

Abstract
Two homologous 29 amino acid-long highly hydrophobic membrane mini-proteins were identified in the Bligh-Dyer lipid extracts of Escherichia coli and Salmonella typhimurium using liquid chromatography/tandem mass spectrometry (LC/MS/MS). The amino acid sequences of the proteins were determined by collision-induced dissociation tandem mass spectrometry, in conjunction with a translating BLAST (tBLASTn) search, i.e. comparing the MS/MS-determined protein query sequence against the six-frame translations of the nucleotide sequences of the E. coli and S. typhimurium genomes. Further MS characterization revealed that both proteins retain the N-terminal initiating formyl-methionines. The methodologies described here may be amendable for detecting and characterizing small hydrophobic proteins in other organisms that are difficult to annotate or analyze by conventional methods.
INTRODUCTION
Only a handful of proteins and peptides that have been reported to be soluble in chloroform, including subunit c of the F1F0 ATP synthase [1], Escherichia coli multidrug resistance protein E (EmrE) [2], myelin proteolipid protein (PLP) [3], lung surfactants [4], and several small peptides [5]. These unusually hydrophobic proteins and peptides play diverse and critical cellular functions. Here, we report the unexpected identification of two small chloroform–soluble proteins (3 kDa) in the Bligh-Dyer lipid extracts [6] of the Gram-negative bacteria E. coli and Salmonella typhimurium. The full sequences of these homologous proteins were determined by using the partial amino acid sequences determined by collision-induced dissociation tandem mass spectrometry in database searches. Further characterization by accurate mass measurement and MS/MS revealed that both proteins retain their N-terminal initiating formyl-methionines.
EXPRIMENTAL PROCEDURES
Materials
All solvents were of HPLC grade and were obtained from VWR (West Chester, PA).
Bacterial Growth Conditions
Wild type strains E. coli W3110 and S. typhimurium χ3761 were grown in Luria-Bertani (LB) broth (1% tryptone, 0.5% yeast extract and 1% NaCl). Typically, a 100 ml cell culture, inoculated from an overnight culture grown to A600 = 0.02, was grown to A600 = 2. The cells were collected by centrifugation and washed with phosphate-buffered saline (PBS, pH 7.4). The cell pellets were resuspended in 152 ml of a single-phase Bligh-Dyer mixture consisting of chloroform/methanol/water (1:2:0.8, v/v/v), incubated at room temperature for 60 min, and centrifuged at 3000 × g to remove insoluble debris. The supernatant was transferred to a new glass tube and converted to a two-phase Bligh-Dyer system by adding chloroform and water to generate a mixture consisting of chloroform/methanol/water (2:2:1.8, v/v/v). The lower phase was dried under a stream of nitrogen and stored at −20 °C until analysis.
Electrospray Ionization/Mass Spectrometry
For negative ion ESI/MS analysis [7], each of the dried Bligh-Dyer lipid extracts of E. coli and S. typhimurium cells was first re-dissolved in 200 μl of chloroform. Typically, 5 μl of the solution was diluted into 200 μl of chloroform/methanol (2:1, v/v), followed by the addition of 1 μl of piperidine (Sigma-Aldrich). The solutions were infused at flow rates of 5–10 μl/min into the ESI source of the high-resolution QSTAR XL quadrupole time-of-flight tandem mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA). The negative electrospray voltage was −4200 V. Other MS settings were as follows: Curtain Gas = 20 psi (pressure), Ion Gas Source = 20 psi, Declustering Potential = −55 V, and Focusing Potential = −265 V. Data acquisition and analyses were performed using Analyst QS software (Applied Biosystems/MDS Sciex).
Liquid Chromatography/Tandem Mass Spectrometry
LC/MS/MS analysis was performed in the positive ion mode using a Shimadzu LC system (comprising a solvent degasser, two LC-10A pumps and a SCL-10A system controller) coupled to a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (as above). LC was operated at a flow rate of 200 μL/min with a linear gradient as follows: 100% A was held isocratically for 2 min and then linearly increased to 100% B over 14 min and held at 100% B for 4 min. Mobile phase A consisted of methanol/acetonitrile/aqueous 1 mM ammonium acetate (60/20/20, v/v/v). Mobile phase B consisted of 100% ethanol containing 1 mM ammonium acetate. A Zorbax SB-C8 reversed-phase column (5 μm, 2.1 × 50 mm) was purchased from Agilent (Palo Alto, CA). The post-column split diverted ~10% of the LC flow to the ESI source. Positive ion mass spectra were acquired with an electrospray voltage of +5500 V. The collision-induced dissociation tandem mass spectra were obtained with collision energy of +50 V (laboratory frame of reference) and with nitrogen as the collision gas.
