

Abstract
Identifying quantitative trait loci (QTL) underlying complex, low-heritability traits is notoriously difficult. Prototypical for such traits, calving ease is an important breeding objective of cattle (Bos taurus)-improving programs. To identify QTL underlying calving ease, we performed a genome-wide association study using estimated breeding values (EBVs) as highly heritable phenotypes for paternal calving ease (pCE) and related traits. The massively structured study population consisted of 1800 bulls of the German Fleckvieh (FV) breed. Two pCE-associated regions on bovine chromosomes (BTA) 14 and 21 (P = 5.72 × 10−15 and P = 2.27 × 10−8, respectively) were identified using principal components analysis to correct for population stratification. The two most significantly associated SNPs explain 10% of the EBV variation. Since marker alleles with negative effect on pCE have positive effects on growth-related traits, the QTL may exert their effects on the birthing process through fetal growth traits. The QTL region on BTA14 corresponds to a human chromosome (HSA) region that is associated with growth characteristics. The HSA region corresponding to the BTA21 pCE QTL is maternally imprinted and involved in the Prader–Willi and Angelman syndromes. Resequencing of positional candidate genes on BTA14 revealed a highly significantly (P = 1.96 × 10−14) associated polymorphism ablating a polyadenylation signal of the gene encoding ribosomal protein S20 (RPS20). Our study demonstrates the leverage potential of EBVs in unraveling the genetic architecture of lowly heritable traits.
THE recent availability of genome-wide SNP panels in cattle and other livestock species enables the mapping of quantitative trait loci (QTL) as well as the prediction of an animal's genetic merit without relying on phenotypic information (Goddard and Hayes 2009). However, the complex genetic architecture of agriculturally important traits renders the systematic identification and characterization of individual QTL a difficult task. The proportion of trait variance explained by an average QTL is very small. First mapping results in cattle seem to validate the classical quantitative genetic model of a large number of loci of small additive effects (Barendse et al. 2007, Daetwyler et al. 2008, Cole et al. 2009) and agree with findings from mapping QTL in the human genome (Manolio et al. 2009). In addition to the relative contribution of a QTL to the trait variation, the heritability of the trait is a major determinant of the mapping power (Goddard and Hayes 2009).
The heritability of calving traits, i. e. traits that describe the birthing process (dystocia in the case of difficulties) and the perinatal viability (stillbirth) of the calf as affected by the birthing process, are low, ranging from 0.04 to 0.15 (Lin et al. 1989, Steinbock et al. 2003, Seidenspinner et al. 2009). Calving traits are of considerable economic importance due to veterinary treatment costs, calf loss and lower production of cows affected by dystocia. Estimated breeding values (EBVs) for calving traits are used as selection criteria in attempts to reduce calving problems both in dairy and beef breeds (e.g., Van Tassell et al. 2003, Freer 2008)). Calving traits are complex since they are influenced by a sire-effect through the size of the calf as well as dam effects consisting mainly of the pelvic dimensions. Routine progeny testing results in highly reliable EBVs for calving traits and thereby boosts the heritability to levels that make them amenable to QTL mapping even with medium-sized samples.
An important prerequisite for unbiased QTL mapping based on linkage disequilibrium (LD) is homogeneity of the mapping population (Devlin and Roeder 1999). The heavy use of genetically superior bulls, facilitated by artificial insemination, and introgression lead to massively stratified populations. We attempted to correct for population stratification by principal components analysis (PCA)-based approaches that have been successful in human genome-wide QTL mapping (Price et al. 2006).
Here we report the mapping of two loci affecting very low heritability calving traits in a heavily structured dual purpose (dairy, beef) cattle population. The mapping approach was facilitated by the use of EBVs and consequent correction of population stratification.
MATERIAL AND METHODS
Animals and phenotypes:
Bulls of the dual purpose breed Fleckvieh (FV, n = 1829) were genotyped using the Illumina BovineSNP 50K Bead chip composed of 54,001 single nucleotide polymorphisms (SNPs). Phenotypes in the form of EBVs for beef (daily gain, DG) and conformation traits (body size, BS) as well as functionality traits such as paternal calving ease (pCE) and paternal stillbirth incidence (pSB) were obtained from the Bavarian State Research Center for Agriculture (http://www.lfl.bayern.de/bazi-rind) (November 2009 version, supporting information, Table S1). Breeding value estimation was based on best linear unbiased prediction (BLUP) animal model. The calving process is described by a score ranging from 1 (unassisted delivery) to 4 (surgical delivery, fetotomy). Stillbirth is recorded as categorical trait (alive or not 48 hr postpartum). Paternal and maternal effects on calving ease and stillbirth incidence are estimated multivariately for the first vs. later parities. Parity-specific EBVs are combined to produce paternal and maternal EBVs, respectively.
Genotypes and quality control:
Of 1829 genotyped FV animals, 6 were excluded from further analyses due to genotype call rates below 90%. The remaining samples exhibited an average genotyping rate of 99.14%. A total of 549 SNPs were omitted because their chromosome position was not known. A total of 728 SNPs were discarded because genotyping failed in more than 10% of animals, 8480 SNPs were excluded due to a minor allele frequency smaller than 1%, and 810 SNPs showed a significant (P < 1 × 10−3) deviation from the Hardy–Weinberg equilibrium, indicating genotyping errors, and were thus not considered for further analyses. Pairwise identity-by-descent (IBD) was calculated on the basis of identity-by-state (IBS) information derived from the remaining 43,863 SNPs using the method-of-moments approach implemented in PLINK (Purcell et al. 2007). The IBD relationship of each pair of animals was compared with the corresponding pedigree relationship calculated using the PyPedal package (Cole 2007). Comparison of the marker with the pedigree relationship revealed several inconsistencies, most likely resulting from mislabeling of DNA samples and false relationships. Unresolved inconsistencies led to the exclusion of 23 animals (Figure S1). The final set consisted of 1800 animals. The phenotype and genotype data are available from the authors upon request.
