

Abstract
Purpose
Although much is known about the safety of an anticancer agent at the time of initial marketing approval, sponsors customarily collect comprehensive safety data for studies that support supplemental indications. This adds significant cost and complexity to the study but may not provide useful new information. The main purpose of this analysis was to assess the amount of safety and concomitant medication data collected to determine a more optimal approach in the collection of these data when used in support of supplemental applications.
Methods
Following a prospectively developed statistical analysis plan, we reanalyzed safety data from eight previously completed prospective randomized trials.
Results
A total of 107,884 adverse events and 136,608 concomitant medication records were reviewed for the analysis. Of these, four grade 1 to 2 and nine grade 3 and higher events were identified as drug effects that were not included in the previously established safety profiles and could potentially have been missed using subsampling. These events were frequently detected in subsamples of 400 patients or larger. Furthermore, none of the concomitant medication records contributed to labeling changes for the supplemental indications.
Conclusion
Our study found that applying the optimized methodologic approach, described herein, has a high probability of detecting new drug safety signals. Focusing data collection on signals that cause physicians to modify or discontinue treatment ensures that safety issues of the highest concern for patients and regulators are captured and has significant potential to relieve strain on the clinical trials system.
INTRODUCTION
For marketing approval, the US Food and Drug Administration (FDA) requires commercial firms to submit data from adequate, well-controlled studies that demonstrate the safety and effectiveness of investigational agents in their intended use populations.1,2 The initial approval of an oncologic agent from a New Drug Application (NDA) or a Biologic License Application (BLA) is based on the results from trials that include approximately 1,000 patients in the aggregate. Approval in a supplemental indication often occurs years after the initial approval, usually after extensive postmarketing evaluations have been conducted in a larger, more general population.
Though considerably more is known about the safety profile of a drug at the time of a supplemental application, sponsors collect extensive safety data, similar to that collected for initial marketing approval. Recent studies indicate that documenting and validating extensive adverse event (AE) data places a substantial burden on the clinical trials infrastructure, especially at the site level.3,4 Similar considerations apply to the collection of data on concomitant medications, where large quantities of data continue to be collected in support of supplemental applications, but are seldom used to change drug labeling. Therefore, it is important to understand the type and extent of clinical data necessary to inform regulatory decisions that lead to changes in drug labeling and to clinical decisions regarding dose modification or discontinuation of treatment.
These considerations have prompted calls for development of specific data collection standards, particularly for supplemental NDAs/BLAs.5–7 Collecting targeted data necessary to inform regulatory and clinical decisions may enhance physician participation in clinical trials and enable more rapid completion of studies. This may result in allowing faster delivery of new drugs to patients, reducing the cost of clinical trials and enhancing data quality.
To address these issues, a working group was formed to provide a forum for the FDA, the National Cancer Institute, academic investigators, and industry to develop AE data collection standards for supplemental NDAs/BLAs in oncology indications. Representatives from the Cancer and Leukemia Group B, Eli Lilly, Genentech, GlaxoSmithKline, and Novartis Pharma AG volunteered to re-evaluate safety data from previously completed clinical trials. The main purpose of this analysis was to determine whether subsets of an AE database used for a supplemental application could adequately identify the new safety signals that would be learned from complete AE collection.
METHODS
A statistical analysis plan (SAP) was prospectively developed and approved by the project stakeholders. The analyses evaluated subsampling methods and their likelihood of missing clinically important AEs or over-representing events, relative to the information known from the established safety profile of the agent. Further, the SAP assessed the extent of data collection and cleaning effort saved through subsampling.
In all subsampling methods evaluated, serious AEs and events leading to drug discontinuation or dose modification, referred to here as “serious+” AEs, were collected in all patients. The SAP focused on the subsampling of grade 3 and higher events, referred to as grade 3+ AEs, and of grade 1 to 2 AEs, both groups distinct from the category of serious+ events.
Eight completed phase III clinical trials were selected for individual reanalysis (Table 1).8–15 These industry-sponsored and publicly funded trials investigated chemotherapy, biologic, and hormonal treatments in the metastatic and adjuvant treatment settings across multiple tumor types. In each case, the investigational agent had been studied in other phase III clinical trials and an established safety profile existed that served as the standard for the reanalysis. Treatment regimens differed substantially between the initial registration trial and the reanalyzed studies in four of the eight trials.
Table 1.
Study Characteristics
| Candidate Study Profile |
Source of Known Safety Profile |
||||||
|---|---|---|---|---|---|---|---|
| Study | Patient Population | Study Treatment | Safety Analysis Population | Precursor Studies | Primary Precursor Study Population | Precursor Study Treatment | Safety Analysis Population |
| AVAiL8 | First-line nonsquamous NSCLC | Arm 1: cisplatin/gemcitabine Arm 2: cisplatin/gemcitabine + bevacizumab | 656 | AVF2107g9 | First-line mCRC | Arm 1: irinotecan/FU/LV (bolus-IFL) + placebo Arm 2: bolus IFL + bevacizumab | 813 |
| ECOG 320016 | Second-line mCRC | FOLFOX4; FOLFOX4 + bevacizumab | 585 | ||||
| AVF2107g9* | First-line mCRC | Arm 1: irinotecan/FU/LV (bolus IFL) + placebo Arm 2: bolus IFL + bevacizumab | 788 | AVF2119g17 | mBC | Arm 1: capecitabine Arm 2: capecitabine + bevacizumab | 462 |
| ECOG 459910 | First-line nonsquamous NSCLC | Arm 1: paclitaxel/carboplatin Arm 2: paclitaxel/carboplatin + bevacizumab | 878 | AVF2107g4 | First-line mCRC | Arm 1: irinotecan/FU/LV (bolus IFL) + placebo Arm 2: bolus IFL + bevacizumab | 813 |
| ECOG 320016 | Second-line mCRC | FOLFOX4; FOLFOX4 + bevacizumab | 585 | ||||
| EGF3000111 | First-line mBC | Arm 1: paclitaxel + placebo Arm 2: paclitaxel + lapatinib | 580 | EGF10015118 | Refractory advanced or mBC | Arm 1: capecitabine Arm 2: capecitabine + lapatinib | 408 |
| JMDB12 | First-line NSCLC | Arm 1: cisplatin plus pemetrexed Arm 2: cisplatin plus gemcitabine | 1,669 | JMCH19 | MPM | Arm 1: cisplatin plus pemetrexed Arm 2: cisplatin | 331 |
| IBCSG BIG 1-9813 | PMP women with HR+ EBC | Arm 1: letrozole Arm 2: tamoxifen; double-blind using double-dummy technique | 7,963 | NCIC MA-17 (PI 11/2004)20 | Extended Adjuvant | Letrozole 2.5 mg orally daily for 5 years Placebo orally daily for 5 years; double-blind using double-dummy technique | 5,136 |
| CALGB 8980314 | Patients with resected adenocarcinoma of the colon | Arm 1: LV + FU Arm 2: irinotecan + LV + FU | 1,264 | Cunningham et al,21 1998 | mCRC | Irinotecan v best supportive care (FU failures) | 279 |
| Rougier et al,22 1998 | mCRC | Irinotecan v FU | 267 | ||||
| HERA15 | HER2+ adj breast cancer | Arm 1: observation Arm 2: trastuzumab | 3,386 | H0648g23 | First-line mBC | Trastuzumab + CT v CT alone; CT was either (1) anthracycline + cyclophosphamide or (2) paclitaxel + cyclophosphamide | 469 |
Abbreviations: AVAiL, Avastin in Lung; NSCLC, non–small-cell lung cancer; ECOG, Eastern Cooperative Oncology Group; mCRC, metastatic colorectal cancer; FU, fluorouracil; LV, leucovorin; IFL, irinotecan plus fluorouracil and leucovorin; FOLFOX4, infusional fluorouracil, leucovorin, and oxaliplatin; mBC, metastatic breast cancer; MPM, malignant pleural mesothelioma; IBCSG, International Breast Cancer Study Group; BIG, Breast International Group; PMP, postmenopausal; HR, hormone receptor; EBC, early breast cancer; NCIC, National Cancer Institute of Canada; PI, package insert (US); CALGB, Cancer and Leukemia Group B; HERA, HERceptin Adjuvant; adj, adjuvant; CT, chemotherapy.
