

Abstract
Bisphosphonates (BPs) are a class of compounds that have been used extensively in the treatment of osteoporosis and malignancy-related hypercalcemia. Some of these compounds act through inhibition of farnesyl diphosphate synthase (FPPS), a key enzyme in the synthesis of isoprenoids. Recently, nitrogen-containing bisphosphonates (N-BPs) used in bone resorption therapy have been shown to be active against Trypanosoma cruzi, the parasite that causes American trypanosomiasis (Chagas disease), suggesting that they may be used as anti-trypanosomal agents. The crystal structures of TcFPPS in complex with substrate (isopentenyl diphosphate, IPP) and five N-BP inhibitors show that the C-1 hydroxyl and the nitrogen-containing groups of the inhibitors alter the binding of IPP and the conformation of two TcFPPS residues, Tyr94 and Gln167. Isothermal titration calorimetry experiments suggest that binding of the first N-BPs to the homodimeric TcFPPS changes the binding properties of the second site. This mechanism of binding of N-BPs to TcFPPS is different to that reported for the binding of the same compounds to human FPPS.
Introduction
Bisphosphonates (BPs) are a class of drugs that have been used extensively in the treatment of osteoporosis and malignancy-related hypercalcemia. BPs can be divided into two classes based on whether or not a nitrogen-containing group is present in the side-chain. The two classes of BPs differ in their mechanisms of action. Older, non-nitrogen-containing BPs such as etidronate are pyrophosphate analogues and are metabolized to cytotoxic ATP analogues, leading to apoptosis of osteoclasts. In contrast, the nitrogen-containing BPs (N-BPs) such as pamidronate are not metabolized. Instead, they act as analogues of isoprenoid diphosphates, inhibiting the enzyme farnesyl diphosphate synthase (FPPS) 1.
FPPSs, homodimeric proteins belonging to the isoprenyl diphosphate synthase family, are key enzymes in the synthesis of isoprenoids. They catalyze the successive condensation of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) to generate geranyl diphosphate (GPP), then farnesyl diphosphate (FPP). GPP and FPP are precursors of most isoprenoid compounds, including cholesterol and other sterols. Moreover, post-translational farnesylation is required for the membrane localization of many proteins, including small GTPases. Through their inhibitory effects on FPPS, N-BPs interfere with many normal functions of cells. However, due to their highly preferred absorption by bone tissue, N-BPs are preferentially cytotoxic to osteoclasts, with limited side effects. Recently, bisphosphonates were demonstrated to possess in vitro and in vivo efficacy against trypanosomes (T. cruzi and T. brucei)2–7, making them promising therapeutic agents against trypanosomal infections. The target of these compounds is the parasites’ FPPS. These enzymes share 36% sequence identity with the human enzyme.
The structures of FPPS from T. cruzi, the causative agent of Chagas disease 8, were recently determined by X-ray crystallography, both alone and complexed with the N-BP inhibitors, risedronate and alendronate 9. These structures provided insight into the interaction of N-BPs with critical TcFPPS residues at the enzyme active site. They also show that binding of substrate and inhibitor results in significant conformational changes, especially in the helical region spanning residues 92 to 102 and the loop extending from residues 103 to 113. Comparison of the structures of trypanosomal and human FPPS revealed that their active site residues are highly conserved, in particular those involved in the interaction with the substrates and the N-BPs, underscoring the difficulty of designing parasite-specific drugs.
To explore other features of N-BPs that might be exploited for the development of TcFPPS-specific inhibitors, we have determined the crystal structures of TcFPPS in complexes with five N-BPs, both in the absence and in the presence of IPP. These structures reveal an unexpected effect of the C-1 hydroxyl of many N-BPs on the binding of the physiological substrate IPP: it causes disordering of the isopentenyl end of the substrate IPP. The conformational changes that accompany substrate and inhibitor binding raise another important question: does the changes that take place upon ligand binding to the first monomer of the homodimer affect the conformation and/or the affinity of the second monomer? To address this question, we investigated the thermodynamics of N-BP binding by isothermal titration calorimetry (ITC). Taken together, the structural and thermodynamic results suggest that binding of the first N-BP to the TcFPPS dimer modifies the binding properties of the second site. This result differs from the situation in human FPPS (hFPPS) and may provide additional clues for the design of TcFPPS-specific inhibitors for the treatment of Chagas disease.
Materials and Methods
Protein expression and purification
TcFPPS was cloned and expressed as reported before 9. Briefly, DNA coding for TcFPPS with an N-terminal His-tag and a thrombin site was cloned into the pET28a vector (Novagen). BL21(DE3) E. coli cells transformed with this plasmid were grown in LB medium. After the culture reached an OD600 of 0.9, expression of TcFPPS was induced with 1 mM IPTG for 3 hours at 37°C. Cells were harvested and resuspended in buffer A (50 mM NaH2PO4/Na2HPO4 pH 8.0, 400 mM NaCl, 10 mM imidazole, 1mM TCEP [tris(2-carboxyethyl)phosphine]) and disrupted with a microfluidizer. Cell debris was pelleted and the supernatant loaded onto a nickel affinity column, which was then washed with buffer A. The protein was eluted with a gradient of 0 to 100% of buffer B (buffer A with 250 mM imidazole). Following gel filtration chromatography in 20 mM Tris HCl pH 8.0, 50 mM NaCl, and 2mM TCEP, the His-tag was removed for TcFPPS samples used for crystallization, by thrombin cleavage overnight at 4°C. A second nickel affinity column was then used to remove uncleaved protein, with a final anion exchange chromatography step (binding buffer: 20 mM Tris, 10 mM NaCl, 1 mM TCEP; elution buffer: 20 mM Tris, 250 mM NaCl, 1 mM TCEP) resulting in TcFPPS of over 95% purity, as judged by SDS-PAGE. The protein was dialyzed against 20 mM Tris HCl pH 8.0, 50 mM NaCl, and 2 mM TCEP and concentrated to 12–20 mg/ml.