Single-marker analysis:
Single-marker analysis was first carried out without considering population stratification. The EBVs were regressed on the number of copies of one of the alleles as implemented by the PLINK–assoc option. Quantile–quantile plots of the expected vs. the observed P-values were inspected for an inflation of small P-values indicating false positive association signals due to a structured population. The genome-wide inflation factor was computed according to Devlin and Roeder (1999).
We next applied a PCA-based approach, implemented in the EIGENSOFT 3.0 package (Price et al. 2006), for eliminating false positive association signals due to ancestry differences and resulting population stratification. One SNP of a pair in LD with r2 > 0.25 was excluded using the PLINK–indep-pairwise option (500 SNP window, shifted at 50 SNP intervals). A smartpca version of the EIGENSOFT 3.0 package (compiled from source code with modifications for the bovine chromosome complement) was run on the pruned data set consisting of 20,000 autosomal SNPs with the following option: the value of each marker is replaced with the residuals from a multivariate regression without intercept on the five preceding markers to further reduce redundancies due to LD. Eigenvalues (λ) and eigenvectors were calculated for all axes of variation. Correlation of ancestry adjusted EBVs and genotypes was calculated using the previously obtained eigenvectors with a smarteigenstrat version of EIGENSOFT 3.0 compiled for the bovine chromosome complement. The resulting test statistic is equal to (N − K − 1) times the squared correlation and χ2 distributed, where N is the number of samples and K the number of axes with an eigenvalue that amounts to at least 70% of the mean eigenvalue (Jolliffe's criterion, Jolliffe 2002) used to adjust for ancestry (Price et al. 2006). Quantile–quantile plots were inspected and the genomic inflation factors were calculated (see above) to judge the extent of false positive signals. SNPs were considered as significantly associated for P-values below the 5% Bonferroni-corrected type I error threshold for 43,863 independent tests. Allele substitution effects were estimated for each significant marker in a linear regression model implemented in R (http://www.r-project.org) with axes of variation with λ ≥ 0.7 as covariables.
Haplotype analysis:
Haplotypes for each chromosome region with significant association signals were reconstructed using default parameters in fastPHASE (Scheet and Stephens 2006) and inspected by means of bifurcation plots obtained with sweep (Sabeti et al. 2002) to visualize recombination events and to define the length of haplotypes. The resulting haplotypes were analyzed for association in a multilinear regression model implemented in R (see above).
Estimating the power of the genome-wide association study:
According to Goddard and Hayes (2009) the correlation (r) between marker and trait, 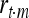 , is equal to
, is equal to 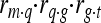 , with m representing the marker genotype, q the QTL, g the genotypic value, and t the phenotypic value (EBV) of an animal.
, with m representing the marker genotype, q the QTL, g the genotypic value, and t the phenotypic value (EBV) of an animal. 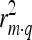 measures the LD between marker and trait,
measures the LD between marker and trait, 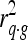 the variance explained by the QTL, and
the variance explained by the QTL, and 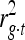 the reliability of the EBV. Using this equation and the formula for the standard error of the correlation coefficient, the number of animals (N) required for identifying a QTL can be calculated as
the reliability of the EBV. Using this equation and the formula for the standard error of the correlation coefficient, the number of animals (N) required for identifying a QTL can be calculated as
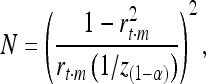 |
where z is the normal score and α the Bonferroni-corrected type I error rate for 43,863 independent tests. Assuming a reliability of the EBV of 0.9, a LD between marker and QTL of r2 = 0.35, and the QTL to explain 4% of the genetic variance, the required number of animals amounts to about 1700. Thus the power of our study with N = 1800 should allow identification of QTL, explaining at least 4% of the genetic variance using EBVs of high reliability.
Annotation and polymorphism analysis of candidate genes:
The GENOMETHREADER software tool (Gremme et al. 2005) was used to predict the genomic structure and localization of the candidate genes based on the University of Maryland UMD3.1 assembly of the bovine genome sequence (Zimin et al. 2009) and the Dana–Farber Cancer Institute bovine gene index release 12.0 (Quackenbush et al. 2001) together with the annotated RNA sequences of the UMD3.1 assembly (Zimin et al. 2009). The GENOMETHREADER output was viewed and edited using the Apollo sequence annotation editor (Lewis et al. 2002). The exons and the promoter regions of the candidate genes were PCR amplified (the primers are listed in Table S2) and resequenced in 12 FV bulls with specific genotypes for the SNP with the most significant signal for the pCE EBV (BTA14–ARS–BFGL–NGS-104268), i.e., in 1 bull with GG and in 11 bulls with AG genotypes.
Genotyping of candidate gene polymorphisms:
Genotypes of selected SNPs were determined by TaqMan genotyping assays (Applied Biosystems Applera, Darmstadt, Germany). DNA samples were available for 810 FV animals only. Candidate gene polymorphisms were genotyped in these animals, and the genotypes of the remaining 990 animals of the study population were inferred using the EM algorithm implemented in fastPHASE.
