HR values are expressed as the average of a 12 hour period to monitor diurnal variation in AP. Additional data expressing 24 hour average is shown in Figure 5. Differences between groups were evaluated using two-way repeated measures ANOVA followed by Bonferroni post-hoc test. Differences were considered significant for P<0.05.
Research Highlights
Blocking AT1-, AT2- or mas-receptors in the PVN prevents IH-induced hypertension
Blocking AT1- but not mas-receptors in the PVN lowered blood pressure in Sham rats
Mas-receptors only appear to regulate blood pressure in pathological conditions
Angiotensin receptors in the PVN are necessary for IH to increase arterial pressure
Supplementary Material
Acknowledgments
Ana Quênia Gomes da Silva was a recipient of a CNPq PhD-fellowship at the Post-Graduation Program in Biological Science: Physiology and Pharmacology, ICB, UFMG. This research was supported by FAPEMIG, EPA grant RD-83186001, HL82799 and a Research Allocations Committee grant from UNM (NLK). NLK is an Established Investigator of the American Heart Association. Special thanks to Dr. Kyan Allahdadi for his technical assistance and critical comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- Allahdadi KJ, Walker BR, Kanagy NL. Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension. 2005;45:705–709. doi: 10.1161/01.HYP.0000153794.52852.04. [DOI] [PubMed] [Google Scholar]
- Allen AM. Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension. 2002;39:275–280. doi: 10.1161/hy0202.104272. [DOI] [PubMed] [Google Scholar]
- Allen AM, Moeller I, Jenkins TA, Zhuo J, Aldred GP, Chai SY, Mendelsohn FA. Angiotensin receptors in the nervous system. Brain research bulletin. 1998;47:17–28. doi: 10.1016/s0361-9230(98)00039-2. [DOI] [PubMed] [Google Scholar]
- Ambuhl P, Felix D, Imboden H, Khosla MC, Ferrario CM. Effects of angiotensin analogues and angiotensin receptor antagonists on paraventricular neurones. Regulatory Peptides. 1992;38:111–120. doi: 10.1016/0167-0115(92)90049-z. [DOI] [PubMed] [Google Scholar]
- Ambuhl P, Felix D, Khosla MC. [7-D-ALA]-angiotensin-(1-7): selective antagonism of angiotensin-(1-7) in the rat paraventricular nucleus. Brain research bulletin. 1994;35:289–291. doi: 10.1016/0361-9230(94)90103-1. [DOI] [PubMed] [Google Scholar]
- Badoer E. Hypothalamic paraventricular nucleus and cardiovascular regulation. Clinical and experimental pharmacology & physiology. 2001;28:95–99. doi: 10.1046/j.1440-1681.2001.03413.x. [DOI] [PubMed] [Google Scholar]
- Becker LK, Etelvino GM, Walther T, Santos RA, Campagnole-Santos MJ. Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am J Physiol Heart Circ Physiol. 2007;293:H1416–H1424. doi: 10.1152/ajpheart.00141.2007. [DOI] [PubMed] [Google Scholar]
- Blair ML, Piekut D, Want A, Olschowka JA. Role of the hypothalamic paraventricular nucleus in cardiovascular regulation. Clinical and experimental pharmacology & physiology. 1996;23:161–165. doi: 10.1111/j.1440-1681.1996.tb02590.x. [DOI] [PubMed] [Google Scholar]
- Block CH, Santos RA, Brosnihan KB, Ferrario CM. Immunocytochemical localization of angiotensin-(1-7) in the rat forebrain. Peptides. 1988;9:1395–1401. doi: 10.1016/0196-9781(88)90208-2. [DOI] [PubMed] [Google Scholar]
- Braga VA, Soriano RN, Machado BH. Sympathoexcitatory response to peripheral chemoreflex activation is enhanced in juvenile rats exposed to chronic intermittent hypoxia. Experimental physiology. 2006;91:1025–1031. doi: 10.1113/expphysiol.2006.034868. [DOI] [PubMed] [Google Scholar]
- Cato MJ, Toney GM. Angiotensin II excites paraventricular nucleus neurons that innervate the rostral ventrolateral medulla: an in vitro patch-clamp study in brain slices. Journal of neurophysiology. 2005;93:403–413. doi: 10.1152/jn.01055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QH, Haywood JR, Toney GM. Sympathoexcitation by PVN-injected bicuculline requires activation of excitatory amino acid receptors. Hypertension. 2003;42:725–731. doi: 10.1161/01.HYP.0000085197.20043.44. [DOI] [PubMed] [Google Scholar]