Crystallization
For cocrystallization of TcFPPS with bisphosphonate inhibitors and IPP, 25 μL of purified TcFPPS (12–20 mg/ml) was first mixed with 2 μL 100 mM MgCl2, 2 μL 10 mM IPP, and 2 μL inhibitor stock solution (25 mM zoledronate [91B], 5 mM minodronate [261A], 10 mM 300B, 100 mM 461 A, 100 mM 476A; the structures of these inhibitors are shown in Figure 1). Crystals were grown in hanging drops by addition of 1 μl of the FPPS-IPP-inhibitor mixture to 1 μl of reservoir containing 0.1 M sodium acetate, pH 4.6–5.2, 0.2 M ammonium sulfate, and 2–10% PEG 4K. Tiny hexagonal crystals appeared within 20 min of the drops being set up (in contrast to TcFPPS apo enzyme crystals, which normally took from hours to days to appear) and reached their final sizes of 100 μm × 100 μm × 50 μm in two to three days. Crystals were frozen with glycerol as cryoprotectant (1:4 v/v).
Figure 1. Compounds used in this study.
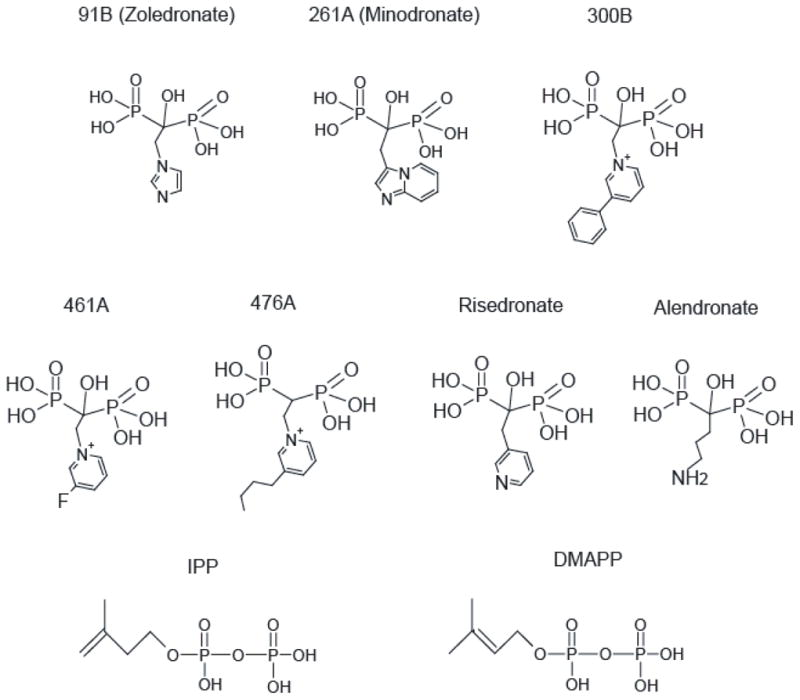
91B, 261A, 300B, 461A, and 476A were used for structure determination and, along with risedronate, in ITC experiments. The structures of alendronate, IPP, and DMAPP are shown for comparison.
Structure determinations
X-ray diffraction data collections were carried out at Beamlines X29A and X6A of the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory and the SGX Collaborative Access Team (SGX-CAT) beamline at the Advanced Photon Source (APS). The structures of TcFPPS-inhibitor-IPP-Mg2+ complexes were determined at resolutions between 2.0 and 2.4 A by molecular replacement with apo TcFPPS (PDB ID: 1YHK) 9 as the search molecule, using the program AMoRe 10. Model building was carried out using the program O 11 and refined with the program REFMAC5 of the CCP4 Suite 12.
ITC (Isothermal Titration Calorimetry)
Purified TcFPPS was diluted to a concentration of 0.9 mg/ml (22.5 μM) and dialyzed against a buffer containing 20 mM HEPES pH 7.5, 1 M NaCl, 2 mM MgCl2, and 2 mM TCEP. Bisphosphonate inhibitors (250 μM) were prepared in the same buffer. ITC experiments were carried out at 26°C with a high-precision VP-ITC titration calorimeter system (Microcal Inc.). Ten microliters of the inhibitor solutions were added every 300 seconds to the cell containing 1.4 ml of dialyzed TcFPPS. The heat of binding was obtained by integrating the calorimetric signal, subtracting the corresponding heat of dilution. The first injections were excluded from the analyses. Data were analyzed using Origin 5.0 (Microcal Software, Inc., Northampton, MA). Details of the analyses are presented in the Results section.
