

Abstract
Regulatory T cells (Tregs) play an important role in maintaining immunological tolerance that is one of the main obstacles to overcome for improving antitumor immunity. Recently, the Treg has been shown to constitutively express OX40 (CD134), which is a member of the TNF receptor family that is transiently expressed on effector T cells after TCR triggering, and through which the signal enhances effector T cell proliferation and memory T cell development. However, little is known about the role of OX40 costimulation to Tregs in tumor immunology. Here we show that OX40 signaling modulates the function of naturally occurring Tregs in vitro and in vivo. Foxp3 expression on Tregs was reduced by OX40 costimulation, but not by IL-2 stimulation. Tregs suppressed the proliferation of naïve CD4+ CD25− T cells after TCR triggering, in contrast, OX40 costimulated Tregs that reduced Foxp3 expression reversed the suppressive function. In addition, Tregs inhibited the proliferation of TCR stimulated (primed) CD4+ T cells and naïve CD8+ T cells after TCR-mediated activation, however, Tregs with OX40 costimulation lost their suppressive function. Interestingly, Tregs minimally suppressed the proliferation or the cytokine secreting of Ag-specific CD8+ T cells after Ag-restimulation. Furthermore, Tregs suppressive function to the antitumor effect was reversed by OX40 costimulation in vivo. Our data indicate that, in addition to controlling effector T cell function, OX40 costimulation directly controls Treg-mediated suppression in tumor immunity.
Keywords: regulatory T cell, OX40 signaling, costimulation, tolerance, tumor immunity
Introduction
Regulatory T cells (Tregs) negatively modulate immune responses (1–4). Tregs exert suppressive effects on the inhibition of interleukin-2 (IL-2) production by effector T cells via an unknown mechanism that requires cell-to-cell contact. They are anergic and do not make productive responses to T-cell-receptor (TCR) triggering in vitro unless exogenous IL-2 is supplied (5, 6). Their suppressive function on effector T cells plays an important role in maintaining immune tolerance to prevent autoimmune diseases and allergies (7–11). On the other hand, immune tolerance to tumor (self)-antigens is one of the most crucial hurdles to overcome for successful antitumor immunotherapy. In cancer patients, Tregs have been shown to increase both in peripheral blood and at the tumor site (12). A depletion of CD4+CD25+ T cells in vivo by means of a antagonistic monoclonal antibody (mAb) to CD25 (PC61) before tumor challenge has been shown to enhance the ability of the host to reject several types of immunogenic tumors in different strains of mice (13, 14). Therefore, one of the important strategies to break immunological tolerance might be to modulate the suppressive function of regulatory T cells against the induction of effector T cells.
There are at least three major types of CD4+ regulatory T cells: Th3, Tr1, and CD4+CD25+ T cells, in which CD4+CD25+ regulatory T cells (Tregs) are the best characterized because a large number of the cells is easily obtained (15). Tregs express Foxp3, which is an X chromosome-encoded member of the forkhead winged-helix family of transcription factors. Recent findings have demonstrated a highly specialized function for Foxp3, a master regulator, in Tregs and a pivotal function for Tregs in the persistence of immunological tolerance (16–18). In addition, another recent report has shown that continuous Foxp3 expression is needed to maintain the developmentally established suppressive program in mature Tregs (19). Thus, manipulation of Foxp3 expression, if it were possible, might be an approach to modulate the Tregs function and to control immune tolerance.
Tregs have been also revealed to express several costimulatory molecules such as CD28, CD40 ligand (CD40L), cytotoxic T lymphocyte antigen 4 (CTLA-4), glucocorticoid-induced TNF receptor (GITR) and OX40 (CD134). CD28 and CD40L are involved in the maintenance of peripheral Treg homeostasis, whereas CTLA-4 and GITR regulate Treg-suppressive activity (5). OX40 (CD134) is a member of the TNF receptor family that is transiently expressed on effector T cells with TCR triggering. OX40 has been shown to costimulate CD4+ T cell responses and is associated with increased T cell expansion and effector function (20, 21), increased T cell survival (22,23), and enhanced memory cell development (24, 25). We have recently demonstrated that OX40 costimulation when combined with a GM-CSF-secreting vaccine overcomes established CD8+ T cell tolerance to an endogenous tumor antigen (26). Recent reports have shown that OX40 signaling on Tregs can control Treg-mediated suppression of effector CD4+ T cell proliferation, IL-2 gene transcription, and cytokine production (5, 27). The role of OX40 costimulation in Tregs-mediated suppression needs to be more precisely clarified because elucidating the mechanism behind the regulatory function of Tregs has been a critical area of research, practical application in the clinical field, and the prevention of immune related diseases and cancer.
We examined whether OX40 costimulation could modulate the Treg-mediated suppressive function in vitro and in vivo. After OX40 costimulation, Tregs reduced Foxp3 expression, whereas, after IL-2 stimulation, Tregs vigorously expanded, but maintained Foxp3 expression. OX40 costimulation, but not IL-2 stimulation, inhibited the suppressive function of Tregs, which was correlated with the decreased Foxp3 expression on Tregs. Adoptively transferred Tregs suppressed antitumor immune effects of a tumor antigen-encoded GM-CSF-secreting vaccine. In contrast, transferred Tregs from mice that had been given agonistic anti-OX40 mAb in vivo did not affect the antitumor vaccine effects, indicating that OX40 signaling abrogated the suppressive function of Tregs in vivo. Our data propose that in addition to controlling effector T cell function, OX40 costimulation can directly control Treg-mediated suppression in tumor immunity.
Materials and Methods
Mice
FVB mice and HER2/neu-transgenic mice (neu-N) were purchased from Jackson River Farms (USA). Animals were housed under pathogen-free conditions at Shiga University of Medical Science (Shiga, Japan). All experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of Shiga University of Medical Science.