- Chen QH, Toney GM. AT(1)-receptor blockade in the hypothalamic PVN reduces central hyperosmolality-induced renal sympathoexcitation. American journal of physiology. 2001;281:R1844–R1853. doi: 10.1152/ajpregu.2001.281.6.R1844. [DOI] [PubMed] [Google Scholar]
- Chen QH, Toney GM. Responses to GABA-A receptor blockade in the hypothalamic PVN are attenuated by local AT1 receptor antagonism. American journal of physiology. 2003;285:R1231–R1239. doi: 10.1152/ajpregu.00028.2003. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Bonagamba LG, Machado BH, Biancardi VC, Stern JE. Intermittent activation of peripheral chemoreceptors in awake rats induces Fos expression in rostral ventrolateral medulla-projecting neurons in the paraventricular nucleus of the hypothalamus. Neuroscience. 2008;157:463–472. doi: 10.1016/j.neuroscience.2008.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler MJ, Swift NM, Keller DM, Wasmund WL, Smith ML. Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol. 2004;96:754–761. doi: 10.1152/japplphysiol.00506.2003. [DOI] [PubMed] [Google Scholar]
- Dampney RA. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev. 1994;74:323–364. doi: 10.1152/physrev.1994.74.2.323. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Fontes MA, Hirooka Y, Horiuchi J, Potts PD, Tagawa T. Role of angiotensin II receptors in the regulation of vasomotor neurons in the ventrolateral medulla. Clinical and experimental pharmacology & physiology. 2002;29:467–472. doi: 10.1046/j.1440-1681.2002.03658.x. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Tan PS, Sheriff MJ, Fontes MA, Horiuchi J. Cardiovascular effects of angiotensin II in the rostral ventrolateral medulla: the push-pull hypothesis. Curr Hypertens Rep. 2007;9:222–227. doi: 10.1007/s11906-007-0040-4. [DOI] [PubMed] [Google Scholar]
- Decavel C, Van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. The Journal of comparative neurology. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- Dick TE, Hsieh YH, Wang N, Prabhakar N. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Experimental physiology. 2007;92:87–97. doi: 10.1113/expphysiol.2006.035758. [DOI] [PubMed] [Google Scholar]
- Egli M, Laurent JP, Mosimann R, Felix D, Imboden H. Morphological and immunocytochemical characterization of electrophysiologically investigated neurons in the PVN of the rat. Journal of neuroscience methods. 2000;95:145–150. doi: 10.1016/s0165-0270(99)00166-1. [DOI] [PubMed] [Google Scholar]
- Felix D, Khosla MC, Barnes KL, Imboden H, Montani B, Ferrario CM. Neurophysiological responses to angiotensin-(1-7) Hypertension. 1991;17:1111–1114. doi: 10.1161/01.hyp.17.6.1111. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Bains JS. Actions of angiotensin in the subfornical organ and area postrema: implications for long term control of autonomic output. Clinical and experimental pharmacology & physiology. 1997;24:96–101. doi: 10.1111/j.1440-1681.1997.tb01790.x. [DOI] [PubMed] [Google Scholar]
- Fletcher EC. Invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. 2001;90:1600–1605. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- Fletcher EC. Sympathetic over activity in the etiology of hypertension of obstructive sleep apnea. Sleep. 2003;26:15–19. doi: 10.1093/sleep/26.1.15. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension. 1999;34:309–314. doi: 10.1161/01.hyp.34.2.309. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension. 1992a;20:612–619. doi: 10.1161/01.hyp.20.5.612. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Qian W, Miller CC, 3rd, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992b;19:555–561. doi: 10.1161/01.hyp.19.6.555. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999;86:298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- Hocht C, Opezzo JA, Gironacci MM, Pena C, Taira CA. Hypothalamic cardiovascular effects of angiotensin-(1-7) in spontaneously hypertensive rats. Regulatory peptides. 2006;135:39–44. doi: 10.1016/j.regpep.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Horn T, Smith PM, McLaughlin BE, Bauce L, Marks GS, Pittman QJ, Ferguson AV. Nitric oxide actions in paraventricular nucleus: cardiovascular and neurochemical implications. Am J Physiol. 1994;266:R306–R313. doi: 10.1152/ajpregu.1994.266.1.R306. [DOI] [PubMed] [Google Scholar]