Results
Binding of bisphosphonate inhibitors to FPPS
Crystal structures of TcFPPS in complex with IPP and each of five bisphosphonate inhibitors were determined at resolutions between 2.0 and 2.4 Å (see Table 1 for statistics of data collection and refinement). In all these structures, two TcFPPS monomers are related by a crystallographic two-fold axis, corresponding to the tightly coupled physiological homodimer. Each TcFPPS monomer in the homodimer contains two pockets at the active site to accommodate its two substrates: the allylic binding site (for DMAPP or GPP), and the homoallylic binding site (for IPP) 13. In all five TcFPPS-bisphosphonate structures determined here, as well as in the two previously determined structures 9, the inhibitors bind to the allylic site, with the bisphosphonate group coordinated by three Mg2+ ions (Figure 2A, 2B). When different inhibitor-bound structures are superimposed, there is an almost perfect overlap of the bisphosphonate geminal carbons and of the phosphonates (r.m.s.d < 0.47 Å), with slight variations in the planes of the nitrogen-containing heterocyclic rings (Figure 2C).The active site cavity, identified by the presence of the inhibitors and IPP, is lined by the two conserved Asp-rich motifs (residues 98–102 and 250–254) characteristic of FPPSs. Compared to the structure of the apo enzyme, these inhibitor-bound structures reveal a hinge-like conformational change that closes the enzyme around the active site, as observed in the structures of TcFPPS in complexes with risedronate/DMAPP (PDB ID: 1YHL) and with alendronate/IPP (PDB ID: 1YHM) 9. The r.m.s.d of C-α between pairs of inhibitor-bound TcFPPS structures vary from 0.2–0.5 Å. In contrast, the C-α r.m.s.d. between apo TcFPPS and TcFPPS-91B (zoledronate)-IPP is 0.72 Å, and the r.m.s.d. between the two Asp-rich active site segments of these two structures is 1.3 Å.
Table 1.
Statistics of data collection and refinement
| 91B+IPP | 261A+IPP | 300B + IPP | 461A + IPP | 476A+IPP | 461A | 91B | |
|---|---|---|---|---|---|---|---|
| Space Group | P6122 | P6122 | P6122 | P6122 | P6122 | P6122 | P6122 |
| Unit Cell | a,b=58.3 Å, c=390.4 Å | a,b=58.1 Å, c=390.6 Å | a,b=58.3 Å, c=394.2 Å | a,b=58.0 Å, c=389.7 Å | a,b=58.4 Å, c=391.8 Å | a,b=58.2 Å, c=390.2 Å | a,b=103.6 Å, c=386.7 Å |
| X-ray Source | BNL-X29 | BNL-X6a | BNL-X6a | BNL-X6a | BNL-X6a | APS SGX-CAT | APS SGX-CAT |
| Resolution(Å) | 2.40 | 2.40 | 2.00 | 2.40 | 2.15 | 2.80 | 3.71 |
| Measured Reflections | 116,898 | 136,943 | 284,397 | 63,888 | 229,077 | 160,609 | 274,040 |
| Unique Reflections | 14,730 | 15,342 | 27,660 | 16,079 | 21,604 | 9382 | 13809 |
| Completeness (%) | 88.4(85.1) | 92.2(71.0) | 96.6(69.8) | 97.0(87.7) | 93.4(73.4) | 88.1(69.9) | 99.2(94.2) |
| Rmerge (%) | 7.9(17.1) | 9.0 (52.6) | 7.5(43.0) | 7.9(23.9) | 9.6(22.7) | 5.7(18.4) | 7.1(11.9) |
| <I>/<sigma> | 45.8(16.6) | 23.4(2.7) | 32.9(1.5) | 14.7(3.5) | 29.8(3.6) | 58.8(9.5) | 83.9(41.8) |
| Redundancy | 7.9(7.3) | 8.9(6.1) | 10.3(3.3) | 4.0(2.2) | 10.6(5.0) | 17.1(9.6) | 19.8(14.0) |
| Refinement | |||||||
| Rcryst | 0.23 | 0.19 | 0.19 | 0.18 | 0.19 | 0.22 | 0.24 |
| Rfree | 0.29 | 0.26 | 0.25 | 0.24 | 0.26 | 0.28 | 0.30 |
| r.m.s.d | |||||||
| Bonds (Å) | 0.006 | 0.017 | 0.020 | 0.018 | 0.021 | 0.007 | 0.006 |
| Angles (°) | 0.9 | 1.9 | 1.8 | 1.7 | 1.8 | 1.0 | 0.9 |
| Total atoms | 3224 | 3175 | 3232 | 3220 | 3224 | 2963 | 8745 |
| Protein atoms | 2891 | 2891 | 2891 | 2891 | 2891 | 2891 | 8673 |
| Water | 294 | 230 | 291 | 284 | 295 | 38 | 0 |
| Ligands | 1 91B | 1 261A | 1 300B | 1 461A | 1 476A | 1 461A | 1 91B |
| 1 IPP | 1 IPP | 1 IPP | 1 IPP | 1 IPP | |||
| Metals | 3 Mg2+ | 3 Mg2+ | 3 Mg2+ | 3 Mg2+ | 3 Mg2+ | 3 Mg2+ | 3 Mg2+ |
Figure 2. The allylic site of TcFPPS in complex with N-BPs.