RESULTS
Single-marker analysis:
In a first attempt to identify QTL for pCE, we applied a linear regression model that did not account for the covariance of related animals. This model yielded 1146 autosomal SNPs exceeding the significance threshold and a genome-wide inflation factor of 4.75. However, an apparent association signal was observed on chromosome 14 (P = 1.64 × 10−55; Figure S2). The inflation of significant association signals most likely results from relatedness of animals leading to a massively structured population. The 1800 bulls within our study descend from 234 sires and 328 maternal grand sires. The paternal half sib families and the maternal grand sire families encompass up to 81 and 137 members, respectively. This is manifested by an average coefficient of relationship of 0.047 and distinct clusters of related animals (Figure S3A and Figure S3B). Recent introgression of Holstein-Friesian (HF) into FV can be uncovered by PCA. A 50% HF sire was broadly used within the FV population in the early 1980s to improve milk performance and udder quality of cows. Of 1800 FV bulls within the study population, 1050 exhibit HF ancestry via two of his sons (both 25% HF), as can be visualized by contrasting the top two axes of variation of the PCA (Figure S3C). Thus, HF admixture and the paternal and maternal sire families lead to a massively structured study population and concomitant inflation of significant association signals.
Therefore, the association study was repeated, now correcting for population stratification using a PCA-based approach implemented in the EIGENSOFT 3.0 package. The correction was based on 773 axes of variation that met the Jolliffe's criterion. In addition to the highly significant association with the pCE EBV on chromosome 14 that was already observed in the analysis without correction for stratification, the PCA-based analysis now also revealed significant association on chromosome 21 (Figure 1A). The Q–Q plot (Figure 1B) and an inflation factor of 0.97 document that the PCA-based analysis successfully eliminated association artifacts resulting from population stratification.
Figure 1.—

Association of 43,863 SNPs with the estimated breeding value (EBV) for paternal calving ease (pCE) in the Fleckvieh breed. (A) Manhattan plot. Red triangles represent SNPs with P < 1.14 × 10−6 (Bonferroni corrected significance level). (B) Quantile–quantile plot. The shaded area represents the 95% concentration band under the null hypothesis of no association. The open black dots represent the P-values of the entire study, open triangles represent SNPs with P < 1 × 10−8, and the solid blue dots indicate the P-values excluding those from the associated regions on chromosomes 14 and 21.
Eight SNPs on chromosome 14 and three SNPs on chromosome 21 meet the Bonferroni-corrected significance threshold (Table 1). Of the eight significant SNPs on chromosome 14, six lie within a 1.4-Mb interval (from 24.06 to 25.4 Mb). Two significant SNPs outside this interval are in LD (r2 = 0.48 and 0.68) with the most significantly associated SNP on chromosome 14. Three significantly associated SNPs in high LD define a second pCE QTL region on chromosome 21 (2.15–2.39 Mb). While the minor allele of the significant SNPs on chromosome 14 has a negative effect on the pCE EBV, it is the major allele of the significant SNPs on chromosome 21 that lowers the pCE EBV. The most significant SNP on chromosome 14 exhibits an allele substitution effect of −7.01, corresponding to 58% of the standard deviation of the EBV. The substitution effect of the major allele of the most significant marker on chromosome 21 is −2.93, i.e., 24% of the standard deviation of the EBV (Figure 2A).
TABLE 1.
SNPs showing significant association with pCE, pSB, DG, and BS EBVs in 1800 Fleckvieh animals
| SNP | Chromosome | Minor allele (minor allele frequency) | Physical position (bp) | EBV | Eigenstrat statistic | P-value | α |
|---|---|---|---|---|---|---|---|
| ARS-BFGL-NGS-104268 | 14 | A (0.12) | 24,057,354 | pCE | 60.99 | 5.72 × 10−15 | −0.58 |
| pSB | 47.12 | 6.69 × 10−12 | −0.54 | ||||
| BTA-91250-no-rs | 14 | A (0.10) | 24,145,838 | pCE | 59.32 | 1.34 × 10−14 | −0.62 |
| pSB | 47.51 | 5.47 × 10−12 | 0.58 | ||||
| BS | 26.89 | 2.15 × 10−7 | 0.45 | ||||
| BTB-01417924 | 14 | G (0.13) | 24,182,406 | pCE | 43.54 | 4.15 × 10−11 | −0.46 |
| pSB | 33.79 | 6.13 × 10−9 | −0.42 | ||||
| Hapmap59686-rs29020689 | 14 | A (0.14) | 24,365,162 | pCE | 39.07 | 4.08 × 10−10 | −0.40 |
| pSB | 38.27 | 6.18 × 10−10 | −0.42 | ||||
| ARS-BFGL-NGS-28867 | 14 | G (0.10) | 20,323,857 | pCE | 38.03 | 6.96 × 10−10 | −0.50 |
| pSB | 33.78 | 6.17 × 10−9 | −0.49 | ||||
| DG | 25.50 | 4.42 × 10−7 | 0.41 | ||||
| UA-IFASA-7112 | 14 | G (0.09) | 16,109,986 | pCE | 36.94 | 1.22 × 10−9 | −0.51 |
| pSB | 32.09 | 1.47 × 10−8 | −0.49 | ||||
| Hapmap46735-BTA-86653 | 14 | G (0.20) | 25,401,722 | pCE | 34.40 | 4.48 × 10−9 | −0.36 |
| pSB | 28.89 | 7.65 × 10−8 | −0.34 | ||||
| ARS-BFGL-NGS-53975 | 21 | G (0.27) | 2,151,256 | pCE | 31.25 | 2.27 × 10−8 | 0.24 |
| BTB-01532239 | 14 | A (0.28) | 24,437,778 | pCE | 28.05 | 1.19 × 10−7 | −0.26 |
| pSB | 26.94 | 2.10 × 10−7 | −0.27 | ||||
| ARS-BFGL-NGS-114372 | 21 | C (0.22) | 2,381,941 | pCE | 25.65 | 4.09 × 10−7 | 0.24 |
| Hapmap52072-rs29018920 | 21 | A (0.22) | 2,333,804 | pCE | 24.12 | 9.01 × 10−7 | 0.23 |
Eleven SNPs meet the genome-wide significance level of P < 1.14 × 10−6. SNPs are arranged in the order of increasing P-values for the association with the paternal calving ease EBV. The P-value for each trait x genotype combination is obtained by a principal components analysis - based approach to account for population stratification. The allelic substitution effect (α) is given for the minor allele in additive genetic standard deviations of the EBV. Physical positions are based on the UMD3.1 assembly of the bovine genome sequence.