Cell lines and media
The GM-CSF-secreting vaccine cell line 3T3neuGM (expressing HER2/neu) was generated and grown as previously described (26, 28, 29). The HER2/neu-expressing NT cells used in tumor-challenge experiments and T2Dq cells used for targets in T cell assays were also previously described. (28). T cells for T cell assays were maintained at 37°C and 5% CO2 in CTL medium [RPMI 1640 supplemented with 10% FBS, 1% L-glutamine, 0.5% penicillin/streptomycin and 0.1 mM 2-mercaptoethanol (SIGMA)].
Peptides and Abs
RNEU420–429 (PDSLRDLSVF) and NP118–126 (RPQASGVYM) peptides were synthesized at >95% purity by the Oncology Peptide Synthesis Facility at Johns Hopkins University and generously provided. An agonist anti-OX40 mAb was produced from OX86 hybridoma cell lines. Purified rat IgG reagent (SIGMA) was used as a control Ab.
APC anti-mouse CD4, FITC rat anti-mouse CD4, APC anti-mouse CD8a, FITC rat anti-mouse IFN-γ, purified rat anti-mouse CD3, purified anti-mouse CD134 (OX40 antigen), and APC labeled goat anti-rat Ig were purchased from BD Pharmingen. Recombinant mouse IL-2 and anti-murine IL-2 neutralizing antibody were obtained from BD Pharmingen and R & D Systems, respectively. APC conjugated anti-mouse/rat Foxp3 and an eBioscience Foxp3 staining buffer set were purchased from e-Bioscience for Foxp3 staining. Intracellular cytokine staining (ICS) was performed as previously described (30) using murine IFN-g and a Cytofix/Cytoperm™ Plus (with Golgistop™) kit obtained from BD Biosciences. Flow cytometric data were collected using a BD FACSCalibur cytometer (BD Biosciences). Data were analyzed using CellQuest (BD Bioscience) and FlowJo software (Tree Star, Inc.).
Purification of T cell subsets and APCs
CD4+ T cells and CD8+ T cells were isolated from spleens of female mice, 6 to 12 weeks of age, by negative selection using an EasySep Mouse CD4+ T cell Enrichment Kit and a Mouse CD8+ T cell Enrichment Kit (StemCell Technologies Inc). The purity of these T cells was confirmed >90%. CD4+CD25+ T cells (regulatory T cells) and CD4+CD25− T cells were purified from the spleens of those mice with a CD4+CD25+ regulatory T cell Isolation Kit (Miltenyi Biotech). The purity of the CD4+CD25+ T cells was confirmed >90%. Primed CD4+ T cells and primed CD8+ T cells were isolated with EasySep from splenocytes incubated with an anti-CD3 Ab (0.1μg/ml) for 4 days. APCs were obtained from splenocytes treated with Red Blood Cell Lysing Buffer (SIGMA) and irradiated with 5,000 rad.
T cell suppression assay in vitro
CD4+ T cells and CD8+ T cells (2.5 ×104 cells/well) isolated from FVB mice were mixed with Tregs or CD4+CD25− T cells (2.5 ×104 cells/well) which had been stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days and washed well, and then plated onto 96-well tissue culture plates. Primed CD4+ T cells and CD8+ T cells were isolated from splenocytes treated with Red Blood Cell Lysing Buffer and incubated with anti-CD3 (0.1μg/ml) for 4 days. Tregs and CD4+CD25− T cells were irradiated with 3000 rad to prevent proliferation. The cells were incubated with APCs (2.5 ×105 cells/well) and anti-CD3 (0.1μg/ml) at 37°C for 60 hours. For the last 10 hours of culture, 1 μCi (0.037 MBq) [methyl-3H]Thymidine/well (Amersham) was added, and incorporation of [methyl-3H]Thymidine was determined using a Liquid Scintillation Analyzer (TRI-CARB 3100TR, Packard). Data were collected as the mean CPM of triplicate assays.
T cell suppression assay by CFSE
CD4+CD25+ T cells (Tregs) were purified from the spleens of FVB/N mice with a CD4+CD25+ regulatory T cell Isolation Kit, and stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days. Naive CD4+ T cells were isolated from spleens of FVB/N mice by negative selection using an EasySep Mouse CD4+ T cell Enrichment Kit. Isolated CD4+ T cells were labeled with 5μM of CFSE (Invitrogen), and the CFSE-labeled CD4+ T cells (2.5 ×104 cells/well) were incubated with non-irradiated Tregs (2.5 ×104 cells/well), irradiated APCs (2.5 ×105 cells/well) and anti-CD3 (0.1μg/ml) at 37°C for 4 days. CFSE dilution was determined by flow cytometry.
Antigen-primed CD8+ T cell suppression assay
To obtain Ag-specific primed CD8+ T cells, NT2 tumor cells (5×106 cells injected s.c. in the mammary fat pad) were injected on day −3 and 3T3 neu/GM vaccine cells (3×106 total cells injected s.c. divided equally among two forelimbs and one hind limb) were injected on day 0 to FVB mice. HER2/neu-primed CD8+ T cells were isolated using an EasySep Mouse CD8+ T cell Enrichment Kit on day 10. Tregs and CD4+CD25− T cells were isolated from naïve FVB mice using a MACS CD4+CD25+ regulatory T cell Isolation Kit on day7, and stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days. On day 10, Ag-primed CD8+ T cells (1×105 cells) and TCR-stimulated Tregs or TCR-CD4+CD25− T cells (1×105 cells) were cocultured at 37°C for 2 hours. After that they were incubated for 10 hours with an equal ratio of T2Dq cells pulsed with either RNEU420–429 or NP118–126. and ICS was performed as previously described (30).
Adoptive transfer of Tregs
HER2/neu-transgenic mice (neu-N), which have been shown to have an established immune tolerance to HER2/neu, were intraperitoneally given anti-OX40 mAb or control IgG (300μg per animal). Two days after administration Tregs were isolated from the spleens of these treated neu-N mice with a MACS CD4+CD25+ regulatory T cell Isolation Kit. These pretreated Tregs in donor mice were adoptively transferred into FVB recipient mice (5×105 cells per animal) that had been given HER2/neu-expresing NT tumor cells (5×106 cells/body) 3 days before transfer (n = 6 per group). Saline was injected into the tail vein of FVB mice as a control. (n=8). After Tregs transfer, 3T3 neu/GM vaccine cells (3×106 cells) were given subcutaneously to all the recipient FVB mice. Tumor size (mm2) was recorded by measuring the tumor diameter along orthogonal axes twice a week.