(A) A 2Fo-Fc electron density map superimposed on the ball-and-stick models of 91B. (B) Magnesium ions in TcFPPS-91B are represented by green spheres, with coordination shown as dashed lines. (C) Stereo view of the superposition of allylic sites from TcFPPS in complex with 91B (green), 261A (cyan), 300B (magenta), 461A (yellow), and 476A (gray), with the N-BPs shown as ball-and-stick models.
Conformations of Tyr94 and Gln167
The nitrogen-containing groups of bisphosphonates are located in a pocket formed by the following residues: Tyr94, Leu95, Thr163, Gln167, Lys207, Thr208, and Gln247. Despite the overall similarities of the conformations of the active sites of TcFPPS bound to different bisphosphonates, the side chains of two residues, Tyr94 and Gln167, show substantial differences that correlate with the overall length of the bisphosphonate side chain. Upon binding of 91B, 261A, or 461A, which have small nitrogen-containing groups, the side chains of Tyr94 and Gln167 are close to each other (Figure 3A), while the longer nitrogen-containing groups of 300B and 476A cause these two residues to become more separated (the distance between the oxygen atoms on their side chains increases by ~2.0 Å, Figure 3B). Since FPPS uses both DMAPP and GPP as substrates, such conformational flexibility may reflect the need of these active site residues to accommodate the different lengths of the two physiological substrates.
Figure 3. Alternative conformations of Tyr94 and Gln167 (Stereo view).

The conformations of Tyr94 and Gln167 are dependent on the size of the nitrogen-containing group in the bisphosphonate inhibitors. Tyr94, Gln167, and N-BPs are shown as sticks. The same color coding as in Figure 2C is used for different N-BPs. (A) In the structures of TcFPPS in complex with 91B, 261A, and 461A, where the nitrogen-containing groups are shorter, Tyr94 and Gln167 are closer to each other. (B) These two residues become more separated (by ~ 2.0 Å)accommodate the longer nitrogen-containing groups of 300B and 476A. (C) Superposition of the two conformational states of residues Tyr94 and Gln167, as well as the structure of human FPPS/zoledronate (PDB ID: 1ZW5, blue) 14.
Binding to the homoallylic site of TcFPPS
Only in the structures of TcFPPS complexed with 476A and IPP is density for an IPP molecule clearly visible in the homoallylic site of an omit electron density difference map. Compound 476A differs from the other four bisphosphonates used in this study in that it lacks the C-1 hydroxyl group (Figure 1). When IPP was built into the homoallylic site of the other four TcFPPS-inhibitor structures, the IPP model fitted poorly into the 2Fo-Fc electron density map. We could not rule out the possibility that bisphosphonates, which are structural analogues of both IPP and DMAPP, might also bind to the homoallylic site. However, attempts to build the bisphosphonate inhibitors into the electron density were not satisfactory either. The possibility remained that the maps reflected partial occupancy of the homoallylic sites IPP and by bisphosphonates.
To answer the question as to whether bisphosphonates are able to bind to the homoallylic site, TcFPPS was cocrystallized with bisphosphonates in the absence of IPP. One of these, TcFPPS cocrystallized with 10 mM 461A, diffracted to 2.8 Å, and the structure was determined and refined to R/Rfree of 0.22/0.28 (Table 1). In addition to density for the inhibitor in the allylic site, the 2Fo-Fc map shows a roughly spherical density at the homoallylic site but it was not big enough to accommodate 461A. Instead, a molecule of sulfate, a component of the crystallization buffer, fits well into this electron density. In support of this model, the crystal structure of the apo TcFPPS contained a sulfate molecule at the same location within the homoallylic site 9. This result clearly shows that this bisphosphonate only binds to the allylic site in TcFPPS. In a 3.7 Å resolution map of TcFPPS complexed with compound 91B in the absence of IPP (Table 1), electron density consistent with a sulfate molecule is also seen at the homoallylic site.
Effect of the C-1 hydroxyl group on IPP binding the homoallylic site
As mentioned in the previous section, IPP could not account for the electron density at the homoallylic site of TcFPPS in complexes with IPP and each of the four bisphosphonates containing the C-1 hydroxyl group (91B, 261A, 300B, or 461A). However, comparisons between electron density maps of TcFPPS-bisphosphonate complexes with or without IPP indicate that IPP must be present at the homoallylic site of these complexes. For example, the Fo-Fc and 2Fo-Fc OMIT maps of TcFPPS complexed with IPP and 461A together contain much larger electron densities at the homoallylic site compared with those of TcFPPS with 461A alone (Figure 4A and 4B). When IPP is modeled into the bulky density, some residual density is seen at the location corresponding to the sulfate molecule in the TcFPPS/461A complex (Figure 4B, lower panel), but a sulfate modeled into the residual density will clash with IPP.
Figure 4. Comparison of electron density maps at the homoallylic site of TcFPPS/461A and TcFPPS/461A/IPP complexes showing the presence of a sulfate molecule.