Figure 2.—

Effect of the most significantly associated markers on the EBV for pCE in the Fleckvieh breed. The boxplots show the effects of the most significantly associated SNPs (A) and haplotypes (B) on chromosomes 14 and 21 separately and combined. The solid line represents the population mean, and the dotted lines indicate one standard deviation of the EBV.
pCE is highly correlated with the paternal stillbirth incidence (pSB) as well as with growth-related EBVs such as for DG and BS (Table S3). Consequently, association signals can also be observed for these EBVs, particularly on chromosome 14 (Table 1 and Figure S4). The QTL alleles that lower the pCE and pSB EBVs have a positive effect on the growth-related EBVs.
Several chromosome regions show suggestive association (P < 1 × 10−3, Table S4), most prominently a second region on chromosome 14 with 5 SNPs located between 58.3 and 59.3 Mb.
Haplotype analysis:
Haplotype analysis was carried out for the associated regions on chromosomes 14 and 21 in an attempt to delineate the chromosomal segment carrying the pCE QTL. On chromosome 14, the allele that lowers the pCE EBV could be pinpointed to a specific haplotype that spans 1.58 Mb (starting at 23.82 Mb) and encompasses 23 SNPs (Table 2). This haplotype version occurs in a frequency of 10% in the study population. Its negative effect on the pCE EBV (P = 1.56 × 10−16) is more prominent than any of the associated SNPs (−0.66σA vs. −0.62σA, Figure 2B). This is a strong indication for the causal variant lowering the pCE EBV to exclusively reside on this haplotype version.
TABLE 2.
SNPs within the haplotype associated with the estimated breeding value (EBV) for paternal calving ease (pCE) on bovine chromosome 14
| SNP | Physical position (bp) | Haplotype allele | Minor allele (allele frequency) | Eigenstrat statistic | P value | α |
|---|---|---|---|---|---|---|
| BTB-01953819 | 23,817,572 | A | G (0.26) | 0.37 | 0.54 | 0.03 |
| Hapmap45796-BTA-25271 | 23,853,811 | T | A (0.07) | 5.39 | 0.02 | −0.18 |
| ss250608741* | 23,884,989 | G | A (0.09) | 1.06 | 0.3 | 0.06 |
| ARS-BFGL-BAC-8052 | 23,893,220 | G | A (0.01) | 6.72 | 9.55 × 10−3 | −0.45 |
| ARS-BFGL-NGS-97821 | 23,946,436 | G | A (0.1) | 0.98 | 0.32 | 0.07 |
| ARS-BFGL-NGS-104268 | 24,057,354 | A | A (0.12) | 61.00 | 5.71 × 10−15 | −0.58 |
| BTA-91250-no-rs | 24,145,838 | A | A (0.1) | 59.32 | 1.34 × 10−14 | −0.62 |
| BTB-01417924 | 24,182,406 | G | G (0.13) | 43.54 | 4.15 × 10−11 | −0.46 |
| ARS-BFGL-NGS-110427 | 24,326,513 | A | G (0.11) | 0.02 | 0.89 | −0.01 |
| Hapmap59686-rs29020689 | 24,365,162 | A | A (0.14) | 36.94 | 1.22 × 10−9 | −0.40 |
| ARS-BFGL-NGS-102351 | 24,407,125 | G | G (0.25) | 18.34 | 1.85 × 10−5 | −0.21 |
| BTB-01532239 | 24,437,778 | A | A (0.28) | 28.04 | 1.19 × 10−7 | −0.26 |
| BTB-01530788 | 24,524,205 | A | G (0.34) | 8.65 | 3.27 × 10−3 | 0.12 |
| BTB-01530836 | 24,573,257 | G | A (0.35) | 4.30 | 0.04 | 0.07 |
| BTB-00557585 | 24,607,527 | A | G (0.35) | 4.75 | 0.04 | 0.08 |
| BTB-00557532 | 24,643,266 | A | G (0.35) | 4.53 | 0.03 | 0.07 |
| ss250608762* | 24,759,177 | G | T (0.01) | 1.00 | 0.32 | −0.14 |
| Hapmap40120-BTA-34288 | 24,787,245 | C | A (0.09) | 0.28 | 0.6 | −0.05 |
| ss250608721* | 24,954,981 | A | A (0.16) | 58.57 | 1.96 × 10−14 | −0.47 |
| ss250608720* | 24,955,318 | T | C (0.32) | 3.56 | 0.06 | 0.06 |
| Hapmap41234-BTA-34285 | 25,107,556 | G | A (0.04) | 13.89 | 1.94 × 10−4 | −0.42 |
| BTB-02056709 | 25,175,950 | A | G (0.18) | 2.55 | 0.11 | −0.08 |
| BTB-00559128 | 25,215,027 | A | G (0.21) | 0.01 | 0.92 | 0.00 |
| BTB-00557354 | 25,254,540 | G | A (0.12) | 1.63 | 0.2 | 0.09 |
| Hapmap46986-BTA-34282 | 25,307,116 | A | G (0.46) | 9.62 | 1.93 × 10−3 | 0.13 |
| BTB-01779799 | 25,351,733 | G | A (0.44) | 19.00 | 1.30 × 10−5 | 0.19 |
| Hapmap46735-BTA-86653 | 25,401,722 | G | G (0.2) | 34.40 | 4.48 × 10−9 | −0.36 |
Twenty-three SNPs belong to the BovineSNP50 Bead chip collection and four additional SNPs designated by * result from resequencing. The P-values were obtained by using a principal components analysis-based approach to account for population stratification. Genotypes for SNPs resulting from resequencing were determined in 810 animals and imputed for the remaining 990 animals of the study population. The allelic substitution effect (α) is given for the minor allele in additive genetic standard deviations of the pCE EBV. SNPs are arranged according to their physical position, on the basis of UMD3.1 assembly of the bovine genome sequence.