Statistical analysis
Analyses were performed with the Students t -test.
Results
Naturally-occurring Tregs express OX40 and Foxp3
To confirm whether the OX40 or Foxp3 expression on Tregs or non-Tregs changed during activation, we examined the OX40 and Foxp3 expression on CD4+CD25+ naturally-occurring Tregs or CD4+CD25− non-Tregs in the resting (naïve) or TCR-stimulation (activated) state. As previously described (31, 32), naïve Tregs expressed OX40 and naïve CD4+CD25− T cells did not (Figure 1A). After stimulation with plate-bound anti-CD3, both Tregs and non-Tregs expressed OX40 (Figure 1B). Foxp3 expression was constantly expressed on Tregs, but not non-Tregs even after TCR-stimulation (Figure 1C, 1D). Thus, naturally-occurring Tregs did not change their OX40 and Foxp3 expression, but CD4+ non-Tregs never expressed Foxp3 during activation by TCR-stimulation.
Figure 1.
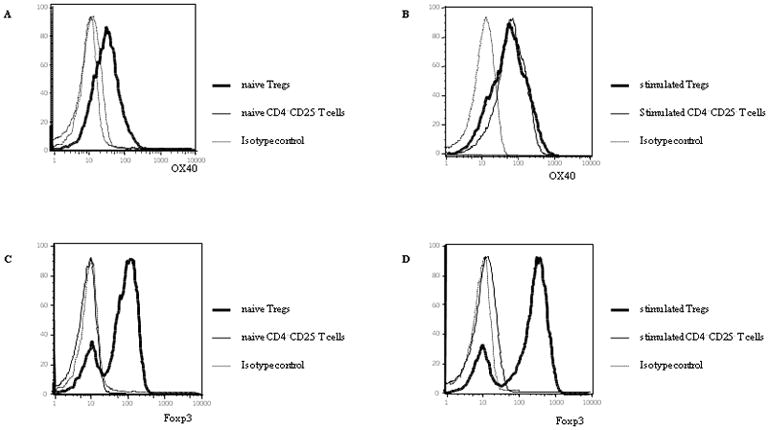
OX40 and Foxp3 expression on Tregs or CD4+CD25− T cells.
Tregs and CD4+CD25− T cells were isolated from the spleens of FVB mice and stained for OX40 with purified rat anti-mouse CD134 (OX40 antigen) and APC labeled goat anti-rat Ig (A, B). We also stained them for Foxp3 with APC conjugated anti-mouse/rat Foxp3 (C, D). They were analyzed by flow cytometry. (A) OX40 expression on naïve Tregs and CD4+CD25− T cells. (B) OX40 expression on Tregs and CD4+CD25− T cells stimulated with plate-bound anti-CD3. (C) Foxp3 expression on naïve Tregs and CD4+CD25− T cells. (D) Foxp3 expression on Tregs and CD4+CD25− T cells stimulated with plate-bound anti-CD3.
OX40 costimulation reduced Foxp3 expression on Tregs
Foxp3 has been shown to be a pivotal molecule for the suppressive function in Tregs (19). However, Foxp3 expression on Tregs during their proliferation remains poorly understood. We next sought to investigate the change of Foxp3 expression on Tregs through IL-2 or OX40 costimulation with TCR-stimulation. Tregs were stimulated with plate-bound anti-CD3 in the presence of anti-OX40 mAb or rat IgG for 3 days. The rat IgG group was divided into two groups, with or without IL-2 (Figure 2C, B). We examined Foxp3 expression at 3 different time points: before anti-CD3 stimulation (day 0), 1 day after anti-CD3 stimulation (day 1) and day 3, and evaluated the change of Foxp3 expression by mean fluorescence intensity (MFI) of positive for Foxp3 expression. As shown in Figure 2A, Foxp3 was not expressed on CD4+CD25− T cells for 3 days. Foxp3 expression did not change for 3 days on Tregs with rIgG (Figure 2B, MFI of positive expression on day 0, 1, and 3 were 335, 340, and 310, respectively.) or on Tregs with rIgG and IL-2 (Figure 2C, MFI of positive expression on day 0, 1, and 3 were 335, 344, and 320, respectively.). In contrast, Foxp3 expression on Tregs with anti-OX40 mAb remained unchanged on day 1 (MFI of positive expression on day 0 and 1 were 335 and 348.), but was clearly, though not uniformly, decreased by day 3 (Figure 2D, MFI of Foxp3+ expression was 208 and ranged from 70 to 300). Although we saw proliferation of Treg cells after stimulation with anti-CD3, the proportion of Foxp3− to Foxp3+ T cells did not change regardless of whether or not OX40 costimulation was provided (On day3 the proportion of Foxp3− to Foxp3+ T cells was 33.4% to 66.6% in Tregs + rIgG, 30.9% to 69.1% in Tregs + IL-2, 32.8% to 67.2% in Tregs + anti-OX40). Thus, OX40 costimulation reduces Foxp3 expression on Tregs during their activation, but IL-2 does not affect Foxp3 expression on Tregs.
Figure 2.