Fo-Fc maps are shown as green meshes. In (A), electron density maps of TcFPPS/461A complex (no IPP) show extra densities (red arrows) in the Fo-Fc OMIT map. This density corresponds to a sulfate molecule at the homoallylic site in the apo enzyme structure9. For the TcFPPS/461A/IPP complex, the Fo-Fc OMIT map (B, upper panel) show much bigger densities than those in the TcFPPS/461A complex, indicating possible occupancy by IPP. However, an IPP molecule cannot fully account for those densities and leaves residual density (red arrows in B, lower panel) at a location corresponding to the density for the sulfate molecule in the TcFPPS/461A complex (A). When both IPP and sulfate are built into the homoallylic site of the TcFPPS/461A/IPP complex with 50% occupancy each (C), the model fits the electron density map well except for some residual density at the isopentenyl end of IPP, indicating disordering of this region. All Fo-Fc maps are contoured 2.8 σ.
Assuming that the homoallylic site can be occupied by either a sulfate or IPP but not both, a model was made by assigning 50% occupancy to both the sulfate and IPP. This led to a greatly improved fit with electron density maps (Figure 4C), and the refined B factors (<B> ~ 30.8 Å2) of the IPP and sulfate are similar to those of the protein atoms. Nevertheless, some residual density is seen near the isopentenyl end of IPP, indicating disordering of this part due to proximity (~ 3 Å) to the C-1 hydroxyl group of the bisphosphonate. We conclude that the presence of a C-1 hydroxyl group in the bisphosphonate interferes with the binding of IPP, reducing its occupancy and causing disordering, particularly at its isopentenyl end.
ITC of bisphosphonates binding to the TcFPPS homodimer
The thermodynamics of N-BP binding to TcFPPS was studied in ITC experiments carried out at 26°C. N-BPs were added to purified TcFPPS in a buffer containing 20 mM HEPES pH 7.5, 1 M NaCl, 2 mM MgCl2, and 2 mM TCEP (Table 2). Binding of N-BPs to TcFPPS resulted in a positive enthalpy change (Figure 5), in agreement with similar experiments done with human FPPS 14. In contrast, bisphosphonate binding to the FPPS from Trypanosoma brucei could be either entropy- or enthalpy-driven 15. Surprisingly, however, for all five N-BPs used in this study, and risedronate, the binding isotherms show a steep slope at the beginning that cannot be fitted with a model of two identical sites per dimer (one per monomer). The results are only compatible with a model that involves two sites (Figure 5).
Table 2.
Thermodynamic parameters of N-BPs binding to TcFPPS
| Compound | 91B | 261A | 300B |
|---|---|---|---|
| K1 (M−1) | 3.5 ± 0.7 × 107 | 5.3 ± 1.4 × 107 | 2.5 ± 0.5 × 106 |
| H1 (kcal/mol) | 12.2 ± 0.1 | 14.1 ± 0.3 | 20.5 ± 0.7 |
| S1 (cal/mol*K) | 75.2 | 82.4 | 97.8 |
| K2 (M−1) | 2.3 ± 0.19 × 107 | 9.2 ± 1.6 × 107 | 1.5 ± 0.1 × 106 |
| H2 (kcal/mol) | 8.6 ± 0.1 | 10.9 ± 0.3 | 11.6 ± 0.7 |
| S2 (cal/mol*K) | 62.3 | 72.8 | 66.9 |
| K1/K2 | 1.56 | 0.58 | 1.65 |
| Compound | 461A | 476A | Ris |
|---|---|---|---|
| K1 (M−1) | 1.6 ± 0.9 × 107 | 1.2 ± 0.1 × 107 | 1.0 ± 0.3 × 107 |
| H1 (kcal/mol) | 17.5 ± 1.0 | 19.0 ± 0.1 | 14.7 ± 0.7 |
| S1 (cal/mol*K) | 91.5 | 96.0 | 81.0 |
| K2 (M−1) | 1.9 ± 0.2 × 107 | 1.9 ± 0.1 × 106 | 1.5 ± 0.2 × 107 |
| H2 (kcal/mol) | 9.3 ± 0.9 | 13.6 ± 0.1 | 6.0 ± 0.7 |
| S2 (cal/mol*K) | 64.3 | 74.1 | 52.9 |
| K1/K2 | 0.84 | 6.45 | 0.68 |
Figure 5. ITC.

(A) Titration of 461A into TcFPPS (a) fits a two-sequential binding site model (c) better than a one-site model (b). (B) Isotherms of ITC experiments by titrating 91B, 261A, 300B, 476A, or risedronate into TcFPPS all fit a two-sequential binding site model. The sharp slope at the very beginning of the isotherm (blue arrows) is only predicted by a two-sequential site but not a two-permanent site binding model.