On chromosome 21, the associated SNPs are contained within a haplotype spanning 0.6 Mb (starting at 1.78 Mb). The most frequent haplotype version occurs in a frequency of 66% and has a negative effect on the pCE EBV (P = 3.15 × 10−7). However, it explains less of the genetic variance than the most significant SNP does (−0.18σA vs. −0.24σA).
Identification and analysis of candidate genes:
The assessment of the transcriptional content of the pCE EBV-associated regions was based on the UMD3.1 assembly and annotation of the bovine genome (Zimin et al. 2009). The 23.82–25.40 Mb interval on chromosome 14 encompasses 13 transcripts/genes (Figure 3A). The associated region on bovine chromosome 14 is conserved in human chromosome 8q21, which has been shown to be associated with adult height (Gudbjartsson et al. 2008). Since adult stature is positively correlated with fetal size and fetal size is an important determinant of the birthing process, we considered PLAG1, MOS, CHCHD7, RDHE2 (alias SDR16C5), RPS20, LYN, TGS1, PENK, as proposed by Gudbjartsson et al. (2008) as positional and functional candidate genes for the pCE QTL in cattle. Of this list, PLAG1, TGS1, RPS20, and LYN together with SOX17, another gene in the critical region that we considered a functional candidate, were resequenced in a panel of 12 animals of our study population. In total, we screened 30.3 kb resulting in the detection of 48 polymorphisms (Table S5). We decided to genotype four putatively functional SNPs, located in SOX17 (ss250608762), RPS20 (ss250608720, ss250608721), and TGS1 (ss250608741), in 810 animals and analyzed the association with the pCE EBV in the complete study population using genotype imputation (Figure 3, B and C). Only ss250608721 produced a highly significant signal (P = 1.96 × 10−14). The polymorphism affects a polyadenylation signal of a cistron encompassing the genes for the ribosomal protein S20 (RPS20, a ribosomal component) and the small nucleolar RNA U54 (SNORD54, a ribosomal RNA modifying RNA) (Figure 4).
Figure 3.—

Detailed view of the region on chromosome 14 delineated by the haplotype associated with the EBV for pCE. (A) Map of genes contained in this region. Red symbols indicate genes resequenced in this study. (B) P-values of 27 SNPs from analysis of association with the pCE EBV. The open black dots indicate results from genotyping of the entire study population, and the blue triangles represent P-values resulting from imputation based on 810 genotyped animals. (C) Heatmap of the pairwise linkage disequilibrium (r2). The triangle delineates a linkage disequilibrium block containing the most significantly associated SNPs, including the potentially functional ss250608721 variant in RPS20.
Figure 4.—

Predicted 3′-UTR of cattle RPS20. The gray-shaded sequence designates the predicted exon 4, while the predicted polyadenylation [poly(A)] sites are denoted by underscoring. The star locates the candidate quantitative trait nucleotide position, ablating a poly(A) site.
The association signals on chromosome 21 result from the most proximal region on the chromosome (Figure S5). The region contains, among other transcripts, those encoding SNURF–SNRPN and UBE3A. These two transcripts are encoded in the human chromosome interval 15q11–15q13 that is subject to imprinting. The lack of a functional paternal copy of 15q11–15q13 causes the Prader–Willi syndrome, while the lack of a functional maternal copy of UBE3A is implicated in the Angelman syndrome (Horsthemke and Wagstaff 2008). The SNURF–SNRPN mRNA is derived from a single large transcriptional unit of which more than 70 snoRNAs of the C/D box type are processed (Bachellerie et al. 2002). Preliminary BLAST analyses indicate the presence of a snoRNA cluster in the proximal region of bovine chromosome 21. However, a systematic annotation has not been attempted. The lack of detailed knowledge of the genomic organization, the imprinting status and transcriptional content of the associated region on chromosome 21 precluded the analysis of candidate genes, although a functional implication of the region in fetal growth and thus pCE seems obvious when considering that fetal growth retardation is symptomatic for the Prader–Willi syndrome.