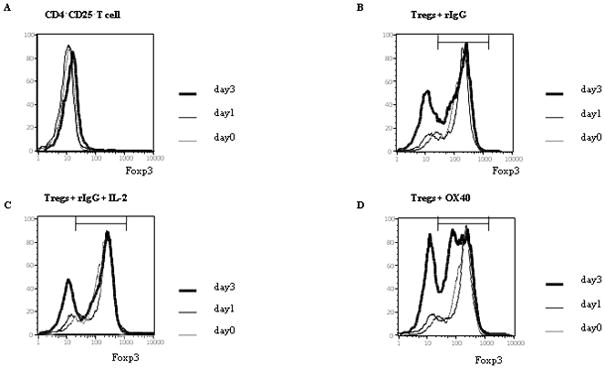
Foxp3 expression on Tregs through OX40 costimulation Tregs were stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days. The rat IgG group was divided into two groups, with or without IL-2 (10 ng/ml). CD4+CD25− T cells were also stimulated with plate-bound anti-CD3 in the presence of rat IgG (A). (B) Tregs stimulated with rat IgG. (C) Tregs stimulated with rat IgG and IL-2. (D) Tregs stimulated with anti-OX40 mAb. We examined the change of Foxp3 expression at 3 different time points: before anti-CD3 stimulation, 1 and 3 days after anti-CD3 stimulation (day 1 and day 3, respectively). In these figures, “OX40” indicates the agonistic anti-OX40 mAb. The data shown are representative data of 3 individual experiments. The black bar indicates the positive range of Foxp3 expression.
OX40 costimulation reduces the suppressive function of Tregs
To evaluate any correlation between reduced Foxp3 expression on Tregs by OX40 costimulation and their suppressive function on T cells, we examined the suppressive function of Tregs in contrast with Foxp3 expression on these Tregs stimulated with TCR-triggering and with or without OX40 ligation. Tregs were stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of agonistic anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days. The rat IgG group was divided into two groups, with or without IL-2. The kinetics of Foxp3 expression on Tregs and their suppressive function on CD4+ CD25− effector T cells were examined for 3 days. Naïve splenic CD4+CD25− T cells were incubated with irradiated APCs, anti-CD3 Ab, and irradiated Tregs that had been stimulated for 0, 1, or 3 days as described above, and Tregs before irradiation were also tested for Foxp3 expression at the same time (Figure 2). Thereafter incorporation of [methyl-3H] Thymidine was determined. As shown in Figure 3A, naïve Tregs without TCR-stimulation suppressed the proliferation of CD4+ CD25− T cells. On day 1, Tregs alone or Tregs with IL-2 or OX40 expressed Foxp3 (Figure 2) and all of them suppressed the proliferation of CD4+CD25− T cells (Figure 3B). On day 3, Foxp3 expression persisted in Tregs alone or Tregs with IL-2 (Figure 2) and the suppression of CD4+CD25− T cells proliferation was maintained (Figure 3C). In contrast, Foxp3 expression was reduced in Tregs with OX40 ligation on day 3 (Figure 2). Importantly, the suppressive effect of Foxp3+ Treg cells after OX40 costimulation was significantly diminished compared to an equal number of Foxp3+ Treg cells after anti-CD3 plus rat IgG or anti-CD3 plus IL-2. Irradiated Tregs with rat IgG, rat IgG and IL-2, or anti-OX40 mAb did not proliferate (data not shown). These data suggest that down-regulation of Foxp3 expression on Tregs through OX40 costimulation correlated with the loss of their suppressive function on CD4+CD25− T cells proliferation.
Figure 3. Down-regulation of Foxp3 expression on Tregs through OX40 costimulation and their suppressive function.
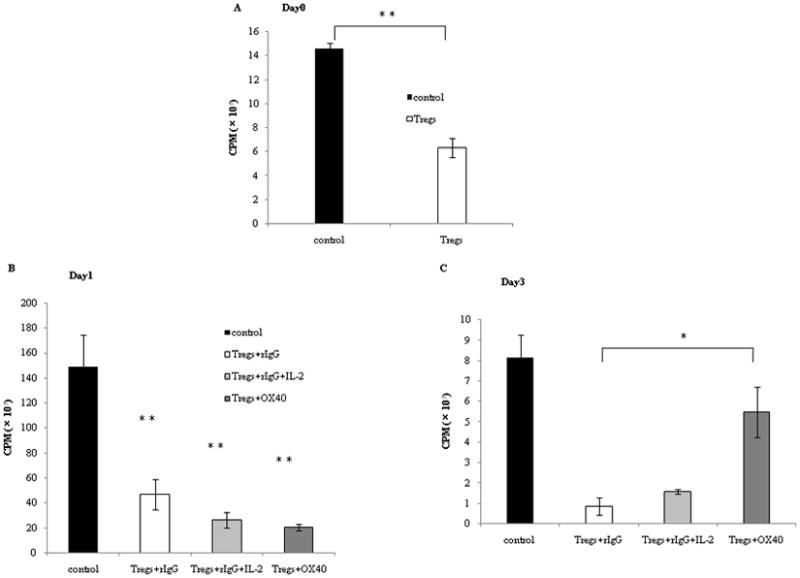
Tregs were stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days.
A, B, C: the rat IgG group was divided into two groups, with or without IL-2 (10 ng/ml). Naive CD4+ T cells (2.5 ×104 cells/well) were mixed with or without those Tregs which had been stimulated with rIgG or anti-OX40 mAb by the same method (2.5 × 104 cells/well) and plated into 96-well tissue culture plates. The cells were incubated with irradiated APCs (2.5 ×105 cells/well) and anti-CD3 (0.25μg/ml) at 37°C for 60 hours. For the last 10 hours of culture, 1 μCi [methyl-3H]Thymidine/well was added, and incorporation of thymidine was determined.
We examined the change of the suppressive function at 3 different time points: (A) before anti-CD3 stimulation, (B) day 1 and (C) day 3. “Control” indicates naïve CD4+ T cells proliferation without Tregs. In these figures, “OX40” indicates the agonistic anti-OX40 mAb. Results are from 1 representative out of 2 independent experiments. The significance of the data was evaluated with the Student’s t -test (*, p<0.05; **, p<0.01).
D: Tregs were stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml) for 3 days. CD4+ T cells were isolated from spleens of FVB/N mice and labeled by 5μM of CFSE. CFSE-labeled CD4+ T cells (2.5 ×104 cells/well) were incubated with irradiated APCs (2.5 ×105 cells/well), with anti-CD3 (0.1μg/ml), and with or without non-irradiated Tregs (2.5 ×104 cells/well), at 37°C for 4 days. Thereafter, dilution of CFSE gated by CD4+ T cells was visualized by flow cytometry.