The structural studies described above shows that one molecule of N-BP binds to the allylic site of each TcFPPS monomer. Furthermore, binding of substrates and/or inhibitors triggers conformational change between the apo and the complexed TcFPPS that results in increased interactions between the enzyme and the ligands 9. Most likely, binding of an N-BP molecule to one FPPS monomer in the dimer induces the observed conformational change in that monomer. Two possibilities exist: either the change in one monomer leaves the other monomer unchanged or it affects the conformation of the other monomer in a way that alters the affinity for N-BP to the other FPPS monomer. In this second case, inhibitor binding should follow a model involving two sequential binding sites, rather than two permanent sites with different affinities. Indeed, two sequential sites describe the isotherm much better as judged by: 1) errors of thermodynamic parameters obtained from this model are smaller than those from the two different permanent site model (data not shown); 2) the sharp downward slope at the very beginning of the titration curve is much better fit by the two-sequential binding site model than the two-permanent site model, especially in ITC experiments with 91B and risedronate (Figure 5B). The better fit of the two-sequential site model is not a result of over-fitting since it actually contains fewer (four) adjustable parameters than those (six) of the two-permanent site model. ITC experiments carried out in various concentrations of salt show that although the actual values of the thermodynamic parameters depend on the experimental condition, the two-sequential binding behavior is clearly visible in all cases (data not shown).
When the affinities of the two sites are compared, we find greater affinities for the second binding site for 261A, 461A, and risedronate, indicating positive cooperative binding for these inhibitors. However, for the three other compounds (91B, 300B, and 476A), the first site has higher affinity.
Discussion
The effects of a C-1 hydroxyl group in bisphosphonates on IPP binding
Most bisphosphonates in clinical use contain a C-1 hydroxyl group, which has been shown to enhance their affinity for bone mineral 16. Our structural studies indicate that presence of the C-1 hydroxyl in N-BPs has two effects on the binding of IPP at the homoallylic site of TcFPPS: (1) it reduces IPP occupancy; (2) it disorders the isopentenyl side chain. More specifically, the homoallylic sites in the four complexes of TcFPPS with hydroxyl-containing bisphosphonates are occupied by either of IPP or sulfate. It is not known to which extent the reduction in IPP occupancy is due to competition by the sulfate used in crystallization, but no evidence of sulfate binding is seen in the structure of TcFPPS in complex with IPP and 476A (no C1-hydroxyl). In the structures of human FPPS in complexes with IPP and N-BPs containing a C-1 hydroxyl group the IPP appears to be ordered 14. Interestingly, ITC results in the same study suggest that IPP competes with N-BP binding at the allylic site. Here we provide evidence for a second mechanism of competition between IPP and N-BPs containing C-1 hydroxyl groups.
Asymmetry within the TcFPPS homodimer in the presence of N-BPs
All known FPPSs, regardless of species, are homodimers. Although the mechanistic benefit of a dimeric FPPS is not clear, it has been suggested that the two subunits do not act independently 13. For example, heterodimerization of two different inactive mutant forms of a bacterial FPPS was shown to partially rescue catalytic activity, indicating a productive interaction between the two subunits 17. From a structural standpoint, the two subunits of the homodimer in most reported crystal structures of FPPS, including those of avian 18 and human enzymes14,19, are related by crystallographic symmetry, and are therefore indistinguishable. Even for those FPPSs that are not crystallographic dimers, such as those from S. aureus, E. coli 20 and a putative FPPS from Plasmodium vivax (PDB ID: 2IHI), only slight structural differences are detected between the two subunits.
Examination of all available crystal structures of TcFPPS bound to inhibitors hints at possible structural differences between the two subunits of the liganded TcFPPS dimer. For all TcFPPS structures determined to date, the crystals belong to the same space group (P6122), but can be divided into two groups, based on the size of their unit cells. In the first group, the unit cell axes are aI = bI = 58 Å, cI = 390 A, with one TcFPPS monomer in the asymmetric unit forming the TcFPPS homodimer by a crystallographic 2-fold axis. In the second group, the unit cell axes are aII = bII = 103 Å, cII = 390 Å, with three TcFPPS monomers in each asymmetric unit (Figure 6A). This cell change results from the loss of a crystallographic two-fold axis, which becomes a local two-fold. As a result, the new a and b cell axes run along the (1, 1, 0) and (2, −1, 0) directions of the group 1 cell. The group 2 aII and bII axis have lengths of ~2 × cos60° × aI [or 2 × (√3/2) × aI], and the c axis remains unchanged, resulting in an increase of the unit cell volumes of crystals roughly three fold (Figure 6B).
Figure 6. Asymmetry of TcFPPS crystals.

(A) Arrangement of TcFPPS monomers in unit cells. The view is down the c-axis. In group 1 crystals (left), each asymmetric unit contains one TcFPPS monomer, which forms a dimer with another monomer from a neighboring cell (not shown). In group 2 crystals (right), unit cells are larger and each asymmetric unit contains three TcFPPS monomer, with two of them (a and b) forming a dimer. The other monomer c forms a dimer with its symmetry mate in a neighboring cell (not shown). (B) The two crystal forms shown in the orientation that highlights the almost identical packings. The only difference between the two forms results from the loss of a crystallographic two-fold axis in group 2 crystals (that becomes a local two-fold), with the two cell axes aII and bII (arrows, right) corresponding to the vectors (2, −1, 0) and (1, 1, 0) of group 1 crystals (see text). (C) Alignment of the three TcFPPS monomers within an asymmetric unit from the group 2 crystal TcFPPS-alendronate-IPP (PDB ID: 1YHM) 9. Color coding of the monomers are the same as in (A).