AVF2107g was a three-arm trial. A third arm with treatment of FU/LVplus recombinant humanized monoclonal antibody vascular endothelial growth factor was omitted from this analysis.
In each candidate trial, cutoffs for AE signal detection were set to capture the smallest changes in AE frequency that oncologists might consider clinically relevant; therefore, drug effects were defined as those grade 3+ events with a ≥ 2% difference in incidence between the treatment and control arms and as those grade 1 to 2 events with a ≥ 5% difference.
We identified AE signals from previous trials that lead to the initial NDA/BLA approval and other studies conducted before the conduct of the candidate trial (ie, AEs from labeling, safety databases, and published literature).16–23 To this list, we added serious+ events that occurred in ≥ 2% excess from the candidate trial to establish the base known safety profile for the grade 3+ event subsampling analysis. For the assessment of the grade 1 to 2 subsamples, the base known safety profile was defined as above with the addition of grade 3+ events in ≥ 2% excess in the candidate trial.
AEs identified as drug effects in the candidate trial that were not listed as part of the base known safety profile were defined as events that could potentially be missed under the subsampling analysis. Noise events were defined as drug effects identified in subsamples that were not identified as drug effects in the candidate trial's full safety database.
Random and systematic sampling methods were applied to each candidate trial. Subsampling simulations on candidate trial data used 1,000 independent replications targeting 200, 300, 400, 500, and 600 patients, equally divided between the treatment arms, selected randomly by patient and randomly by treatment center. For each sample size and random subsampling method, we tabulated both the rate of event detection of potentially missed events and the mean number of noise events across the replications.
The systematic subsampling methods selected the target numbers of patients from the biggest centers and the first patients enrolled. From each subsample, we calculated the incidence differences of those AEs identified as drug effects, thus determining the missed signals and the noise AEs.
For each of the candidate studies, we determined the number of distinct AEs in the database for serious+ events, grade 1 to 2 events not serious+, and grade 3+ events not serious+. The mean number of AEs per patient was reported for each category.
We also determined the number of database records and fields needed to store concomitant medication data and whether the results were noted in subsequent FDA-approved labels.
RESULTS
Toxicity records from the candidate trials included 17,184 patients. The metastatic disease trials ranged in patient safety population size from 580 to 1,669 patients, whereas the adjuvant disease trials ranged from 1,264 to 7,963 patients (Table 1).
In the eight studies, 43 grade 3+ events were detected as drug effects (Table 2); however, 34 of these events were previously identified as part of their corresponding base known safety profile. The subsampling analysis focused on detection of the remaining nine events that could potentially be missed. Because so few AEs could be missed, known events from several trials representing varying full trial incidence differences were selected for subsampling analysis. This allowed us to observe AE detection trends as the incidence differences increased beyond the 2% cutoff rate.
Table 2.
AEs Detected as Drug Effects in the Analysis of All Patients in the Candidate Studies
| Trial | Grade 3+ Events Detected in≥ 2% Incidence Difference |
|---|---|
| AVAiL | |
| AVF2107g | |
| ECOG 4599 | |
| EGF30001 | |
| JMDB | |
| BIG 1-98 | No events identified in excess of 2% |
| CALGB 89803 | |
| HERA | Ejection fraction decreased† |
| Trial | Grade 1 to 2 Events Detected in≥ 5% Incidence Difference |
| AVAiL | |
| BIG 1-98 | Hypercholesterolemia |
| CALGB 89803 | |
| HERA |
NOTE. Adverse events in italics could be missed under AE subsampling. Trial JMDB was a head-to-head study. Drug signals were determined where the pemetrexed arm had a 2% excess of incidence over the gemcitabine arm. For trial CALGB 89803, serious AEs and AEs leading to dose modifications were not identified as such in the case report forms. The determination of known events from this study was based only on a 2% excess of AEs leading to drug discontinuation or death. For study ECOG 4599, serious AEs and AEs leading to drug discontinuation or dose modifications were not identified as such in the case report forms. Therefore, there was no separate determination of known events from this study.
Abbreviations: AE, adverse event; AVAiL, Avastin in Lung; ECOG, Eastern Cooperative Oncology Group; BIG, Breast International Group; CALGB, Cancer and Leukemia Group B; HERA, HERceptin Adjuvant.
Known from previous trials.
Identified as known in candidate trial from analysis of AEs to be collected in all patients (see Methods).
Likewise, there were 24 grade 1 to 2 AEs identified as drug effects in the four relevant studies, with 20 of them represented in the corresponding base known safety profile (Table 2). The subsampling analysis focused on detection of the remaining four events that could potentially be missed, along with known grade 1 to 2 events illustrating trends across varying incidence rates.
Subsampling examples are presented and discussed in detail to illustrate overarching trends in the data across the metastatic and adjuvant studies. Reanalysis results of the adjuvant trials are reported in the Appendix (online only).
Rates of detection of grade 3+ events for the metastatic trials ranged from 62% to 94% for the 200-patient subsamples and from 61% to 100% for the 600-patient subsamples when using the random-by-patient selection method (Table 3). For the grade 1 to 2 events, rates of detection ranged from 76% to 99.5% for the 200-patient subsamples and from 99.5% to 100% for the 600-patient subsamples (Table 3). The chance of detecting these events increased with increasing subsample size. The rates of detection for the centers-at-random subsamples were consistent with, although slightly lower than, those using the random-by-patient method.
Table 3.