RESULTS
Two unknown species, each with a molecular weight of 3 kDa, were observed upon ESI/MS analysis of the total lipid extracts of E. coli and S. typhimurium cells. Figures 1A and 1B show the negative ion ESI mass spectra of the species present in the Bligh-Dyer extracts of the E. coli and S. typhimurium cells, respectively. The major ion species, with mass-to-charge (m/z) ratios ranging from 600 to 800, were identified by MS/MS to be the [M-H]− ions of the major bacterial phospholipids, including phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) [8]. In both spectra, there exist unknown 3 kDa species, whose triply charged ions appeared near m/z 1000. As shown in the magnified ion peaks in the insets, the charge state (3-) was determined as the reciprocal of the spacing between two adjacent isotopic peaks (differing by 1 atomic mass unit) [9]. The monoisotopic masses of these species, determined by accurate mass measurements of the [M-3H]3− ions, were 3023.740 Da and 2981.686 Da for the E. coli and S. typhimurium proteins, respectively. These unknown species were also detected by LC/MS operated in the positive ion mode. Figure 2 shows the zoomed-in spectra of the triply charged (protonated) [M+3H]3+ ions at m/z 1008.9 and m/z 994.9 for the unknown protein species from E. coli and S. typhimurium, respectively.
Figure 1.

Negative ion ESI mass spectra of the Bligh-Dyer extracts of E. coli (A) and S. typhimurium (B). The insets show the magnified ion peaks for the [M-3H]3− ions of the small 3 kDa proteins.
Figure 2.

Positive ion ESI mass spectra showing the [M+3H]3+ ions of the small proteins in the Bligh-Dyer extracts of E. coli (A) and S. typhimurium (B). These positive ion mass spectra are averaged from the spectra acquired by LC/MS (during the LC elution times indicated in the figures).
To identify these unknown 3 kDa species, collision-induced dissociation MS/MS was performed on the triply charged ions (in both positive and negative ion modes). While the negative ion MS/MS spectra of the [M-3H]3− ions were not structurally informative (data not shown), the positive ion MS/MS spectra of the [M+3H]3+ ions (acquired through targeted LC/MS/MS) showed fragmentation patterns characteristic of a peptide. As illustrated in Figure 3, the partial amino acid sequences deduced from the product ion spectra of [M+3H]3+ ions at m/z 1008.9 (E. coli) and m/z 994.9 (S. typhimurium) are: GL(I)L(I)VAVGLTAALH (E. coli) and GVL(I)VAGGL(I)TAALH (S. typhimurium). The isobaric I and L residues could not be discerned by the low-energy CID MS/MS used in this study. Our initial attempt at searching these partial sequences against the protein sequence database (prior to annotation) yielded no matches. We then performed a translating BLAST (tBLASTn) search, i.e. comparing the partial amino acid sequences against the six-frame translations of the nucleotide sequences of the bacterial genomes. This yielded matches for both proteins. The full sequences of the two proteins were identified as: MSTDLKFSLVTTIIVLGLIVAVGLTAALH (E. coli) and MSTDLKFSLITTLIVLGVIVAGGLTAALH (S. typhimurium). Differing by only three amino acid residues, these proteins are apparently derived from orthologous genes. The E. coli protein was later annotated as YnhF in the EcoGene database (http://www.ecogene.org), containing updated information on the E. coli K-12 genome and proteome sequences.
Figure 3.

Assignment of partial amino acid sequences based on the collision-induced dissociation MS/MS spectra of the small 3 kDa proteins from E. coli and S. typhimurium. LC/MS/MS spectra of [M+3H]3+ ions at m/z 1008.9 (A and C) for E. coli, and m/z 994.9 (B and D) for S. typhimurium. Peaks labeled with iv, iL/I and iH correspond to the immonium ions of valine, leucine/isoleucine and histidine.