- Kannan H, Hayashida Y, Yamashita H. Increase in sympathetic outflow by paraventricular nucleus stimulation in awake rats. Am J Physiol. 1989;256:R1325–R1330. doi: 10.1152/ajpregu.1989.256.6.R1325. [DOI] [PubMed] [Google Scholar]
- Kannan H, Yamashita H. Connections of neurons in the region of the nucleus tractus solitarius with the hypothalamic paraventricular nucleus: their possible involvement in neural control of the cardiovascular system in rats. Brain research. 1985;329:205–212. doi: 10.1016/0006-8993(85)90526-8. [DOI] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes--physiology and clinical implications. Acta physiologica Scandinavica. 2003;177:377–384. doi: 10.1046/j.1365-201X.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, Oldfield BJ, Mendelsohn FA, Chai SY. The brain renin-angiotensin system: location and physiological roles. The international journal of biochemistry & cell biology. 2003;35:901–918. doi: 10.1016/s1357-2725(02)00306-0. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Crabtree DC, Palta M, Skatrud JB. Combined hypoxia and hypercapnia evokes long-lasting sympathetic activation in humans. J Appl Physiol. 1995;79:205–213. doi: 10.1152/jappl.1995.79.1.205. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, Kato M, Phillips BG, Davison DE, Somers VK. Baroreflex control of sympathetic nerve activity and heart rate in obstructive sleep apnea. Hypertension. 1998;32:1039–1043. doi: 10.1161/01.hyp.32.6.1039. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Somers VK. Sympathetic nerve activity in obstructive sleep apnoea. Acta physiologica Scandinavica. 2003;177:385–390. doi: 10.1046/j.1365-201X.2003.01091.x. [DOI] [PubMed] [Google Scholar]
- Olivan MV, Bonagamba LG, Machado BH. Involvement of the paraventricular nucleus of the hypothalamus in the pressor response to chemoreflex activation in awake rats. Brain research. 2001;895:167–172. doi: 10.1016/s0006-8993(01)02067-4. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Dick TE, Nanduri J, Kumar GK. Systemic, cellular and molecular analysis of chemoreflex-mediated sympathoexcitation by chronic intermittent hypoxia. Experimental physiology. 2007;92:39–44. doi: 10.1113/expphysiol.2006.036434. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Jacono FJ, Kumar GK, Dick TE. Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clinical and experimental pharmacology & physiology. 2005;32:447–449. doi: 10.1111/j.1440-1681.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Reddy MK, Patel KP, Schultz HD. Differential role of the paraventricular nucleus of the hypothalamus in modulating the sympathoexcitatory component of peripheral and central chemoreflexes. American journal of physiology. 2005;289:R789–R797. doi: 10.1152/ajpregu.00222.2005. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelat SG, Reagan LP, King JL, Fluharty SJ, Flanagan-Cato LM. Analysis of angiotensin type 2 receptors in vasopressinergic neurons and pituitary in the rat. Regulatory peptides. 1998;73:103–112. doi: 10.1016/s0167-0115(97)01066-5. [DOI] [PubMed] [Google Scholar]
- Sheriff MJ, Fontes MA, Killinger S, Horiuchi J, Dampney RA. Blockade of AT1 receptors in the rostral ventrolateral medulla increases sympathetic activity under hypoxic conditions. American journal of physiology. 2006;290:R733–R740. doi: 10.1152/ajpregu.00410.2005. [DOI] [PubMed] [Google Scholar]
- Sica AL, Greenberg HE, Ruggiero DA, Scharf SM. Chronic-intermittent hypoxia: a model of sympathetic activation in the rat. Respir Physiol. 2000;121:173–184. doi: 10.1016/s0034-5687(00)00126-2. [DOI] [PubMed] [Google Scholar]
- Silva AQ, Santos RA, Fontes MA. Blockade of endogenous angiotensin-(1-7) in the hypothalamic paraventricular nucleus reduces renal sympathetic tone. Hypertension. 2005;46:341–348. doi: 10.1161/01.HYP.0000179216.04357.49. [DOI] [PubMed] [Google Scholar]