In each group 2 asymmetric unit, two monomers form a TcFPPS dimer, while the third monomer forms an equivalent dimer with a symmetry mate in a neighboring asymmetric unit (Figure 6B). This means that one third of the dimers in the crystal are symmetric, while two thirds may be asymmetric. When the three monomers in the asymmetric unit are aligned, regions of the active site do not overlap, particular around the isopentenyl part of IPP and the nitrogen-containing group of alendronate, where corresponding atoms can be over 1.0 Å apart (Figure 6C).
Notably, the crystals of the apo enzyme belong to group 1, while those of most TcFPPS plus N-BPs and without IPP belong to group 2. The only exception is the crystal of TcFPPS in complex with 461A, which belongs to the first group. However, the ratio between the affinities of the two binding sites for 461A is closest to 1 among all the compounds (Table 2), which may explain the absence of symmetry breaking in this crystal. It should be noted that despite the similar affinities of the two sites for 461A, the enthalpic difference is large enough to account for the two-site character of the binding isotherm. The presence of IPP restores the symmetry of most crystals. Although these observations may be the result of non-functional, accidental breakage of symmetry, they may reflect the structural differences that result from the sequential binding of N-BPs to TcFPPS, which is most clearly demonstrated by the ITC experiments.
Model of bisphosphonate binding to TcFPPS
Comparison of the crystal structures of apo TcFPPS with those of the complexes with N-BPs and/or IPP shows a hinge-like displacement within each monomer that results in a “closed” conformation. Moreover, addition of inhibitors to TcFPPS shortens the time for detectable crystals to form, from several days to within an hour, reflecting a possible change in conformation or flexibility of TcFPPS induced by binding of N-BPs. Since this conformational change reflects differences between the apo and the bound forms, binding in solution of N-BPs to the first subunit of the dimer must cause a conformational change that may be transmitted to the tightly coupled second subunit, altering the affinity of the other site as well as its inhibitor-bound conformation.
These considerations are in complete agreement with the ITC experiments. The observed isotherms could only be fit by two unequal binding sites that belong to different subunits in the dimer. The significantly better fit of the isotherms to a model involving two sequential binding sites (as opposed to two permanent and different sites; the sequential model has fewer parameters) suggest that the hinge-like motion that results in the “closed” binding site takes place when binding to the first site occurs.
Based on the two sequential binding sites inferred from the ITC experiments and the asymmetry in the crystals of TcFPPS in complex with N-BPs, we propose the following model of N-BP binding to TcFPPS: (1) the two monomers of the dimeric TcFPPS apo enzyme are indistinguishable; (2) binding of N-BPs to one monomer of TcFPPS induces the enzyme to close around the active site; (3) this conformational change is transmitted to the other TcFPPS monomer in the dimer, resulting in its altered affinity for the N-BPs; (4) the dimer with both sites occupied may exist as an equilibrium between symmetric and asymmetric dimers. This is shown schematically in Figure 7, in which K1 and K2 denote the two observed binding affinities in ITC experiments.
Figure 7. Thermodynamic model of N-BP binding to TcFPPS dimer.
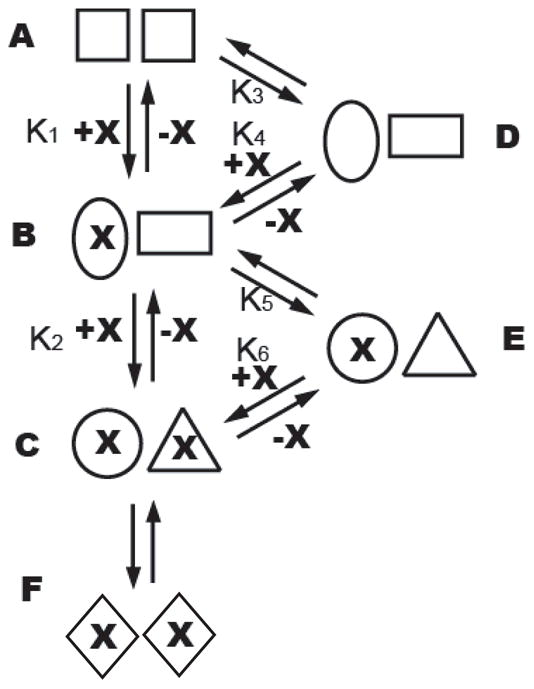
Before addition of N-BP (denoted by X), TcFPPS exist mainly as symmetric dimers (A), though a small population of asymmetric dimers (D) is present as required by thermodynamic equilibrium. Addition of N-BP shifts the equilibrium to asymmetric dimers (B) as N-BP binds to the first monomer (binding constant K1), with a change of conformation in both monomers. The second monomer then binds N-BP with a different binding constant (K2) than that of the first monomer, resulting in the formation of an asymmetric TcFPPS-N-BP dimer (C), which may be in equilibrium with symmetric dimers (F).
Interestingly, the symmetry breaking event is not seen in the human FPPS dimer, for which fifteen crystal structures of enzyme-inhibitor complexes are available in the Protein Data Bank. In these human FPPS-inhibitor structures, all the dimers are crystallographically symmetric. Notably, binding of risedronate and zoledronate to human FPPS fits well with a one-binding site model 14, in stark contrast to the two-site binding of these and other N-BPs to TcFPPS. This difference between the human and trypanosomal enzymes may provide clues for the development of TcFPPS-specific inhibitors for the treatment of Chagas disease.