Probability of Detecting AEs Under Random Subsampling Methods for Metastatic Disease Studies
| Study, AE, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|---|
| 200 (%) | 300 (%) | 400 (%) | 500 (%) | 600 (%) | ||
| Grade 3+ events | ||||||
| JMDB, anorexia*†, 2.1% | Random by patient | 62.4 | 61 | 63.3 | 60.4 | 61 |
| Random by center‡ | 50.6 | 49.9 | 52.7 | 56.7 | 56.4 | |
| AVAiL, weight decreased,2.1% | Random by patient | 63 | 65 | 66 | 68 | 79 |
| Random by center‡ | 51 | 54 | 52 | 59 | 65 | |
| ECOG 4599, infection without neutropenia, 2.4% | Random by patient | 63 | 67 | 68 | 72 | 75 |
| Random by center‡ | 57 | 60 | 63 | 68 | 70 | |
| EGF30001, leukopenia,2.4% | Random by patient | 68 | 70 | 79 | 86 | NA |
| Random by center‡ | 58 | 61 | 66 | 79 | NA | |
| EGF30001, nausea,2.4% | Random by patient | 66 | 68 | 73 | 84 | NA |
| Random by center‡ | 54 | 57 | 64 | 71 | NA | |
| ECOG 4599, proteinuria,* 3.0% | Random by patient | 87 | 91 | 94 | 96 | 98 |
| Random by center‡ | 78 | 85 | 90 | 93 | 98 | |
| AVF2107g, abdominal pain, 3.4% | Random by patient | 72 | 77 | 80 | 85 | 92 |
| Random by center‡ | 65 | 72 | 75 | 80 | 90 | |
| JMDB, nausea,*† 3.5% | Random by patient | 72.9 | 74.5 | 78.1 | 79.7 | 82 |
| Random by center‡ | 68.7 | 69.6 | 75.5 | 77.2 | 80.5 | |
| AVAiL, epistaxis,† 4.3% | Random by patient | 94 | 98 | 99.4 | 100 | 100 |
| Random by center‡ | 91 | 97 | 99.6 | 100 | 100 | |
| AVF2107g, leukopenia,6.7% | Random by patient | 79 | 88 | 92 | 97 | 99.4 |
| Random by center‡ | 77 | 85 | 90 | 96 | 98 | |
| Grade 1 to 2 events | ||||||
| AVAiL, stomatitis,* 6.4% | Random by patient | 76 | 76 | 88 | 92 | 99.5 |
| Random by center‡ | 67 | 70 | 78 | 87 | 97 | |
| AVAiL, headache,* 15.4% | Random by patient | 99.5 | 100 | 100 | 100 | 100 |
| Random by center‡ | 98 | 99.9 | 100 | 100 | 100 | |
NOTE. AEs in italics could be missed under patient subsampling. The other events are known events but are included here because the magnitude of the active arm rate excess versus the control arm illustrates the properties of AE subsampling. Results from the two missable events from ECOG 4599—hyponatremia with 2.4% excess and febrile neutropenia with 2.6% excess—were omitted because the subsampling results were similar to those for infection without neutropenia, which had a rate excess of 2.4%. Grade 3+ events are detected in a simulation when they appear in 2% excess over the control arm; grade 1 to 2 events are detected when they appear in 5% excess over the control arm. Rates of detection in bold are ≥ 75%.
Abbreviations: AE, adverse event; AVAiL, Avastin in Lung; ECOG, Eastern Cooperative Oncology Group; NA, not applicable.
Known from previous trials.
Identified as known in candidate trial from analysis of AEs to be collected in all patients (see Methods).
Sample sizes are approximate. The number of centers selected at random was determined to achieve an average number of patients at or above the target level.
Further, the chance of event detection was larger the greater the AE rate excess in the full study. For example, “leukopenia,” with a 6.7% excess in the full AVF2107g trial, was detected in 92% of the 400 random-by-patient subsets, whereas “weight decreased,” with a 2.1% excess in the full AVAiL (Avastin in Lung) study, was detected in 66% of the corresponding 400-patient subsets. Across the metastatic studies, all grade 3+ AEs analyzed that had at least 3% excess in the full study were detected in at least 75% of the simulations with subsamples of 400 patients, regardless of the random selection method used. The two grade 1 to 2 events from the AVAiL trial had full-trial incidence rate differences of 6.4% and 15.4%. Both events were likely to be detected with random subsampling of 400 patients (Table 3).
With the systematic subsampling methods for the metastatic trials, the observed AE rate difference varied around the full study value and generally converged to the full study value with larger subsample size (Table 4). Similar to the trend observed with random sampling methods, the grade 3+ events with full trial rate differences close to the cutoff rate of 2% were sometimes missed with subsampling. However, the chance of detecting events increased as the full study event rate difference increased above 2%. Events with full-study incidence excess of 3% or greater were detected in 88% of the subsamples. As with the random subsampling methods, AE detection was more likely in the larger subsamples and as full-study event rate differences increased beyond the 2% cutoff rate. For both grade 3+ and grade 1 to 2 events using the systematic subsampling methods, AE rate differences were similar to the full study rates with subsamples of 400 patients or more.
Table 4.
AE Incidence Differences Under Systematic Subsampling Methods for Metastatic Disease Studies
| Study, AE, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|---|
| 200 (%) | 300 (%) | 400 (%) | 500 (%) | 600 (%) | ||
| Grade 3+ events | ||||||
| JMDB, anorexia,*† 2.1% | Biggest centers‡ | 0.7 | 1.0 | 1.1 | 2.5 | 2.3 |
| First patients enrolled | 1.6 | 1.6 | 2.1 | 2.4 | 2.0 | |
| AVAiL, weight decreased, 2.1% | Biggest center‡ | 2.2 | 3.2 | 2.9 | 2.3 | 2.4 |
| First patients enrolled | 0.1 | 0.7 | 1.6 | 2.3 | 2.3 | |
| ECOG 4599 infection without neutropenia, 2.4% | Biggest centers‡ | 3.5 | 3.1 | 2.3 | 3.4 | 3.0 |
| First patients enrolled | 2.9 | 1.4 | 1.5 | 2.4 | 2.4 | |
| EGF30001, leukopenia, 2.4% | Biggest centers‡ | 4.7 | 4.2 | 4.5 | 3.6 | NA |
| First patients enrolled | 3.9 | 3.3 | 3.0 | 2.8 | NA | |
| EGF30001, nausea, 2.4% | Biggest centers‡ | 2.1 | 1.0 | 1.0 | 1.2 | NA |
| First patients enrolled | 3.0 | 2.6 | 1.4 | 2.0 | NA | |
| ECOG 4599, proteinuria,* 3.0% | Biggest centers‡ | 2.8 | 3.9 | 4.4 | 3.9 | 3.7 |
| First patients enrolled | 2.0 | 2.0 | 4.0 | 3.6 | 3.4 | |
| AVF2107g, abdominal pain, 3.4% | Biggest centers‡ | 2.3 | 3.3 | 3.5 | 2.7 | 1.5 |
| First patients enrolled | 4.6 | 3.2 | 3.9 | 4.3 | 4.9 | |
| JMDB, nausea,*† 3.5% | Biggest centers‡ | 1.9 | 1.9 | 2.0 | 1.2 | 1.1 |
| First patients enrolled | 0.8 | 2.4 | 2.8 | 2.1 | 2.4 | |
| AVAiL, epistaxis,† 4.3% | Biggest centers‡ | 4.2 | 3.9 | 2.9 | 4.7 | 4.7 |
| First patients enrolled | 5.1 | 5.4 | 5.6 | 4.7 | 4.7 | |
| AVF2107g, leukopenia, 6.7% | Biggest centers‡ | 7.9 | 10.6 | 9.6 | 7.3 | 6.4 |
| First patients enrolled | 2.4 | 4.8 | 7.1 | 7.3 | 6.2 | |
| Grade 1 to 2 events | ||||||
| AVAiL, stomatitis,* 6.4% | Biggest centers‡ | 4.3 | 7 | 7.2 | 7.1 | 6.7 |
| First patients enrolled | 10.3 | 6.8 | 6.1 | 6.2 | 7.0 | |
| AVAiL, headache,* 15.4% | Biggest centers‡ | 8.5 | 9.7 | 12.6 | 13.5 | 14.9 |
| First patients enrolled | 18.9 | 19.3 | 18.9 | 17.7 | 16.0 | |
NOTE. AEs in italics could be missed under patient subsampling. The other events are known events but are included here because the magnitude of the active arm rate excess versus the control arm illustrates the properties of the subsampling. Results from the two missable events from ECOG 4599—hyponatremia with 2.4% excess and febrile neutropenia with 2.6% excess—were omitted because the subsampling results were similar to those for infection without neutropenia, which had a rate excess of 2.4%. Incidence differences in bold are ≥ 2 for grade 3+ AEs and ≥ 5 for grade 1 to 2 AEs.