To confirm these results, Treg suppression assays were performed using non-irradiated Tregs and CFSE-labeled effector CD4+ T cells. Irradiation was used to prevent the proliferation of contaminating Foxp3− CD4+ T cells within the Treg cell population; however the possible effects of irradiation on other functional properties of Tregs have not been characterized. As shown in Figure 3D, CFSE dilution was seen in CD4+ T cells stimulated with anti-CD3 in the presence of APC, indicated active proliferation by the effector CD4+ T cells (Figure 3D, top panel). In contrast, co-incubation of activated CD4+ T cells with Treg cells significantly inhibited effector T cells proliferation, as indicated by negligible CFSE dilution (Figure 3D, middle panel). Treg-mediated suppression of effector CD4+ T cell proliferation was abrogated by costimulation of Treg cells with OX40 (Figure 3D, bottom panel), though effector CD4+ T cell proliferation did not reach levels seen in the absence of Treg cell coculture (top panel). These findings are consistent with that of the [methyl-3H] Thymidine incorporation experiments, indicating again that Tregs through OX40 costimulation correlated with the reduction of their suppressive function on CD4+CD25− T cells proliferation.
Tregs can suppress the proliferation of CD8+ T cells and primed CD4+CD25− T cells, and OX40 costimulation reduces their suppressive function
Tregs are well known to have a suppressive function on the proliferation of CD4+CD25− T cells. However, suppressive function of Tregs on CD8+ T cells or antigen-primed CD4+ or CD8+ T cells has not been well clarified. We therefore next sought to determine whether Tregs suppressed the proliferation of naïve CD8+ T cells with TCR-stimulation or of TCR-stimulated (primed) CD4+ T cells or primed CD8+ T cells, and whether OX40 costimulation affected the function of Tregs in these T cells. Naïve CD8+ T cells, primed CD4+ T cells, or primed CD8+ T cells were cocultured at 37°C for 60 hours with APCs, anti-CD3, and the Tregs or the CD4+CD25− T cells which had been stimulated with plate-bound anti-CD3 in the presence of control rat IgG, anti-OX40 mAb, or IL-2 for 3 days, and then the incorporation of [methyl-3H]Thymidine was determined. Tregs suppressed the proliferation of naïve CD8+ T cells, primed CD4+ T cells, but not primed CD8+ T cells after TCR-stimulation (Figure 4A, 4B, 4C). OX40 costimulation protected the proliferation of naïve CD8+ T cells and primed CD4+ T cells after TCR-stimulation from suppression by Tregs.
Figure 4.
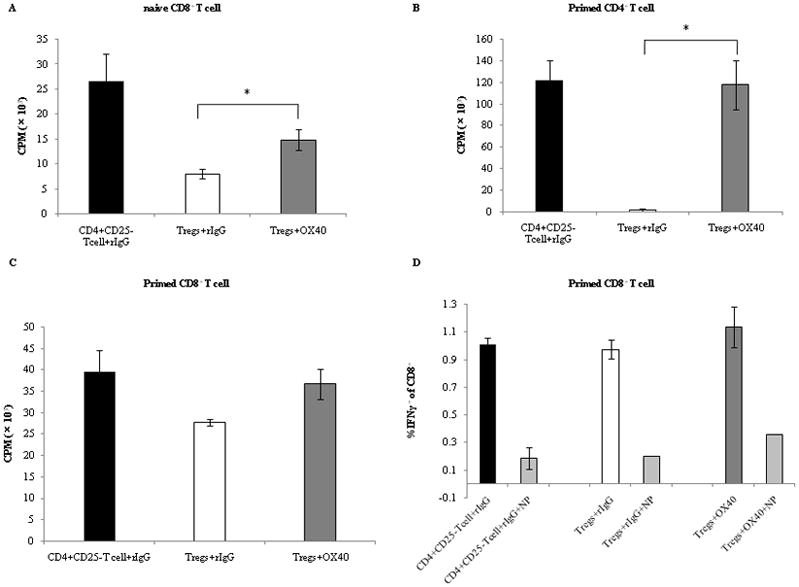
Decrease of the suppressive function of Tregs through OX40 costimulation. CTR-stimulated (primed) CD4+ T cells and naïve CD8+ T cells (2.5 ×104 cells/well) were mixed with Tregs or CD4+CD25− T cells (2.5 ×104 cells/well) which had been stimulated with plate-bound anti-CD3 (5 μg/ml) in the presence of anti-OX40 mAb (5 μg/ml) or IL-2 (10 ng/ml) or rat IgG (5 μg/ml) for 3 days, and then plated onto 96-well tissue culture plates. The cells were incubated with irradiated APCs (2.5 ×105 cells/well) and anti-CD3 (0.25μg/ml) at 37°C for 60 hours. For the last 10 hours of culture, 1 μCi [methyl-3H]Thymidine/well was added, and incorporation of thymidine was determined. (A) naïve CD8+ T cells proliferation cocultured with CD4+CD25− T cells with rIgG, Tregs with rIgG, or Tregs with anti-OX40 mAb. (B) primed CD4+ T cells proliferation cocultured with CD4+CD25− T cells with rIgG, Tregs with rIgG, Tregs with IL-2, or Tregs with anti-OX40 mAb. (C) IFN-g production of tumor antigen-specific primed CD8+ T cells. CD8+ T cells (1×105 cells) primed with NT2 tumor cells and 3T3 neu/GM vaccine cells were cocultured for 2 hours with Tregs or CD4+CD25− T cells (1×105 cells) which had been stimulated with plate-bound anti-CD3 in the presence of anti-OX40 mAb (5 μg/ml) or rat IgG (5 μg/ml). They were incubated for 12 hours with T2Dq cells (1×105 cells) that had been pulsed with either RNEU420–429 (named T2p50) or NP118–126. (named T2NP) and then intracellular cytokine staining (ICS) was performed. In this figure, “NP” indicates primed CD8+ T cells that were stimulated with T2NP cells as a negative control. In other groups, primed CD8+ T cells were stimulated with T2p50. In these figures, “OX40” indicates the agonistic anti-OX40 mAb. Results are from 1 representative out of 3 independent experiments. The significance of the data was evaluated with the Student’s t -test (*, p<0.05).