It was noted, for both the E. coli and S. typhimurium proteins, that there is a 28 Da difference between the sequence-predicated monoisotopic masses and those determined by MS. The sequence-predicated monoisotopic masses are 2995.740 Da (E. coli) and 2953.693 Da (S. typhimurium), while the MS-determined monoisotopic masses were 3023.740 Da (E. coli) and 2981.686 (S. typhimurium). This 28 Da difference implies that both proteins might be modified by a formyl (CO) group. Indeed, this possibility was confirmed by accurate mass measurements and MS/MS. The MS-determined monoisotopic masses (above) are in agreement with the predicated monoisotopic masses of 3023.735 Da (E. coli) and 2981.688 Da (S. typhimurium) for the formylated forms of both proteins. Furthermore, MS/MS showed that the formyl group is attached to the N-terminal methionine, as evidenced by the presence in the MS/MS spectra of the b2 product ions at m/z 247.1 (the predicated b2 ion mass for each of the N-formylated peptides is 247.1). Figure 4 depicts all inter-residual cleavage sites observed in the positive ion MS/MS spectra (Figure 3) of the [M+3H]3+ ions. All sequence fragment ions (b and y) and their masses are listed in Table 1. The nomenclature used for naming the fragment ions is according to Roepstorff and Fohlman [10].
Figure 4.
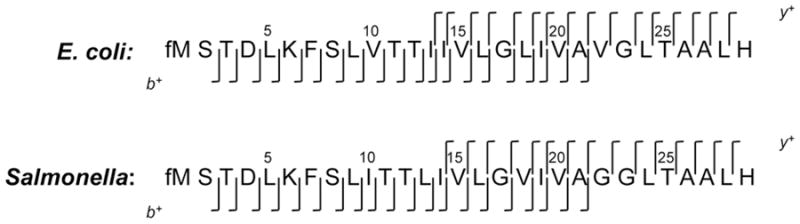
Inter-residual cleavage sites observed in the MS/MS spectra of the [M+3H]3+ ions at m/z 1008.9 and m/z 994.9 for the small proteins from E. coli and S. typhimurium, respectively.
Table 1.
Sequence ions (b and y) observed in the collision-induced dissociation MS/MS spectra of the [M+3H]3+ ions at m/z 1008.9 and m/z 994.9 of the small proteins from E. coli and Salmonella, respectively. The listed ion masses correspond to singly charged species. Abbreviation: f, formyl.
| fMSTDLKFSLVTTIIVLGLIVAVGLTAALH (E. coli) | fMSTDLKFSLITTLIVLGVIVAGGLTAALH (S. typhimurium) | ||||||
|---|---|---|---|---|---|---|---|
| Observed | Observed | Observed | Observed | ||||
| y1 | 156.1 | y1 | 156.1 | ||||
| y2 | 269.2 | y2 | 269.2 | ||||
| y3 | 340.2 | y3 | 340.2 | ||||
| y4 | 411.2 | y4 | 411.2 | ||||
| y5 | 512.3 | y5 | 512.3 | ||||
| y6 | 625.3 | y6 | 625.3 | ||||
| y7 | 682.4 | y7 | 682.4 | ||||
| y8 | 781.4 | b21 | 2244.2 | y8 | 739.4 | b21 | 2244.2 |
| y9 | 852.4 | b20 | 2173.1 | y9 | 810.4 | b20 | 2173.1 |
| y10 | 951.5 | b19 | 2074.1 | y10 | 909.5 | b19 | 2074.1 |
| y11 | 1064.6 | b18 | 1961.1 | y11 | 1022.6 | b18 | 1961.1 |
| y12 | 1177.7 | b17 | 1847.8 | y12 | 1178.6 | b17 | 1861.8 |
| y13 | 1234.7 | b16 | 1790.8 | y13 | 1291.7 | b16 | 1804.9 |
| y14 | 1347.8 | b15 | 1677.8 | y14 | 1390.7 | b15 | 1691.8 |
| y15 | 1446.9 | b14 | 1578.8 | y15 | 1503.8 | b14 | 1592.8 |
| y16 | 1559.9 | b13 | 1465.5 | b13 | 1479.4 | ||
| b12 | 1352.6 | b12 | 1366.6 | ||||
| b11 | 1251.6 | b11 | 1265.6 | ||||
| b10 | 1150.6 | b10 | 1164.6 | ||||
| b9 | 1051.5 | b9 | 1051.5 | ||||
| b8 | 938.4 | b8 | 938.4 | ||||
| b7 | 851.4 | b7 | 851.4 | ||||
| b6 | 704.3 | b6 | 704.3 | ||||
| b5 | 576.2 | b5 | 576.2 | ||||
| b4 | 463.1 | b4 | 463.1 | ||||
| b3 | 348.1 | b3 | 348.1 | ||||
| b2 | 247.1 | b2 | 247.1 | ||||
Although formyl-methione initiates all protein synthesis in Bacteria [11–13], in most cases the formyl group is subsequently removed by deformylase [14; 15]. This is often followed by removal of the N-terminal methionine by methionine aminopeptidase [16]. Nonetheless, the retention of N-terminal formyl-methiones has been reported for several proteins and peptides, such as the aspartate chemoreceptor [17], the membrane-bound F0 subunit c of ATP synthetase [18], and the chemotactic peptide, formyl-methinyl-leucyl-phenyanaline (f-MLF) [19].