Abbreviations: AE, adverse event; AVAiL, Avastin in Lung; ECOG, Eastern Cooperative Oncology Group; NA, not applicable.
Known from previous trials.
Identified as known in candidate trial from analysis of AEs to be collected in all patients (see Methods).
Sample sizes are approximate. Enough centers were selected to meet or exceed the target subsample size.
Regarding noise event detection among subsamples of the metastatic disease trials, fewer were detected for simulations with larger subsample sizes. For example, an average of 13.2 noise events were detected in the random-by-patient subsamples of 200 patients for AVF2107g (Table 5). For simulations of the 400 random-by-patient subsamples in that trial, the average number of noise events decreased to 4.9. This trend was observed across metastatic studies in grade 3+ AE subsets selected by either random method. The number of grade 3+ noise events fluctuated across trials for the systematic subsets of size 200, but was consistently low with subsets of 400 patients, ranging between zero to three noise events. Similar trends were observed for the grade 1 to 2 events.
Table 5.
Number of Noise Events Detected Under Subsampling for Metastatic Disease Studies
| Study and Sampling Method | No. of Noise Events |
||||
|---|---|---|---|---|---|
| 200 Patients | 300 Patients | 400 Patients | 500 Patients | 600 Patients | |
| Grade 3+ events | |||||
| AVAiL | |||||
| Random by patient* | 9 | 4.3 | 2.4 | 1.2 | 0.3 |
| Random by center*† | 5.4 | 2.8 | 1.5 | 0.7 | 0.2 |
| Biggest centers† | 5 | 0 | 0 | 0 | 0 |
| First patients enrolled | 16 | 9 | 2 | 0 | 0 |
| AVF2107g | |||||
| Random by patient* | 13.2 | 7.6 | 4.9 | 3.3 | 2.3 |
| Random by center*† | 8.8 | 5.1 | 3.6 | 2.5 | 1.6 |
| Biggest centers† | 13 | 5 | 1 | 1 | 1 |
| First patients enrolled | 8 | 3 | 2 | 2 | 0 |
| ECOG 4599 | |||||
| Random by patient* | 9.9 | 5.8 | 3.9 | 2.6 | 1.6 |
| Random by center*† | 6.7 | 4.3 | 2.8 | 2 | 1.2 |
| Biggest centers† | 3 | 2 | 1 | 2 | 2 |
| First patients enrolled | 5 | 4 | 1 | 2 | 1 |
| EGF30001 | |||||
| Random by patient* | 12 | 7 | 4 | 2 | NA |
| Random by center*† | 8 | 5 | 3 | 1 | NA |
| Biggest centers† | 6 | 4 | 1 | 1 | NA |
| First patients enrolled | 4 | 4 | 3 | 0 | NA |
| JMDB | |||||
| Random by patient* | 0.448 | 0.39 | 0.422 | 0.528 | 0.139 |
| Random by center*† | 0.534 | 0.387 | 0.449 | 0.373 | 0.17 |
| Biggest centers† | 1 | 2 | 1 | 1 | 1 |
| First patients enrolled | 1 | 2 | 3 | 3 | 2 |
| Grade 1 to 2 events | |||||
| AVAiL | |||||
| Random by patient* | 5 | 1.8 | 1 | 0.2 | 0 |
| Random by center*† | 3.9 | 1.9 | 0.8 | 0.3 | 0 |
| Biggest centers† | 1 | 0 | 1 | 0 | 0 |
| First patients enrolled | 7 | 1 | 1 | 0 | 0 |
NOTE. Noise event quantities in bold are ≤ 3.
Abbreviations: AVAiL, Avastin in Lung; ECOG, Eastern Cooperative Oncology Group; NA, not applicable.
Noise events for random sampling methods are reported as mean numbers determined from 1,000 simulations.
Sample sizes are approximate due to analysis structure for sampling by center methods.
Adjuvant studies Cancer and Leukemia Group B 89803 and HERA (HERceptin Adjuvant) were subsampled for grade 3+ and 1 to 2 AEs, whereas trial Breast International Group 1-98 was subsampled only for grade 1 to 2 events because no missable grade 3+ events were identified (Table 2). Under the random sampling methods, the rates of AE detection increased as subset size increased (Appendix Table A3, online only). Events with high incidence rate differences in the full trial analyses were detected in higher frequencies than those close to their associated cutoff (2% for grade 3+ and 5% for grade 1 to 2). These trends were also observed in the systematic patient subsets selected from largest centers and by enrollment order, regardless of the adjuvant trial analyzed (Appendix Table A4, online only). Noise event detection patterns for grade 3+ and 1 to 2 AE subsamples from the adjuvant disease trials mirrored those from the metastatic trials (Appendix Table A5, online only).
The overall number of AEs contained in the safety databases for seven of the trials was 107,884 (Table 6). Of these AEs, 19,621 were serious+. There were 72,801 grade 1 to 2 events not classified as serious+, ranging from an average of 2.3 to 12.0 per patient. Further, grade 1 to 2 events were from 4.2 to 9.6 times as numerous as the serious+ events in the metastatic trials and from 2.2 to 14.4 times as numerous in the adjuvant trials.
Table 6.