We further studies the inability of Tregs to exert their suppressive function on tumor Ag-primed CD8+ T cells after tumor Ag-restimulation. HER2/neu Ag-primed CD8+ T cells taken from the FVB mice that had been inoculated with HER2/neu-expressing NT tumor cells and HER2/neu-expressing and GM-CSF-secreting 3T3/neuGM vaccine cells have been shown to contain a HER2/neu-specific and high avidity CD8+ T cell repertoire (28). These Ag-primed CD8+ T cells were cocultured for 2 hours with the Tregs or the CD4+CD25− T cells that had already been stimulated with plate-bound anti-CD3 Ab in the presence of anti-OX40 mAb or control IgG for 3 days. T2Dq cells that had been pulsed with either RNEU420–429, an immunodominant peptide of HER2/neu, or NP118–126. were added into this cell mixed culture, and cocultured for more for 10 hours. Thereafter, the intra-cellular IFN-g secretion of the CD8+ T cells response to RNEU420–429 was determined. Interestingly, the expansion and IFN-g secretion of tumor Ag-primed CD8+ T cells after Ag-restimulation were not suppressed by Tregs (Figure 4D). Taken together, these findings indicate that Tregs can suppress not only the proliferation of naïve CD4+ T cells after TCR-stimulation, but also the proliferation of naïve CD8+ T cells after TCR-stimulation and primed CD4+ T cells after TCR-restimulation. However, Tregs cannot fully suppress the proliferation and the cytokine secretion of Ag-primed CD8+ T cells after Ag-restimulation. On the other hand, OX40 costimulation inhibits the suppressive function of Tregs, resulting in the restoration of the proliferation of naïve CD4+ T cells or naïve CD8+ T cells after TCR-stimulation and primed CD4+ T cells after TCR-restimulation.
Adoptive transferred Tregs with OX40 costimulation lose their suppressive function on the antitumor effect in vivo
It has shown that elimination of Tregs by an anti-CD25 blocking Ab before tumor challenge enhances antitumor immune effects (13, 33). Administration of an agonistic anti-OX40 mAb to tumor bearing mice to enhance the Ag-specific effector T cells function has been shown to improve antitumor immune effects and enhance antitumor vaccine effects (26, 34, 35). However, the antitumor effect of OX40 costimulation regarding the modulation of the Tregs suppressive function in vivo has not yet been clarified. To elucidate whether the modulation of the suppressive function of Tregs by OX40 costimulation in vitro reflects on antitumor effects in vivo, we examined the suppressive antitumor effect of Tregs with or without OX40 costimulation in vivo. Tregs separated from the donor mice that had a pretreatment with or without agonistic anti-OX40 mAb were adoptively transferred into the NT tumor-bearing recipient mice. The anti-OX40 mAb was not administered into the recipient mice to avoid OX40 costimulation of their effector T cells. After transfer of Tregs, 3T3 neu/GM vaccine cells were also administered to the recipient mice, and tumor development was observed. The tumor size decreased after the 17-post-tumor-challenge-day in the control vaccinated mice without transfer of Tregs (vaccine alone) (Figure 5). On the other hand, tumor growth significantly developed beyond 24 days after tumor challenge in the vaccinated mice which had received Tregs with pretreatment of control rat IgG (vaccine + Tregs treated with rIgG) compared with the control vaccinated mice (vaccine alone), (55.7±5.2 mm2 vs. 38.0±9.7 mm2 on day 24, p < 0.01) (Figure 5). However, the tumor size was reduced after the 24-post-tumor-challenge-day in the vaccinated mice which had received Tregs with pretreatment of OX40 costimulation (vaccine + Tregs treated with OX40), and the tumor size was not significantly different from that of the control vaccinated mice at any time point observed (41.8±10.8 mm2 vs. 33.3±8.3 mm2 on day 28, p > 0.12) (Figure 5). Full tumor regression was seen in mice treated with vaccine alone or vaccine + Tregs treated with OX40), whereas tumors grew progressively in mice treated with vaccine + Tregs treated with rIgG. These results indicate that Tregs can suppress antitumor vaccine effects in vivo, in contrast, the suppressive function of Tregs with OX40 costimulation is abrogated in vivo, resulting in no diverse antitumor effect generated by the vaccine.
Figure 5.
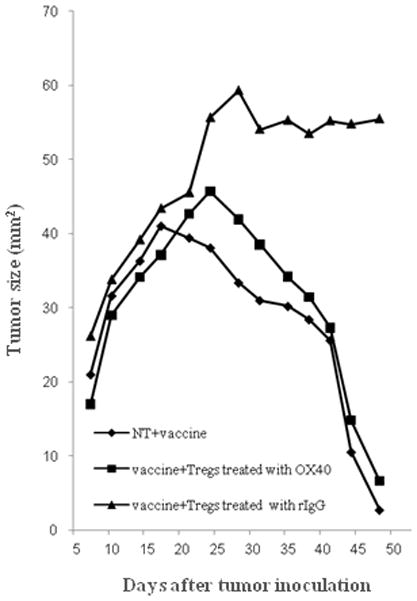
Decrease of the suppressive function of Tregs on antitumor effect through OX40 costimulation in vivo. NT2 tumor cells (5×106 cells per animal) were inoculated on day −3 to FVB mice (n=20). Anti-OX40 mAb or rat IgG (300μg per animal injected i.p.) was injected on day −2 to neu-N mice (n=6 per group). On day 0, Tregs were isolated from the spleens of the injected neu-N mice using a MACS CD4+CD25+ regulatory T cell Isolation Kit. The Tregs (5×105 cells/body) were adoptively transferred to the NT tumor inoculated FVB mice (n=6 per group) via their tail veins. As a control saline was injected into tail veins of FVB mice instead of adoptive transfer (n=8). 3T3 neu/GM vaccine cells (3×106 total cells) were also given on day 0 to all the tumor inoculated FVB mice. Tumor size (mm2) was determined by measuring the tumor diameter along orthogonal axes. The mean tumor size is reported. “vaccine alone” indicates the tumor inoculated and vaccinated mice with saline injection. “vaccine + Tregs treated with rIgG” indicates the tumor inoculated and vaccinated mice which underwent adoptive transfer of Tregs with pretreatment of rIgG, and “vaccine + Tregs treated with OX40” indicates the tumor inoculated and vaccinated mice that underwent adoptive transfer of Tregs with pretreatment of OX40 costimulation. Statistical analysis was performed with the Students t -test.