Acknowledgments
The work is supported by NIH grant GM066895 to L.M.A, and GM65307 to E.O. X-ray diffraction experiments were carried out at beamlines X29 and X6A at the National Synchrotron Light Source at Brookhaven National Laboratory, as well as the SGX Collaborative Access Team (SGX-CAT) at the Advanced Photon Source.
Footnotes
Supporting Information Available
The coordinates of the structures of TcFPPS-bisphosphonate complexes have been deposited in the Protein Data Bank (www.rcsb.org) with the accession numbers 3IBA (TcFPPS-IPP-91B), 3ICK (TcFPPS-IPP-261A), 3ICM (TcFPPS-IPP-300B), 3ID0 (TcFPPS-IPP-461A), 3ICZ (TcFPPS-IPP-476A), and 3ICN (TcFPPS-461A).
References
- 1.Rogers MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des. 2003;9(32):2643–2658. doi: 10.2174/1381612033453640. [DOI] [PubMed] [Google Scholar]
- 2.Garzoni LR, Caldera A, Meirelles Mde N, de Castro SL, Docampo R, Meints GA, Oldfield E, Urbina JA. Selective in vitro effects of the farnesyl pyrophosphate synthase inhibitor risedronate on Trypanosoma cruzi. Int J Antimicrob Agents. 2004;23(3):273–285. doi: 10.1016/j.ijantimicag.2003.07.020. [DOI] [PubMed] [Google Scholar]
- 3.Garzoni LR, Waghabi MC, Baptista MM, de Castro SL, Meirelles Mde N, Britto CC, Docampo R, Oldfield E, Urbina JA. Antiparasitic activity of risedronate in a murine model of acute Chagas’ disease. Int J Antimicrob Agents. 2004;23(3):286–290. doi: 10.1016/j.ijantimicag.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 4.Montalvetti A, Fernandez A, Sanders JM, Ghosh S, Van Brussel E, Oldfield E, Docampo R. Farnesyl pyrophosphate synthase is an essential enzyme in Trypanosoma brucei. In vitro RNA interference and in vivo inhibition studies. J Biol Chem. 2003;278(19):17075–17083. doi: 10.1074/jbc.M210467200. [DOI] [PubMed] [Google Scholar]
- 5.Montalvetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, Docampo R. Bisphosphonates are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. J Biol Chem. 2001;276(36):33930–33937. doi: 10.1074/jbc.M103950200. [DOI] [PubMed] [Google Scholar]
- 6.Martin MB, Grimley JS, Lewis JC, Heath HT, 3rd, Bailey BN, Kendrick H, Yardley V, Caldera A, Lira R, Urbina JA, Moreno SN, Docampo R, Croft SL, Oldfield E. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: a potential route to chemotherapy. J Med Chem. 2001;44(6):909–916. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 7.Szajnman SH, Montalvetti A, Wang Y, Docampo R, Rodriguez JB. Bisphosphonates derived from fatty acids are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. Bioorg Med Chem Lett. 2003;13(19):3231–3235. doi: 10.1016/s0960-894x(03)00663-2. [DOI] [PubMed] [Google Scholar]
- 8.Urbina JA, Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 2003;19(11):495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, Amzel LM. Structure and mechanism of the farnesyl diphosphate synthase from Trypanosoma cruzi: implications for drug design. Proteins. 2006;62(1):80–88. doi: 10.1002/prot.20754. [DOI] [PubMed] [Google Scholar]
- 10.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr A. 1994;A50:157–163. [Google Scholar]
- 11.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 12.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 13.Szkopinska A, Plochocka D. Farnesyl diphosphate synthase; regulation of product specificity. Acta Biochim Pol. 2005;52(1):45–55. [PubMed] [Google Scholar]
- 14.Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, Rogers MJ, Russell RG, Oppermann U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A. 2006;103(20):7829–7834. doi: 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin F, Cao R, Goddard A, Zhang Y, Oldfield E. Enthalpy versus entropy-driven binding of bisphosphonates to farnesyl diphosphate synthase. J Am Chem Soc. 2006;128(11):3524–3525. doi: 10.1021/ja0601639. [DOI] [PubMed] [Google Scholar]
- 16.Russell RG, Croucher PI, Rogers MJ. Bisphosphonates: pharmacology, mechanisms of action and clinical uses. Osteoporos Int. 1999;9 (Suppl 1):S66–80. doi: 10.1007/pl00004164. [DOI] [PubMed] [Google Scholar]
- 17.Koyama T, Gotoh Y, Nishino T. Intersubunit location of the active site of farnesyl diphosphate synthase: reconstruction of active enzymes by hybrid-type heteromeric dimers of site-directed mutants. Biochemistry. 2000;39(2):463–469. doi: 10.1021/bi991621b. [DOI] [PubMed] [Google Scholar]
- 18.Tarshis LC, Yan M, Poulter CD, Sacchettini JC. Crystal structure of recombinant farnesyl diphosphate synthase at 2.6-A resolution. Biochemistry. 1994;33(36):10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 19.Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem. 2006;1(2):267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- 20.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279(10):8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]