Number of AEs
| Study (safety population analyzed) | Distinct No. of AEs (average No. of AEs per patient) |
|||||
|---|---|---|---|---|---|---|
| Grade 1 to 2 AEs Not Serious+ |
Grade 3+ AEs Not Serious+ |
SAEs and AEs Leading to Dose Discontinuation/Change Serious+ |
||||
| No. | Avg | No. | Avg | No. | Avg | |
| Metastatic studies | ||||||
| AVF2107g (n = 788) | NA | 1,297 | 1.6 | 1,187 | 1.5 | |
| AVAiL (n = 656) | 6,245 | 9.5 | 1,030 | 1.6 | 849 | 1.3 |
| EGF30001 (n = 580) | 6,943 | 12.0 | 377 | 0.6 | 725 | 1.2 |
| JMDB (n = 1,669) | 10,514 | 6.3 | 835 | 0.5 | 2,504 | 1.5 |
| Adjuvant studies | ||||||
| BIG 1-98 (n = 7,963) | 28,098 | 3.5 | 9,612 | 1.2 | 12,845 | 1.6 |
| CALGB 89803 (n = 1,264) | 13,300 | 10.5 | 2,150 | 1.7 | 976 | 0.8 |
| HERA (n = 3,386) | 7,701 | 2.3 | 161 | 0.05 | 535 | 0.2 |
| Total | 72,801 | 4.7 | 15,462 | 0.9 | 19,621 | 1.2 |
NOTE. SAEs were not identified in study Eastern Cooperative Oncology Group 4599.
Abbreviations: AEs, adverse events; SAEs, serious adverse events; NA, grade 1 to 2 adverse events were not analyzed for trial; AVAiL, Avastin in Lung; BIG, Breast International Group; CALGB, Cancer and Leukemia Group B; HERA, HERceptin Adjuvant; Avg, average.
The average number of concomitant medications reported per patient ranged from 14 to 27 in the metastatic trials and from four to seven in the adjuvant trials (Appendix Table A6, online only). Of the 136,608 concomitant medication records included in the summary tabulations for these studies, none contributed to labeling changes for the supplemental indications.
DISCUSSION
The collection of all AEs in all patients in a study designed to support a supplemental NDA/BLA has the potential to identify new drug safety signals (Table 2). Weighing the number and clinical significance of these signals against the effort required to collect them leads us to recommend that, although AEs should be collected comprehensively for the initial NDA/BLA to establish the drug's basic safety profile, toxicity data collection for subsequent supplemental trials should be limited to serious+ AEs in all patients and grade 3+ AEs in a subset of patients. The asymmetric collection of AEs (ie, only in patients on the investigational arm) should be avoided, as there is then no concurrent control for the accurate assessment of safety signals.
For the collection of grade 3+ events in the metastatic trials, a 400-patient subsample selected at random provided adequate probability, averaging 85%, of detecting events that would be notable in the full study (ie, those with an active to control rate excess of ≥ 3%). For AEs with 2% to 3% excess in the full study, there is an approximate 30% chance of missing the signal with a subset of 400 patients. Importantly, AEs close to the data cutoff are hard to detect, regardless of sample size. For example, even in a trial of 3,000 patients, there is a 50% chance of missing an event that has a true 2% excess frequency at a cutoff of 2%. Also, with a 400-patient subsample, the number of noise events is acceptably low, generally in the range of three events or fewer.
For larger metastatic trials and for adjuvant trials, the subsample size should be larger than 400, but it need not be proportionately so; our analysis suggests that subsample sizes from 400 to 800 patients should be sufficient. Based on our results, two approaches were formulated to allow the prospective determination of subsample sizes (Appendix: Sample Size Rationale, online only).24 Subsampling may not be worthwhile in studies that have fewer than 600 patients total, given the effort required to set up the process.
In the adjuvant setting, the benefit/risk profile of a drug is different than in the metastatic setting. Patients and physicians are less willing to tolerate risk. Grade 1 to 2 events may play a larger role in establishing the safety profile of the drug, causing one to question whether it is wise to omit collection of grade 1 to 2 events in this setting. It is important to note that all events meeting the serious+ criteria would still be collected. Therefore, clinically important grade 1 to 2 events—ones that cause a physician to modify or discontinue dosing—would be collected. Using this data collection strategy would have saved the collection of 72,801 grade 1 to 2 AEs across six of our trials, averaging 4.7 AEs per patient, while still not missing any clinically significant events (Table 6).
A notable feature of our reanalysis is that the indication and control-arm medications used in the candidate study differed from the studies used to define the drug's known safety profile. Despite these differences, there were few AEs that would be missed by subsampling. Therefore, our recommendations should apply broadly across supplemental applications except for the first submission after an accelerated approval (or full approval in smaller disease populations) or where the patient population to be studied in the supplemental indication is substantially different in clinical characteristics as to be at substantially higher risk of AEs than were seen in the trials we reanalyzed.
The systematic subsampling methods revealed no consistent bias in the estimates of full-trial AE rate differences in the trials we analyzed. However, random subsampling methods would ensure the absence of bias in general. Sampling centers at random provides the best balance of statistical legitimacy and operational feasibility, for both the site and the sponsor. Limiting sites to one data collection system reduces confusion and potential impact on data quality. A random selection of sites, although unbiased, may not adequately represent the study population. Therefore, we recommend stratification of the sample based on relevant site characteristics. In order to ensure enough patients are included in the subsample, the number of sites selected should be overestimated and ongoing enrollment should be monitored.
The comprehensive collection of concomitant medications is resource intensive and within a supplemental application contributes little to defining the safety profile of a drug. Therefore, although full data collection should continue for clinical trials supporting an initial indication, we recommend that for trials designed to support supplemental applications, collection of concomitant medications should be limited to specific targeted collections based on the known safety and pharmacologic profile of the investigational agent, medications that exhibit anticancer properties, and ones that meet a specific objective of the trial (eg, health economics or costing). Concomitant medications should continue to be reported in the narrative section of serious AE forms.
In conclusion, doctors want to recognize drug safety issues that lead them to change or discontinue a patient's treatment. Collection of large quantities of data that do not inform regulatory or clinical practice decisions taxes resources that could be used to improve the collection of more relevant data and thus risks obfuscating important safety signals. For phase III trials supporting supplemental applications, an optimized AE and concomitant medication collection strategy can identify clinically significant safety information and preserve resources otherwise spent collecting uninformative information. Once several applications that use this methodology have been reviewed by the FDA, it will be important to determine the benefit of subsampling itself and assess whether collection of only serious+ events may be sufficient.
Although this project focused on collection of AE and concomitant medication data, steps could be taken in other areas to further simplify study conduct. For example, significant resources were expended to perform an independent radiologic review of progression events in the Eastern Cooperative Oncology Group 2100 trial that only served to validate the original trial results.25
For clinical trials intended to support supplemental NDAs/BLAs, symmetric collection of events, regardless of grade, that are serious or lead to dose modification/discontinuation or death should occur in all patients. Grade 1 to 2 events and complete concomitant medication records need not be collected. Grade 3+ events should be collected in a subsample of the full trial. This optimized data collection strategy leads to a high probability of capturing events that matter most to patients and their physicians.
Acknowledgment
We thank Joshua Benner, PharmD, ScD, and Mark McClellan, MD (Brookings Institution); Jonathan Denne, PhD (Eli Lilly); Jeffrey Allen, PhD, Rasika Kalamegham, PhD, and Ellen Sigal, PhD (Friends of Cancer Research); Chin-Yu Lin, PhD (Genentech); Melissa Stutts and Susan Bertino (GlaxoSmithKline); Robert Erwin (Marti Nelson Cancer Foundation); Carol Brannan (Nodality); David Carr, BSc, Lynne McGrath, MPH, PhD, and Antonella Maniero, PhD (Novartis); and Richard Pazdur, MD, Rajeshwari Sridhara, PhD, Robert Temple, MD, Karen Weiss, MD, and Janet Woodcock, MD (US Food and Drug Administration).