DISCUSSION
Our genome-wide association study based on a dense SNP marker map provides strong evidence for two QTL on chromosomes 14 and 21, respectively, that together explain at least 10% of the variation of the pCE EBV in the German FV breed. The two QTL also explain a substantial fraction of the pSB EBV as well as of EBVs of postnatal growth such as DG and BS. Stillbirth can be considered as the dichotomic manifestation of the calving-ease score, as dystocia is a major cause of perinatal mortality. The correlation of pCE with growth-related traits and the coincident QTL point to fetal growth and the resulting birth weight as major determinant for the ease of delivery (Meijering 1984, Johanson and Berger 2003). Thus, the two QTL might primarily affect fetal growth. One could expect that they would explain a larger fraction of the genetic variation of birth weight, a trait that is not routinely measured in dairy cattle. Improving postnatal growth along with lactation traits is a major breeding objective of the FV breed. This dual purpose selection is likely to act on the two QTL identified in our study. Animals known to carry favorable alleles for the chromosome 14 and 21 QTL could now be more stringently selected with regard to beef traits. However, the identification of QTL that either affect prenatal or postnatal growth but not both would facilitate the efficient improvement of postnatal beef performance without antagonistically compromising calving ease. In any case, conventional selection schemes seem to allow favorable selection responses for calving ease and postnatal growth despite the genetic antagonism (MacNeil 2003, Bennett 2008, Bennett et al. 2008).
A key factor for successfully mapping a QTL for a complex trait with very low heritability such as pCE was the use of reliably estimated breeding values for calving traits. If one assumes a heritability of 0.08, a LD between marker SNPs and QTL of r2 = 0.35 and 4% of the genetic variation explained by the QTL, one would require approximately 20,000 individuals for the successful identification of a QTL (see material and methods). Using EBVs with a reliability of 90%, i.e., a quasi-heritability of 0.9, requires less than 1800 animals to detect association. Breeding values are routinely estimated for many traits and are thus indispensable for dissecting complex trait variation in livestock species.
Another key factor for successfully mapping the two QTL was careful correction for extensive relationship among the study animals. The adjustment along 773 axes of variation allowed us to account for major as well as for more subtle relationships that can possibly not be revealed by pedigree analyses. The association signal on chromosome 21 became apparent only when population structure was corrected for. Thus, PCA-based elimination of false positive association signals might enable the detection of QTL with a smaller impact on the trait variation that would otherwise be “buried” in the false positive signals. Suggestive signals (P < 1 × 10−3, Table S4) are thus more likely to represent real QTL.
Our findings about two highly significant QTL for pCE as well as about additional suggestive QTL are supported by several previous studies on calving ease and growth trait QTL, based on microsatellite marker analyses. Kneeland et al. (2004) identified three regions on chromosome 14 to affect birth weight in a composite breed. The proximal region from 26.0 to 26.7 cM most likely corresponds to the highly significant QTL region identified in our study, the more distal region between 36.2 and 46.2 cM may corroborate a suggestive QTL region resulting from our study. Davis et al. (1998) also identified a QTL affecting birth weight at 42 cM. Koshkoih et al. (2006) provide additional evidence for two birth weight QTL on chromosome 14 at 26 and 50 cM, respectively, in a cross of Limousin and Jersey animals. Maltecca et al. (2009) recently identified a birth weight QTL at 19 cM on chromosome 14 in a Jersey–Holstein cross. There are also reports on QTL for postnatally measured growth traits in Wagyu (Mizoshita et al. 2004, Takasuga et al. 2007) and a Jersey–Limousin cross (Morris et al. 2002), indirectly supporting our suggestive evidence for a secondary pCE QTL on chromosome 14. Casas et al. (2003) and Davis et al. (1998) identified a QTL for birth weight in the very proximal region of chromosome 21 in crosses of Brahman with Hereford and Charolais, respectively, providing supportive evidence for the pCE QTL identified in this study.
There is also support in the literature for suggestive QTL on other chromosomes: Olsen et al. (2009) and Holmberg and Andersson-Eklund (2006) identified in a Swedish and Norwegian dairy cattle population, respectively, a dystocia/stillbirth QTL at 36–37 cM on chromosome 6. We observe a suggestive pCE QTL at about 40 Mb on chromosome 6. Gutierrez-Gil et al. (2009) identified a fetal growth/birth weight QTL in the same region on the basis of a Charolais–Holstein cross. Eberlein et al. (2009) provide evidence for the gene (NCAPG) encoding the Non-SMC Condensin I Complex, Subunit G, to encompass this QTL, also based on a Charolais–Holstein cross. However, a prominent calving-ease QTL in the Holstein breed on chromosome 18 (Cole et al. 2009) could not be detected in this study or is not segregating in the Fleckvieh breed.