DISCUSSION
Identification and structural characterization of hydrophobic proteins and peptides are challenging, as they are typically membrane-associated, and are difficult to extract or purify. The predominant MS-based proteomic techniques have, in fact, been developed for analyzing soluble proteins and peptides. As a result, hydrophobic proteins and peptides are under-represented in global proteomic analyses. However, encouraging technical advances have been made in recent years in the MS analysis of membrane-associated proteins [20–24]. In particular, the feasibility of high-resolution top-down MS/MS for determining post-translational modifications (PTM) of integral membrane proteins has been demonstrated [24].
The identification here of small membrane proteins from E. coli and S. typhimurium benefited unexpectedly from methods that were developed for lipid analysis. We envision that the methodologies described here could be equally amendable for the identification of unusually hydrophobic proteins in other organisms or cells. It is possible that by subjecting total Bligh-Dyer extracts (or other organic solvent extracts) to additional steps of fractionation, more hydrophobic proteins, especially those of very low abundance, could be uncovered.
A second distinctive feature of these small proteins is that they are encoded by genes defined by very small open reading frames. Small proteins and peptides play key roles in cellular processes including transport, intermediary metabolism, translation regulation, chromosome segregation, genome stability, and other biological and physiological functions [25]. However, correct annotation of genes encoded by small open reading frames (smORFs) is difficult. First, computational methods do not reliably predict small genes, as there are an enormous number of meaningless short ORFs. Second, their small size makes smORFs elusive targets for mutagenic screens [25; 26]. As a result, knowledge of smORF function is rather limited, as compared to their longer counterparts. Detection by MS at the protein or peptide level, as demonstrated in this study, provides the most direct and convincing evidence for identifying the products of genes defined by small open reading frames. This is yet another example illustrating the important roles of proteogenomics, which typically utilizes MS-determined protein/peptide sequences to define novel open reading frames [27–29]. Currently, there are less than 60 proteins comprising fewer than 50 amino acids identified or predicted in the E. coli K-12 genome [30].
The fact that these membrane mini-proteins could be detected by direct infusion ESI/MS in the total lipid extracts of E. coli and S. typhimurium without any pre-fractionation is quite remarkable, implying that they may exist in significant quantities, playing crucial roles in these bacteria. The E. coli protein was annotated as YnhF. In the E. coli genome, ynhF is located upstream to ydhP, encoding a 389 amino acid-long protein predicted to be a transporter, containing 12 trans-membrane helices (Figure 5). It is possible that ynhF, predicted to have one trans-membrane segment, may function as a subunit of this transporter complex, similar to the role played by the membrane-bound subunit c (8 kDa) of the F1F0 ATP synthase complex, or the role of the KdpF subunit (3 kDa) in the K(+) translocating Kdp complex [31].
Figure 5.

Prediction of trans-membrane regions in YnhF and YdhP, encoded by adjacent genes. It is possible, given their genomic and membrane locations, that YnhF forms a complex with YdhP, a putative transporter.
A recent study by Storz and co-workers [32] showed that YnhF is one of the small stress response proteins in E. coli. The expression level of ynhF, measured using a sequential peptide affinity (SPA) tag [30], is over 4-fold higher when cells were grown under aerobic versus low oxygen conditions, and is lower in minimal glycerol-grown cells, as compared to what is seen in minimal glucose-grown cells. These observations call for study on the functions of these membrane mini-proteins, possibly to be approached initially through gene deletion.