Appendix
Subsample Size Rationale
Specified power approach to adverse event subsampling in oncology trials.
For supplemental trials in the adjuvant disease setting and for those metastatic supplemental trials tested in a population very different from the initial setting, subsampling methodology should account for anticipated rates of detection of adverse reactions. Suppose a subsample should capture an event with a 6% incidence rate in active arm patients as compared with 3% incidence in the control group, but display less sensitivity toward events occurring in 46% versus 43% in active versus control groups because physicians would already be primed to address the latter of such events. An approach using normal approximation with continuity correction is recommended to determine subsample sizes for such trials due to its adaptability to specific adverse event (AE) incidence characteristics. For a study with N patients per arm, a subsample of n patients per arm should be selected, according to the following equation:
 |
Because the continuity correction cc is a function of n, equation 1 should be solved numerically. This subsampling method may be applied to all supplement trials eligible for adverse event subsampling, as outlined in the associated guidance. An example calculation of the Specified Power Approach can be found below, and a range of recommended subsample sizes can be found in Appendix Table A1.
Example.
Drug X, having received full approval in its initial metastatic setting, is to be studied in an add-on adjuvant disease trial of 6,000 patients, divided evenly between active and control arms. A subsample of patients should be selected for comprehensive toxicity data collection, powered to identify AEs occurring in a 6% incidence rate in active arm patients, as compared with a 3% incidence rate in control arm patients. Events occurring in 2% incidence rate excess within the subsample will be identified as drug signals. Ensuring an 80% probability that such events are detected:
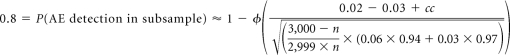 |
where
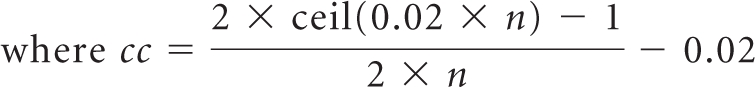 |
Therefore,
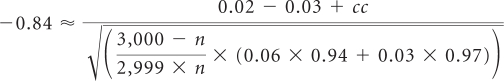 |
Solving numerically for n results in a value of 445, rounded up to the nearest five patients. A total of 890 patients should receive comprehensive AE collection; toxicity data collection on the remaining 5,110 patients may be limited to a targeted strategy, as outlined in the guidance.
Equal standard error AE subsampling approach for metastatic trials with ≥ 800 patients.
In the results of the Data Optimization re-analysis, for metastatic disease trials with 800 subjects, a subsample size of approximately 400, equally distributed between arms, would be sufficient to adequately describe the safety profile for a drug in its supplemental indication. For metastatic trials of similar character with at least 800 patients, a second and simpler approach to subsampling may be used. Applying finite-population sampling formulas, one can extrapolate larger subsample sizes while preserving the 400 of 800 patient subsampling precision. For a study with N patients per arm, a subsample of n patients per arm should be selected, according to the following equation:
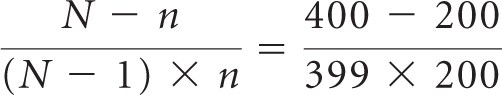 |
Therefore,
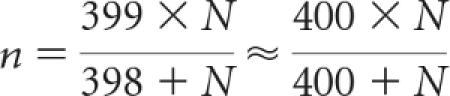 |
An example calculation of the Equal Standard Error approach can be found below, and a range of recommended subsample sizes can be found in Appendix Table A2.
Example.
Drug Y, approved in an initial metastatic disease setting, is to be studied in an add-on metastatic trial of 1,000 patients, equally distributed between active and control arms. To preserve precision of subsampling relative to a 400-patient subset from an 800-patient trial, statisticians should isolate a subsample of size n, according to the following equation:
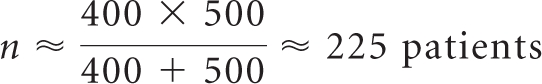 |
A total of 450 patients (225 patients per arm) should receive comprehensive adverse event analysis; toxicity data collection on the remaining 550 patients may be limited to a targeted strategy, as outlined in the associated guidance.
Additional Data Tables
Table A1.
Guideline for Adverse Event Subsampling for Oncology Trials With ≥ 800 Patients to Achieve Similar Power to That of a 400-Patient Subsample of an 800-Patient Trial (specified power approach)
| Total Study Size (1:1) | Total Subsample Size (1:1) |
|---|---|
| 800 | 400 |
| 1,200 | 500 |
| 1,500 | 590 |
| 2,000 | 680 |
| 3,000 | 780 |
| 6,000 | 890 |
Table A2.
Guideline for Adverse Event Subsampling for Metastatic Oncology Trials With ≥ 800 Patients (equal standard error approach)
| Total Study Size (1:1) | Total Subsample Size (1:1) |
|---|---|
| 800 | 400 |
| 1,200 | 480 |
| 1,500 | 530 |
| 2,000 | 580 |
| 3,000 | 640 |
| 6,000 | 710 |
Table A3.
Probability of Detecting Adverse Events Under Random Subsampling Methods for Adjuvant Disease Studies
| CALGB 89803 (n = 1,264), Adverse Event, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|---|
| 200 | 300 | 400 | 500 | 600 | ||
| Grade 3+ events | ||||||
| Thrombosis embolism, 4.2% | Random by patient | 78 | 84 | 88 | 92 | 94 |
| Random by center* | NA | NA | NA | NA | NA | |
| Grade 1 to 2 events | ||||||
| Sweating (diaphoresis), 5.8% | Random by patient | 68 | 63 | 73 | 71 | 77 |
| Random by center* | NA | NA | NA | NA | NA | |
| Constipation, 6.9% | Random by patient | 68 | 69 | 74 | 73 | 83 |
| Random by center* | NA | NA | NA | NA | NA | |
| HERA (n = 3,386), Adverse Event, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) | ||||
| 600 | 900 | 1,200 | 1,500 | 1,800 | ||
| Grade 3+ events | ||||||
| Ejection fraction decreased,†‡ 2.4% | Random by patient | 75 | 77 | 84 | 85 | 92 |
| Random by center* | 62 | 70 | 74 | 79 | 82 | |
| Grade 1 to 2 events | ||||||
| Fatigue,5.1% | Random by patient | 55 | 55 | 56 | 54 | 54 |
| Random by center* | 51 | 50 | 54 | 54 | 53 | |
| Headache,† 6.8% | Random by patient | 86 | 91 | 96 | 97 | 99.1 |
| Random by center* | 84 | 89 | 92 | 96 | 98 | |
| BIG 1-98 (n = 7,963), Adverse Event, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
| 600 | 1,200 | 2,000 | 4,000 | 6,000 | ||
| Grade 1 to 2 events: Hypercholesterolemia,21.2% | Random by patient | 100 | 100 | 100 | 100 | 100 |
| Random by center* | 99.9 | 100 | 100 | 100 | 100 | |
NOTE. To account for their substantially larger safety databases, the HERA study (n = 3,386) and BIG 1-98 study (n = 7,963) were evaluated using larger subsamples. Adverse events in italics could be missed under patient subsampling. The other events are known events but are included here because the magnitude of the active arm rate excess versus the control arm illustrates the properties of adverse event subsampling. From the HERA trial, known adverse events ejection fraction decreased (grade 3 to 4) and headache (grade 1 to 2) were included to demonstrate trends of finding adverse events as the rates of incidence increase from the cutoff rate. Grade 3+ events are detected in a simulation when they appear in 2% excess over the control arm; grade 1 to 2 events are detected when they appear in 5% excess over the control arm. Rates of detection in bold are ≥ 75%.