Discussion
The role of OX40 in the enhancement of the effector T cell function is well characterized. OX40 costimulation is known to increase CD4+ T cell proliferation and effector function(20, 21), enhance the development of the CD4+ T cell memory pool (22, 23), and can prevent or reverse CD4+ T cell tolerance (36). OX40 costimulation also enhances the CD8+ effector T cell function, and can overcome established CD8+ T cell tolerance to an endogenous tumor antigen in vivo when combined with a GM-CSF tumor antigen-encoded vaccine (26). CD4+CD25+ naturally occurring regulatory T cells are known to be important to the induction of self-tolerance and acquired tolerance (37, 38) as deficiency of this cell type leads to the development of autoimmunity and the failure to establish acquired tolerance (18, 39). Recently it has been shown that the elimination of CD4+CD25+ Tregs through low-dose cyclophosphamide permits the activation of tumor antigen-specific CD8+ T cells in established immune tolerant mice (neu-N) (30). These findings suggest that breaking CD4+ or CD8+ T cell tolerance through OX40 costimulation may be associated with attenuating T cell suppression by Tregs (31). Recently, it has been shown that naïve Tregs express several costimulatory molecules including OX40, and OX40 signaling has been shown to control their suppressive function against effector CD4+ T cells (5, 27). It is important to clarify whether OX40 costimulation can modulate Tregs function and could lead to overcoming immune tolerance against tumor antigens.
Our results highlight two important findings regarding the role of OX40 costimulation in Treg-mediated suppression of antitumor immunity. First, OX40 signaling reduces Foxp3 expression on Tregs, which correlates with inhibition of Treg suppressive function and results in the proliferation of effector CD4+ T cells after TCR-stimulation. Second, costimulation of Treg cells with OX40 abrogates the Treg-mediated suppression of both CD4+ and CD8+ T cell effector function, which correlates with an enhanced in vivo response to antitumor vaccination.
A key question in cancer immunotherapy is how to deplete or reverse the Tregs-mediated suppressive function. Approaches based on CD25+ Treg cell depletion have shown some promise (13, 33, 40); however, in the tumor treatment setting this approach might eliminate both Tregs and activated effector T cells because the CD25 marker is expressed both these T cell populations (40, 41). Studies have also shown that a natural ligand for human TLR8 directly reverses the suppressive function of Tregs (42, 43) and that activation of TLR2 with its ligand directly increases the proliferation of murine Tregs and temporally reverses their suppressive function (44, 45). Other approaches to reverse the Tregs-mediated suppression include signaling through GITR, CTLA-4, and OX40. However, compared with the signals mediated by other such surface molecules on Tregs, OX40 costimulation has some unique features in regulating the function of Tregs that may make OX40 a better clinical target for immune modulation. For example, whereas CTLA-4-deficient mice develop autoimmunity in multiple organs and die soon (46), OX40-deficient mice are healthy and do not have any autoimmune diseases (47).
OX40 signaling has been shown to directly inhibit Foxp3 gene transcripts in Tregs (27). Tregs expressing Foxp3, a master regulator of Treg suppressor function, play a crucial role in preventing autoimmune diseases and allergies. On the other hand, immune tolerance is an important hurdle to overcome to enhance antitumor immune effects. Therefore, Foxp3 expression may be a critical control point for breaking immune tolerance and building potent antitumor. Vu et al. have recently shown that OX40 costimulation inhibits Foxp3 gene expression in naturally arising Foxp3+ Tregs (27). In the present study we have shown that OX40 costimulation likewise reduces Foxp3 protein expression on Tregs and, moreover, that reduced Foxp3 expression correlates with the abrogation of Tregs-mediated suppression of antitumor immunity. Although several signals might be associated with the Tregs-mediated function, it is important to clarify what are the pathways upstream of Foxp3 signaling that mainly control the function of Tregs.
Several reports have shown that Tregs have an ability to suppress the proliferation of CD4+CD25− effector T cells that were stimulated through TCR ligation. However, it is still unclear whether or not Tregs also suppress the activation of CD8+ T cells and the proliferation of Ag-primed CD4+ or Ag-primed CD8+ T cells upon Ag-restimulation. Our data show that Treg-mediated suppression of naïve and previously-activated CD4+CD25− T cell proliferation is abrogated when Tregs received OX40 costimulation. These findings suggest that OX40 costimulation of Tregs may contribute to the restoration of tumor-targeted CD4+ helper T cell activation and memory CD4+ T cell function in a tumor-bearing host. These results are consistent with those of Bansal-Pakala et al., who reported that OX40-mediated costimulation can prevent and even overcome established peptide-specific CD4+ T cell tolerance in vitro and in vivo (36).
Our data extend investigations OX40 costimulation and Treg suppressive action to include the effects OX40 costimulation on Treg-mediated suppression of CD8+ T cell effector function. The proliferation of naïve CD8+ T cells or previously activated CD8+ T cells were suppressed by Tregs, though not to the same extent as CD4+ T cells. As with CD4+ T cell effector function, OX40 costimulation of Treg cells abrogated their suppressive effects on CD8+ T cell proliferation. Interestingly, Treg cells did not suppress the proliferation and the IFN-g production of tumor Ag-primed CD8+ T cells, though our studies did not address CD8+ T cell lytic function. Many studies have shown that OX40 costimulation enhances CD4+ or CD8+ T cells effector function (20, 21), memory development (24, 25) and survival (22, 23), resulting in enhancement of antitumor immune effects in vivo (26). Recent studies show that OX40-mediated abrogation of Treg cell function boosts adaptive immune response and intratumor injection of anti-OX40 mAb increases tumor rejection (48). This dual functionality of OX40 signaling, namely enhancing the function of effector T cells and inhibiting the suppressive function of Tregs, may contribute to improving antitumor immunity and overcoming tumor immune tolerance.