Acknowledgments
The mass spectrometry facility in the Department of Biochemistry of the Duke University Medical Center and Z. Guan were supported by the LIPID MAPS Large Scale Collaborative Grant from the National Institutes of Health (GM-069338). We thank Dr. Robert Murphy for advice on LC/MS analysis of lipids, and Dr. Jerry Eichler for critically reading the manuscript.
ABBREVIATIONS
- ESI/MS
electrospray ionization/mass spectrometry
- LC/MS
liquid chromatography/mass spectrometry
- MS/MS
tandem mass spectrometry
- ORF
open reading frame
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Girvin ME, Rastogi VK, Abildgaard F, Markley JL, Fillingame RH. Solution structure of the transmembrane H+-transporting subunit c of the F1F0 ATP synthase. Biochemistry. 1998;37:8817–8824. doi: 10.1021/bi980511m. [DOI] [PubMed] [Google Scholar]
- 2.Yerushalmi H, Lebendiker M, Schuldiner S. EmrE, an Escherichia coli 12-kDa multidrug transporter, exchanges toxic cations and H+ and is soluble in organic solvents. J Biol Chem. 1995;270:6856–6863. doi: 10.1074/jbc.270.12.6856. [DOI] [PubMed] [Google Scholar]
- 3.Greer JM, Lees MB. Myelin proteolipid protein--the first 50 years. Int J Biochem Cell Biol. 2002;34:211–215. doi: 10.1016/s1357-2725(01)00136-4. [DOI] [PubMed] [Google Scholar]
- 4.Curstedt T, Johansson J, Barros-Soderling J, Robertson B, Nilsson G, Westberg M, Jornvall H. Low-molecular-mass surfactant protein type 1. The primary structure of a hydrophobic 8-kDa polypeptide with eight half-cystine residues. Eur J Biochem. 1988;172:521–525. doi: 10.1111/j.1432-1033.1988.tb13918.x. [DOI] [PubMed] [Google Scholar]
- 5.Lees MB, Chao BH, Laursen RA, L’Italien JJ. A hydrophobic tryptic peptide from bovine white matter proteolipid. Biochim Biophys Acta. 1982;702:117–124. doi: 10.1016/0167-4838(82)90034-6. [DOI] [PubMed] [Google Scholar]
- 6.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 7.Guan Z, Breazeale SD, Raetz CR. Extraction and identification by mass spectrometry of undecaprenyl diphosphate-MurNAc-pentapeptide-GlcNAc from Escherichia coli. Anal Biochem. 2005;345:336–339. doi: 10.1016/j.ab.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Murphy RC, Fiedler J, Hevko J. Analysis of nonvolatile lipids by mass spectrometry. Chem Rev. 2001;101:479–526. doi: 10.1021/cr9900883. [DOI] [PubMed] [Google Scholar]
- 9.Henry KD, Mclafferty FW. Electrospray Ionization with Fourier-Transform Mass-Spectrometry - Charge State Assignment from Resolved Isotopic Peaks. Org Mass Spectrom. 1990;25:490–492. [Google Scholar]
- 10.Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 11.Marcker K, Sanger F. N-Formyl-Methionyl-S-Rna. J Mol Biol. 1964;8:835–840. doi: 10.1016/s0022-2836(64)80164-9. [DOI] [PubMed] [Google Scholar]
- 12.Adams JM, Capecchi MR. N-formylmethionyl-sRNA as the initiator of protein synthesis. Proc Natl Acad Sci U S A. 1966;55:147–155. doi: 10.1073/pnas.55.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webster RE, Engelhardt DL, Zinder ND. In vitro protein synthesis: chain initiation. Proc Natl Acad Sci U S A. 1966;55:155–161. doi: 10.1073/pnas.55.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams JM. On the release of the formyl group from nascent protein. J Mol Biol. 1968;33:571–589. doi: 10.1016/0022-2836(68)90307-0. [DOI] [PubMed] [Google Scholar]
- 15.Takeda M, Webster RE. Protein chain initiation and deformylation in B. subtilis homogenates. Proc Natl Acad Sci U S A. 1968;60:1487–1494. doi: 10.1073/pnas.60.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller CG, Strauch KL, Kukral AM, Miller JL, Wingfield PT, Mazzei GJ, Werlen RC, Graber P, Movva NR. N-terminal methionine-specific peptidase in Salmonella typhimurium. Proc Natl Acad Sci U S A. 1987;84:2718–2722. doi: 10.1073/pnas.84.9.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milligan DL, Koshland DE., Jr The amino terminus of the aspartate chemoreceptor is formylmethionine. J Biol Chem. 1990;265:4455–4460. [PubMed] [Google Scholar]