Abbreviations: CALGB, Cancer and Leukemia Group B; NA, not applicable; HERA, HERceptin Adjuvant; BIG, Breast International Group.
Sample sizes are approximate. No. of centers selected at random was determined to achieve an average No. of patients at or above the target level.
Known from previous trials.
Identified as known in candidate trial from analysis of adverse events to be collected in all patients (see Methods).
Table A4.
Adverse Event Incidence Differences Under Systematic Subsampling Methods for Adjuvant Disease Studies
| CALGB 89803 (n = 1,263), Adverse Event, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|---|
| 200 | 300 | 400 | 500 | 600 | ||
| Grade 3+ events | ||||||
| Thrombosis embolism, 4.2% | Biggest centers* | NA | NA | NA | NA | NA |
| First patients enrolled | 5.2 | 7.3 | 5.6 | 5.9 | 5.2 | |
| Grade 1 to 2 events | ||||||
| Sweating (diaphoresis), 5.8% | Biggest centers* | NA | NA | NA | NA | NA |
| First patients enrolled | 6.2 | 6.0 | 4.6 | 5.9 | 6.3 | |
| Constipation, 6.9% | Biggest centers* | NA | NA | NA | NA | NA |
| First patients enrolled | 3.4 | 8.7 | 8.6 | 7.6 | 9.4 | |
| HERA (n = 3,386), Adverse Event, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|---|
| 600 | 900 | 1,200 | 1,500 | 1,800 | ||
| Grade 3+ events | ||||||
| Ejection fraction decreased,†‡ 2.4% | Biggest centers* | 2.2 | 1.7 | 1.9 | 1.6 | 1.9 |
| First patients enrolled | 2.7 | 3.4 | 3.4 | 3.2 | 2.9 | |
| Grade 1 to 2 events | ||||||
| Fatigue, 5.1% | Biggest centers* | 4.6 | 3.6 | 5.5 | 5.1 | 5.4 |
| First patients enrolled | 7.1 | 6.8 | 5.8 | 5.7 | 6.6 | |
| Headache,† 6.8% | Biggest centers* | 5.3 | 4.4 | 5.9 | 6.3 | 6.7 |
| First patients enrolled | 10.1 | 10.2 | 9.6 | 9.2 | 8.6 | |
| BIG 1-98 (n = 7,963), Adverse Event, Active Arm Rate Excess in Full Study | Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|---|
| 600 | 1,200 | 2,000 | 4,000 | 6,000 | ||
| Grade 1 to 2 events: Hypercholesterolemia, 21.2% | Biggest centers* | 18.2 | 20.9 | 16.9 | 19.8 | 21 |
| First patients enrolled | 28.4 | 23.7 | 24.1 | 19.3 | 20.1 | |
NOTE. To account for their substantially larger safety databases, the HERA study (n = 3,386) and BIG 1-98 study (n = 7,963) were evaluated using larger subsamples. Adverse events in italics could be missed under patient subsampling. The other events are known events but are included here because the magnitude of the active arm rate excess versus the control arm illustrates the properties of the subsampling. Results from two missable events from ECOG 4599—hyponatremia with 2.4% excess and febrile neutropenia with 2.6% excess—were omitted because the subsampling results were similar to those for infection without neutropenia, which had a rate excess of 2.4%. Incidence differences in boldare ≥ 2 for grade 3+ adverse events and ≥ 5 for grade 1 to 2 adverse events.
Abbreviations: CALGB, Cancer and Leukemia Group B; NA, not applicable; HERA, HERceptin Adjuvant; BIG, Breast International Group; ECOG, Eastern Cooperative Oncology Group.
Sample sizes are approximate. Enough centers were selected to meet or exceed the target subsample size.
Known from previous trials.
Identified as known in candidate trial from analysis of adverse events to be collected in all patients (see Methods).
Table A5.
Number of Noise Events Detected Under Subsampling Methods for Adjuvant Disease Studies
| CALGB 89803 (n = 1,264): Grade and Sampling Method | Subsample Size (total No. of patients) |
||||
|---|---|---|---|---|---|
| 200 | 300 | 400 | 500 | 600 | |
| Grade 3+ events | |||||
| Random by patient* | 7.5 | 3.9 | 2.1 | 1.2 | 0.7 |
| Random by center*† | NA | NA | NA | NA | NA |
| Biggest centers† | NA | NA | NA | NA | NA |
| First patients enrolled | 8 | 4 | 1 | 0 | 0 |
| Grade 1 to 2 events | |||||
| Random by patient* | 7.6 | 3.9 | 1.0 | 1.9 | 1.7 |
| Random by center*† | NA | NA | NA | NA | NA |
| Biggest centers† | NA | NA | NA | NA | NA |
| First patients enrolled | 7 | 4 | 2 | 3 | 1 |
| HERA (n = 3,386): Grade and Sampling Method | Subsample Size (total No. of patients) |
||||
| 600 | 900 | 1,200 | 1,500 | 1,800 | |
| Grade 3+ events | |||||
| Random by patient* | 0.1 | 0 | 0 | 0 | 0 |
| Random by center *† | 0.1 | 0 | 0 | 0 | 0 |
| Biggest centers† | 0 | 0 | 0 | 0 | 0 |
| First patients enrolled | 0 | 0 | 0 | 0 | 0 |
| Grade 1 to 2 events | |||||
| Random by patient* | 0.5 | 0.1 | 0.1 | 0 | 0 |
| Random by center*† | 0.5 | 0.2 | 0.1 | 0 | 0 |
| Biggest centers† | 1 | 0 | 0 | 0 | 0 |
| First patients enrolled | 3 | 2 | 2 | 0 | 0 |
| BIG 1-98 (n = 7,963): Grade and Sampling Method | Subsample Size (total No. of patients) |
||||
| 600 | 1,200 | 2,000 | 4,000 | 6,000 | |
| Grade 3+ events | |||||
| Random by patient* | 0.9 | 0.5 | 0.4 | 0.2 | 0.1 |
| Random by center*† | 0.8 | 0.4 | 0.3 | 0.3 | 0.1 |
| Biggest centers† | 2 | 1 | 0 | 1 | 0 |
| First patients enrolled | 0 | 0 | 1 | 0 | 0 |
| Grade 1 to 2 events | |||||
| Random by patient* | 0.6 | 0.5 | 0.4 | 0.4 | 0.3 |
| Random by center*† | 0.6 | 0.5 | 0.4 | 0.4 | 0.3 |
| Biggest centers† | 1 | 1 | 1 | 1 | 1 |
| First patients enrolled | 0 | 0 | 0 | 0 | 0 |
NOTE. To account for their substantially larger safety databases, the HERA study (n = 3,386) and BIG 1-98 study (n = 7,963) were evaluated using larger subsamples. Noise event quantities in bold are ≤ 3.