- Smith ML, Niedermaier ON, Hardy SM, Decker MJ, Strohl KP. Role of hypoxemia in sleep apnea-induced sympathoexcitation. Journal of the autonomic nervous system. 1996;56:184–190. doi: 10.1016/0165-1838(95)00062-3. [DOI] [PubMed] [Google Scholar]
- Snow JB, Kitzis V, Norton CE, Torres SN, Johnson KD, Kanagy NL, Walker BR, Resta TC. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol. 2008;104:110–118. doi: 10.1152/japplphysiol.00698.2005. [DOI] [PubMed] [Google Scholar]
- Stern JE, Li Y, Zhang W. Nitric oxide: a local signalling molecule controlling the activity of pre-autonomic neurones in the paraventricular nucleus of the hypothalamus. Acta Physiol Scand. 2003;177:37–42. doi: 10.1046/j.1365-201X.2003.01045.x. [DOI] [PubMed] [Google Scholar]
- Stoll M, Unger T. Angiotensin and its AT2 receptor: new insights into an old system. Regulatory peptides. 2001;99:175–182. doi: 10.1016/s0167-0115(01)00246-4. [DOI] [PubMed] [Google Scholar]
- Tagawa T, Dampney RA. AT(1) receptors mediate excitatory inputs to rostral ventrolateral medulla pressor neurons from hypothalamus. Hypertension. 1999;34:1301–1307. doi: 10.1161/01.hyp.34.6.1301. [DOI] [PubMed] [Google Scholar]
- Veerasingham SJ, Raizada MK. Brain renin-angiotensin system dysfunction in hypertension: recent advances and perspectives. Br J Pharmacol. 2003;139:191–202. doi: 10.1038/sj.bjp.0705262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Bohlen und Halbach O, Walther T, Bader M, Albrecht D. Interactions between Mas and the angiotensin AT1 receptor in the amygdala. Journal of Neurophysiology. 2000;83:2012–2021. doi: 10.1152/jn.2000.83.4.2012. [DOI] [PubMed] [Google Scholar]
- Weiss JW, Liu MD, Huang J. Physiological basis for a causal relationship of obstructive sleep apnoea to hypertension. Experimental physiology. 2007;92:21–26. doi: 10.1113/expphysiol.2006.035733. [DOI] [PubMed] [Google Scholar]
- Wolk R, Shamsuzzaman AS, Somers VK. Obesity, sleep apnea, and hypertension. Hypertension. 2003;42:1067–1074. doi: 10.1161/01.HYP.0000101686.98973.A3. [DOI] [PubMed] [Google Scholar]
- Wright JW, Harding JW. Brain angiotensin receptor subtypes in the control of physiological and behavioral responses. Neuroscience and biobehavioral reviews. 1994;18:21–53. doi: 10.1016/0149-7634(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Morgan BJ. Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol. 2001;91:1555–1562. doi: 10.1152/jappl.2001.91.4.1555. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li YF, Patel KP. Blunted nitric oxide-mediated inhibition of renal nerve discharge within PVN of rats with heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H995–H1004. doi: 10.1152/ajpheart.2001.281.3.H995. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li YF, Patel KP. Reduced endogenous GABA-mediated inhibition in the PVN on renal nerve discharge in rats with heart failure. American journal of physiology. 2002;282:R1006–R1015. doi: 10.1152/ajpregu.00241.2001. [DOI] [PubMed] [Google Scholar]
- Zhang K, Mayhan WG, Patel KP. Nitric oxide within the paraventricular nucleus mediates changes in renal sympathetic nerve activity. The American journal of physiology. 1997;273:R864–R872. doi: 10.1152/ajpregu.1997.273.3.R864. [DOI] [PubMed] [Google Scholar]
- Zhu GQ, Patel KP, Zucker IH, Wang W. Microinjection of ANG II into paraventricular nucleus enhances cardiac sympathetic afferent reflex in rats. Am J Physiol Heart Circ Physiol. 2002;282:H2039–H2045. doi: 10.1152/ajpheart.00854.2001. [DOI] [PubMed] [Google Scholar]
- Zucker IH. Brain angiotensin II: new insights into its role in sympathetic regulation. Circulation research. 2002;90:503–505. doi: 10.1161/01.res.0000014287.96335.21. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Schultz HD, Li YF, Wang Y, Wang W, Patel KP. The origin of ympathetic outflow in heart failure: the roles of angiotensin II and nitric oxide. Progress in biophysics and molecular biology. 2004;84:217–232. doi: 10.1016/j.pbiomolbio.2003.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.