- 18.Walker JE, Lutter R, Dupuis A, Runswick MJ. Identification of the subunits of F1F0-ATPase from bovine heart mitochondria. Biochemistry. 1991;30:5369–5378. doi: 10.1021/bi00236a007. [DOI] [PubMed] [Google Scholar]
- 19.Bokoch GM, Reed PW. Stimulation of arachidonic acid metabolism in the polymorphonuclear leukocyte by an N-formylated peptide. Comparison with ionophore A23187. J Biol Chem. 1980;255:10223–10226. [PubMed] [Google Scholar]
- 20.Whitelegge JP. HPLC and mass spectrometry of intrinsic membrane proteins. Methods Mol Biol. 2004;251:323–340. doi: 10.1385/1-59259-742-4:323. [DOI] [PubMed] [Google Scholar]
- 21.Zabrouskov V, Whitelegge JP. Increased coverage in the transmembrane domain with activated-ion electron capture dissociation for top-down Fourier-transform mass spectrometry of integral membrane proteins. J Proteome Res. 2007;6:2205–2210. doi: 10.1021/pr0607031. [DOI] [PubMed] [Google Scholar]
- 22.Carroll J, Altman MC, Fearnley IM, Walker JE. Identification of membrane proteins by tandem mass spectrometry of protein ions. Proc Natl Acad Sci U S A. 2007;104:14330–14335. doi: 10.1073/pnas.0706817104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thangaraj B, Ryan CM, Souda P, Krause K, Faull KF, Weber AP, Fromme P, Whitelegge JP. Data-directed top-down Fourier-transform mass spectrometry of a large integral membrane protein complex: photosystem II from Galdieria sulphuraria. Proteomics. 2010;10:3644–3656. doi: 10.1002/pmic.201000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryan CM, Souda P, Bassilian S, Ujwal R, Zhang J, Abramson J, Ping P, Durazo A, Bowie JU, Hasan SS, Baniulis D, Cramer WA, Faull KF, Whitelegge JP. Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation. Mol Cell Proteomics. 2010;9:791–803. doi: 10.1074/mcp.M900516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basrai MA, Hieter P, Boeke JD. Small open reading frames: beautiful needles in the haystack. Genome Res. 1997;7:768–771. doi: 10.1101/gr.7.8.768. [DOI] [PubMed] [Google Scholar]
- 26.Kastenmayer JP, Ni L, Chu A, Kitchen LE, Au WC, Yang H, Carter CD, Wheeler D, Davis RW, Boeke JD, Snyder MA, Basrai MA. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 2006;16:365–373. doi: 10.1101/gr.4355406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ansong C, Purvine SO, Adkins JN, Lipton MS, Smith RD. Proteogenomics: needs and roles to be filled by proteomics in genome annotation. Brief Funct Genomic Proteomic. 2008;7:50–62. doi: 10.1093/bfgp/eln010. [DOI] [PubMed] [Google Scholar]
- 28.Ferguson JT, Wenger CD, Metcalf WW, Kelleher NL. Top-down proteomics reveals novel protein forms expressed in Methanosarcina acetivorans. J Am Soc Mass Spectrom. 2009;20:1743–1750. doi: 10.1016/j.jasms.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanner S, Shen Z, Ng J, Florea L, Guigo R, Briggs SP, Bafna V. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007;17:231–239. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol. 2008;70:1487–1501. doi: 10.1111/j.1365-2958.2008.06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gassel M, Mollenkamp T, Puppe W, Altendorf K. The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J Biol Chem. 1999;274:37901–37907. doi: 10.1074/jbc.274.53.37901. [DOI] [PubMed] [Google Scholar]
- 32.Hemm MR, Paul BJ, Miranda-Rios J, Zhang A, Soltanzad N, Storz G. Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J Bacteriol. 2010;192:46–58. doi: 10.1128/JB.00872-09. [DOI] [PMC free article] [PubMed] [Google Scholar]