New strategies that simultaneously stimulate effector T cells while inhibiting or depleting Tregs are needed to improve the outcome of cancer immunotherapy. In the current study, we demonstrate that the suppressive function of Tregs can be reversed through OX40 costimulation that is known to be able to durably enhance the function of effector T cells. These findings suggest that OX40 signaling could modulate the balance between Tregs and effector T cells, resulting in tipping the balance towards antitumor immunity.
Acknowledgments
We thank Mrs. Arikawa, Mrs. Ito, Mrs. Yamamoto and Miss Kamuro for technical assistance, Mr. Yamamoto and Mr. Mori in Central Research Laboratory at Shiga University of Medical Science for technical support with analysis of flow cytometric data.
Footnotes
The novelty and impact of this paper: Here we show that the suppressive function of Foxp3+ regulatory T cells (T regs) can be reversed in vitro and in vivo through OX40 costimulation that is known to be able to durably enhance the function of effector T cells. These findings suggest that OX40 signaling could modulate the balance between T regs and effector T cells, tipping the balance towards antitumor immunity.
References
- 1.Mason D, Powrie F. Control of immune pathology by regulatory T cells. Curr Opin Immunol. 1998;10:649–55. doi: 10.1016/s0952-7915(98)80084-8. [DOI] [PubMed] [Google Scholar]
- 2.Maloy KJ, Poerie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2:816–22. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- 3.Curotto de Lafaille MA, Lafaille JJ. CD4+ regulatory T cells in autoimmunity and allergy. Curr Opin Immunol. 2002;14:771–8. doi: 10.1016/s0952-7915(02)00408-9. [DOI] [PubMed] [Google Scholar]
- 4.Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–7. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 5.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4+CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–51. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 6.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–23. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 7.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocriopathy, entropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 8.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 9.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic dysregulation syndrome. J Clin Invest. 2000;106:R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 11.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 12.Woo EY, Chu CS, Goletz TJ. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–72. [PubMed] [Google Scholar]
- 13.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity and by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–8. [PubMed] [Google Scholar]
- 14.Golgher D, Jones E, Powrie F, Elliott T, Gallimore A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32:3267–75. doi: 10.1002/1521-4141(200211)32:11<3267::AID-IMMU3267>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+CD25+ regulatory T cells. PNAS. 2006;103:7048–53. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 17.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 18.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–7. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 19.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;3:277–84. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 20.Gramaglia I, Weinberg AD, Lemon M, Croft M. OX-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–7. [PubMed] [Google Scholar]
- 21.Song J, So T, Cheng M, Tang X, Croft M. Sustained surviving expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity. 2005;22:621–31. doi: 10.1016/j.immuni.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van Parijs L, croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol. 2004;5:150–8. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 23.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expansion and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–55. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 24.Maxwell JR, Weinberg A, Prell RA, Vella AT. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral depletion. J Immunol. 2000;164:107–12. doi: 10.4049/jimmunol.164.1.107. [DOI] [PubMed] [Google Scholar]
- 25.Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol. 2000;165:3043–50. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- 26.Murata S, Ladle BH, Kim PS, Lutz ER, Wolpoe ME, Ivie SE, Smith HM, Armstrong TD, Emens LA, Jaffee EM, Reilly RT. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol. 2006;176:974–83. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 27.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Chang Li X. OX40 costimulation turns off Foxp3+ T regs. Blood. 2007;110:2501–10. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ercolini AM, Machiels JP, Chen YC, Slansky JE, Giedlen M, Reilly RT, Jaffee EM. Identification and characterization of the immunodominant rat HER-2/neu MHC class I epitope presented by spontaneous mammary tumors from HER-2/neu-transgenic mice. J Immunol. 2003;170:4273–80. doi: 10.4049/jimmunol.170.8.4273. [DOI] [PubMed] [Google Scholar]
- 29.Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH, Jaffee EM. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569–76. [PubMed] [Google Scholar]
- 30.Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JP, Bieler JG, Emens LA, Reilly RT, Jaffee EM. Recruitment of latent pools of high avidity CD8+ T cells to the antitumor immune response. J Exp Med. 2005;201:1591–602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T cell costimulatory molecule OX40. Nat Rev Immunol. 2004;4:420–31. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 32.Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, Qin FX, Liu YJ, Duramad O, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:13138–43. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J, Nomura T, Chiba T, Sakaguchi S. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–91. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000;60:5514–21. [PubMed] [Google Scholar]
- 35.Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, Urba WJ, Alvord G, Bunce C, Shields J. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164:2160–9. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 36.Bansal-Pakala P, Jember AG, Croft M. Signaling through OX40 (CD134) breaks peripheral T-cell tolerance. Nat Med. 2001;7:907–12. doi: 10.1038/90942. [DOI] [PubMed] [Google Scholar]
- 37.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz RH. Natural regulatory T cells and self-tolerance. Nat Immunol. 2005;6:327–30. doi: 10.1038/ni1184. [DOI] [PubMed] [Google Scholar]
- 39.Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 40.Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582–92. doi: 10.1097/01.cji.0000175468.19742.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, Vieweg J. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–33. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 43.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Wang RF. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–4. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 44.Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the function of CD4+CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2006;103:7048–53. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 47.Pippig SD, Pena-Rossi C, Long J, Godfrey WR, Fowell DJ, Reiner SL, Birkeland ML, Locksley RM, Barclay AN, Killeen N. Robust B cell immunity but impaired T cell proliferation in the absence of CD134 (OX40) J Immunol. 1999;163:6520–29. [PubMed] [Google Scholar]
- 48.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilities tumor rejection. J Exp Med. 2008;205:825–39. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]