A preliminary candidate gene analysis identified a highly significantly pCE-associated SNP in a cistron encoding a ribosomal protein (RPS20) and an internally nested small nucleolar RNA (SNORD54). The SNP affects a polyadenylation site. Alternative polyadenylation at tandem poly(A) sites yields transcripts with different 3′-UTR sequences providing the potential of differential regulation of mRNA expression by RNA binding proteins and/or miRNAs (Sandberg et al. 2008, Licatalosi and Darnell 2010). The marker allele causing the gain of an upstream polyadenylation signal is associated with a lower pCE EBV, i.e., a higher incidence of calving difficulties. This is hypothetically compatible with a shorter and more highly expressed mRNA encoding ribosomal components, leading to a higher ribosome assembly rate and concomitantly stronger fetal growth. Thus we consider the polymorphism as a candidate quantitative trait nucleotide position. Interestingly, the pCE QTL on BTA21 is also in a chromosome region encoding factors involved in ribosomal assembly, specifically small nucleolar RNAs. It is therefore possible that both QTL affect ribosomal biogenesis. Mutations disturbing the ribosome assembly are often associated with abnormal fetal growth (Lempiäinen and Shore 2009, Freed et al. 2010).
Acknowledgments
This study is part of the project Funktionelle GenomAnalyse im Tierischen Organismus (FUGATO)-plus GenoTrack and was financially supported by the German Ministry of Education and Research, Bundesministerium für Bildung und Forschung (BMBF; grants 0315134A and 0315134D), the Förderverein Biotechnologieforschung e.V. (F.B.F.), Bonn, and Lohmann Tierzucht GmbH, Cuxhaven.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.124057/DC1.
References
- Bachellerie, J., J. Cavaillé and A. Hüttenhofer, 2002. The expanding snoRNA world. Biochimie 84 775–790. [DOI] [PubMed] [Google Scholar]
- Barendse, W., A. Reverter, R. J. Bunch, B. E. Harrison, W. Barris et al., 2007. A validated whole-genome association study of efficient food conversion in cattle. Genetics 176 1893–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, G. L., 2008. Experimental selection for calving ease and postnatal growth in seven cattle populations. I. Changes in estimated breeding values. J. Anim. Sci 86 2093–2102. [DOI] [PubMed] [Google Scholar]
- Bennett, G. L., R. M. Thallman, W. M. Snelling and L. A. Kuehn, 2008. Experimental selection for calving ease and postnatal growth in seven cattle populations. II. Phenotypic differences. J. Anim. Sci. 86 2103–2114. [DOI] [PubMed] [Google Scholar]
- Casas, E., S. D. Shackelford, J. W. Keele, M. Koohmaraie, T. P. L. Smith et al., 2003. Detection of quantitative trait loci for growth and carcass composition in cattle. J. Anim. Sci 81 2976–2983. [DOI] [PubMed] [Google Scholar]
- Cole, J., 2007. PyPedal: a computer program for pedigree analysis. Comput. Electronics Agric. 57 107–113. [Google Scholar]
- Cole, J. B., P. M. VanRaden, J. R. O'Connell, C. P. Van Tassell, T. S. Sonstegard et al., 2009. Distribution and location of genetic effects for dairy traits. J. Dairy Sci. 92 2931–2946. [DOI] [PubMed] [Google Scholar]
- Daetwyler, H. D., F. S. Schenkel, M. Sargolzaei and J. A. B. Robinson, 2008. A genome scan to detect quantitative trait loci for economically important traits in Holstein cattle using two methods and a dense single nucleotide polymorphism map. J. Dairy Sci. 91 3225–3236. [DOI] [PubMed] [Google Scholar]
- Davis, G. P., D. J. S. Hetzel, N. J. Corbet, S. Scacheri, S. Lowden et al., 1998. The mapping of quantitative trait loci for birth weight in tropical beef herd. Proceedings of the 6th World Congress on Genetics Applied to Livestock Production, Armidale, N.S.W., Australia, Vol. 26, pp. 441–446.
- Devlin, B., and K. Roeder, 1999. Genomic control for association studies. Biometrics 55 997–1004. [DOI] [PubMed] [Google Scholar]
- Eberlein, A., A. Takasuga, K. Setoguchi, R. Pfuhl, K. Flisikowski et al., 2009. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics 183 951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed, E. F., F. Bleichert, L. M. Dutca and S. J. Baserga, 2010. When ribosomes go bad: diseases of ribosome biogenesis. Mol. Biosyst. 6 481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freer, B., 2008. Easy calving: not so difficult. Hereford Breed J. 2008 176–177. [Google Scholar]
- Goddard, M. E., and B. J. Hayes, 2009. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10 381–391. [DOI] [PubMed] [Google Scholar]
- Gremme, G., V. Brendel, M. E. Sparks and S. Kurtz, 2005. Engineering a software tool for gene structure prediction in higher organisms. Inform. Software Technol. 47 965–978. [Google Scholar]
- Gudbjartsson, D. F., G. B. Walters, G. Thorleifsson, H. Stefansson, B. V. Halldorsson et al., 2008. Many sequence variants affecting diversity of adult human height. Nat. Genet. 40 609–615. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Gil, B., J. L. Williams, D. Homer, D. Burton, C. S. Haley et al., 2009. Search for quantitative trait loci affecting growth and carcass traits in a cross population of beef and dairy cattle. J. Anim. Sci. 87 24–36. [DOI] [PubMed] [Google Scholar]
- Holmberg, M., and L. Andersson-Eklund, 2006. Quantitative trait loci affecting fertility and calving traits in Swedish dairy cattle. J. Dairy Sci. 89 3664–3671. [DOI] [PubMed] [Google Scholar]
- Horsthemke, B., and J. Wagstaff, 2008. Mechanisms of imprinting of the Prader–Willi/Angelman region. Am. J. Med. Genet. A 146A 2041–2052. [DOI] [PubMed] [Google Scholar]
- Johanson, J. M., and P. J. Berger, 2003. Birth weight as a predictor of calving ease and perinatal mortality in Holstein cattle. J. Dairy Sci. 86 3745–3755. [DOI] [PubMed] [Google Scholar]
- Jolliffe, I. T., 2002. Principal Component Analysis, Ed. 2. Springer, New York.
- Kneeland, J., C. Li, J. Basarab, W. M. Snelling, B. Benkel et al., 2004. Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J. Anim. Sci. 82 3405–3414. [DOI] [PubMed] [Google Scholar]
- Koshkoih, A. E., W. S. Pitchford, C. D. K. Bottema, A. P. Verbyla and A. R. Gilmour, 2006. Mapping multiple QTL for birth weight using a mixed model approach. Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, MG, Brazil, August 13–18, 2006.
- Lempiäinen, H., and D. Shore, 2009. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 21 855–863. [DOI] [PubMed] [Google Scholar]
- Lewis, S. E., S. M. J. Searle, N. Harris, M. Gibson, V. Lyer et al., 2002. Apollo: a sequence annotation editor. Genome Biol. 3 RESEARCH0082. [DOI] [PMC free article] [PubMed]
- Licatalosi, D. D., and R. B. Darnell, 2010. RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 11 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H. K., P. A. Oltenacu, L. D. Van Vleck, H. N. Erb and R. D. Smith, 1989. Heritabilities of and genetic correlations among six health problems in Holstein cows. J. Dairy Sci. 72 180–186. [DOI] [PubMed] [Google Scholar]
- MacNeil, M. D., 2003. Genetic evaluation of an index of birth weight and yearling weight to improve efficiency of beef production. J. Anim. Sci. 81 2425–2433. [DOI] [PubMed] [Google Scholar]
- Maltecca, C., K. A. Weigel, H. Khatib, M. Cowan and A. Bagnato, 2009. Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein × Jersey crossbred population. Anim. Genet. 40 27–34. [DOI] [PubMed] [Google Scholar]
- Manolio, T. A., F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorff et al., 2009. Finding the missing heritability of complex diseases. Nature 461 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering, A., 1984. Dystocia and stillbirth in cattle: a review of causes, relations and implications. Livestock Prod. Sci. 11 143–177. [Google Scholar]
- Mizoshita, K., T. Watanabe, H. Hayashi, C. Kubota, H. Yamakuchi et al., 2004. Quantitative trait loci analysis for growth and carcass traits in a half-sib family of purebred Japanese Black (Wagyu) cattle. J. Anim. Sci. 82 3415–3420. [DOI] [PubMed] [Google Scholar]
- Morris, C. A., W. S. Pitchford, N. G. Cullen, S. M. Hickey, D. L. Hyndman et al., 2002. Additive effects of two growth QTL on cattle chromosome 14. Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, August 19–23, 2002.
- Olsen, H. G., B. J. Hayes, M. P. Kent, T. Nome, M. Svendsen et al., 2009. A genome-wide association study for QTL affecting direct and maternal effects of stillbirth and dystocia in cattle. Anim. Genet. 41 273–280. [DOI] [PubMed] [Google Scholar]
- Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick et al., 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 904–909. [DOI] [PubMed] [Google Scholar]
- Purcell, S., B. Neale, K. Todd-Brown, L. Thomas, M. A. R. Ferreira et al., 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush, J., J. Cho, D. Lee, F. Liang, I. Holt et al., 2001. The TIGR gene indices: analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 29 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti, P. C., D. E. Reich, J. M. Higgins, H. Z. P. Levine, D. J. Richter et al., 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419 832–837. [DOI] [PubMed] [Google Scholar]
- Sandberg, R., J. R. Neilson, A. Sarma, P. A. Sharp and C. B. Burge, 2008. Proliferating cells express mRNAs with shortened 3′ UTRs and fewer microRNA target sites. Science 320 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheet, P., and M. Stephens, 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenspinner, T., J. Bennewitz, F. Reinhardt and G. Thaller, 2009. Need for sharp phenotypes in QTL detection for calving traits in dairy cattle. J. Anim. Breed. Genet. 126 455–462. [DOI] [PubMed] [Google Scholar]
- Steinbock, L., A. Nasholm, B. Berglund, K. Johansson and J. Philipsson, 2003. Genetic effects on stillbirth and calving difficulty in Swedish Holsteins at first and second calving. J. Dairy Sci. 86 2228–2235. [DOI] [PubMed] [Google Scholar]
- Takasuga, A., T. Watanabe, Y. Mizoguchi, T. Hirano, N. Ihara et al., 2007. Identification of bovine QTL for growth and carcass traits in Japanese black cattle by replication and identical-by-descent mapping. Mamm. Genome 18 125–136. [DOI] [PubMed] [Google Scholar]
- Van Tassell, C. P., G. R. Wiggans and I. Misztal, 2003. Implementation of a sire-maternal grandsire model for evaluation of calving ease in the United States. J. Dairy Sci. 86 3366–3373. [DOI] [PubMed] [Google Scholar]
- Zimin, A. V., A. L. Delcher, L. Florea, D. R. Kelley, M. C. Schatz et al., 2009. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 10 R42. [DOI] [PMC free article] [PubMed] [Google Scholar]