Abbreviations: CALGB, Cancer and Leukemia Group B; NA, not applicable; HERA, HERceptin Adjuvant; BIG, Breast International Group.
Noise events for random sampling methods are reported as mean numbers determined from 1,000 simulations.
Sample sizes are approximate due to analysis structure for sampling by center methods.
Table A6.
Number of Concomitant Medication Records
| Study (safety population analyzed) | Concomitant Medication Records (average No. per patient) |
Concomitant Medication Data Fields (average No. per patient) |
||
|---|---|---|---|---|
| No. | Avg | No. | Avg | |
| Metastatic studies | ||||
| AVF2107g (n = 788) | 20,998 | 26.6 | 83,992 | 106.6 |
| AVAiL (n = 656) | 11,957 | 18.2 | 47,828 | 72.9 |
| EGF30001 (n = 580) | 9,270 | 16.0 | 94,245 | 163.05 |
| JMDB (n = 1,669) | 24,168 | 14.5 | 120,840 | 72.4 |
| Adjuvant studies | ||||
| BIG 1-98 (n = 7,963) | 56,966 | 7.1 | 1,841,572 | 230 |
| HERA (n = 3,386) | 13,249 | 3.9 | 52,996 | 15.7 |
| Total | 136,608 | 9.1 | 2,241,473 | 148.6 |
NOTE. Concomitant medications were not collected in all patients in studies Eastern Cooperative Oncology Group 4599 and Cancer and Leukemia Group B 89803. Because of safety signals identified in previous aromatase inhibitor trials, concomitant medication collection in the BIG 1-98 trial included several additional targeted collections medications.
Abbreviations: AVAiL, Avastin in Lung; BIG, Breast International Group; HERA, HERceptin Adjuvant.
Footnotes
See accompanying editorial on page 5019
Presented in part at the Brookings Institution's 2009 Conference on Clinical Cancer Research, September 14, 2009, Washington, DC.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Lee D. Kaiser, Genentech (C); Allen S. Melemed, Eli Lilly (C); Alaknanda J. Preston, GlaxoSmithKline (C); Hilary A. Chaudri Ross, Novartis (C); Jacqueline M. Gough, Eli Lilly (C); William D. Bushnell, GlaxoSmithKline (C) Consultant or Advisory Role: Hilary A. Chaudri Ross, Novartis (C) Stock Ownership: Lee D. Kaiser, Roche; Allen S. Melemed, Eli Lilly; Alaknanda J. Preston, GlaxoSmithKline; Hilary A. Chaudri Ross, Novartis; Jacqueline M. Gough, Eli Lilly; William D. Bushnell, GlaxoSmithKline Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Lee D. Kaiser, Allen S. Melemed, Gwendolyn A. Fyfe, Jacqueline M. Gough, Jeffrey S. Abrams, Richard L. Schilsky
Administrative support: Allen S. Melemed, Cynthia L. Stephens, M. Kelsey Mace
Collection and assembly of data: Lee D. Kaiser, Allen S. Melemed, Alaknanda J. Preston, Hilary A. Chaudri Ross, Donna Niedzwiecki, Jacqueline M. Gough, William D. Bushnell
Data analysis and interpretation: Lee D. Kaiser, Allen S. Melemed, Alaknanda J. Preston, Hilary A. Chaudri Ross, Donna Niedzwiecki, Gwendolyn A. Fyfe, Jacqueline M. Gough, William D. Bushnell, Cynthia L. Stephens, M. Kelsey Mace, Jeffrey S. Abrams, Richard L. Schilsky
Manuscript writing: Lee D. Kaiser, Allen S. Melemed, Alaknanda J. Preston, Hilary A. Chaudri Ross, Donna Niedzwiecki, Gwendolyn A. Fyfe, Jacqueline M. Gough, William D. Bushnell, Cynthia L. Stephens, M. Kelsey Mace, Jeffrey S. Abrams, Richard L. Schilsky
Final approval of manuscript: Lee D. Kaiser, Allen S. Melemed, Alaknanda J. Preston, Hilary A. Chaudri Ross, Donna Niedzwiecki, Gwendolyn A. Fyfe, Jacqueline M. Gough, William D. Bushnell, Cynthia L. Stephens, M. Kelsey Mace, Jeffrey S. Abrams, Richard L. Schilsky
REFERENCES
- 1.United States Code of Federal Regulations. Washington, DC: US Government Printing Office; 1985. Title 21-Food and Drugs. Part 314.50. [Google Scholar]
- 2.US Food and Drug Administration. Guidance for Industry: Cancer Drug and Biological Products—Clinical Data in Marketing Applications. 2001. Oct, http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071323.pdf.
- 3.Mahoney MR, Sargent DJ, O'Connell MJ, et al. Dealing with a deluge of data: An assessment of adverse event data on North Central Treatment Group trials. J Clin Oncol. 2005;23:9275–9281. doi: 10.1200/JCO.2004.00.0588. [DOI] [PubMed] [Google Scholar]
- 4.Roche K, Paul N, Smuck B, et al. Factors affecting workload of cancer clinical trials: Results of a multicenter study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2002;20:545–556. doi: 10.1200/JCO.2002.20.2.545. [DOI] [PubMed] [Google Scholar]
- 5.Doroshow J, Fyfe G, Comis R. Session 3: Data collection standards to establish safety and efficacy. Presented at the National Cancer Policy Forum Workshop on Multi-site Clinical Trials and the National Cancer Institute Cooperative Group Program; July 1-2, 2008; Washington, DC. [Google Scholar]
- 6.Schilsky R, Abrams J, Fyfe G, et al. Panel 1: Data submission standards and evidence requirements. Presented at the 2008 Conference on Clin Cancer Res, hosted by the Engelberg Center for Health Care Reform and Friends of Cancer Res; September 26, 2008; Washington, DC. [Google Scholar]
- 7.Schilsky R, Fyfe G, Abrams J, et al. Panel 1: Data submission standards and evidence requirements. Presented at the Engelberg Center for Health Care Reform and Friends of Cancer Res 2009 Conference on Clin Cancer Res; September 14, 2009; Washington, DC. [Google Scholar]
- 8.Reck M, von Pawel J, Zatloukal P, et al. Phase III Trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAiL. J Clin Oncol. 2009;27:1227–1234. doi: 10.1200/JCO.2007.14.5466. [DOI] [PubMed] [Google Scholar]
- 9.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 10.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 11.Di Leo A, Gomez H, Aziz Z, et al. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol. 2008;26:5544–5552. doi: 10.1200/JCO.2008.16.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 13.Thürlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer: Breast International Group 1-98 Collaborative Group. N Engl J Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 14.Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan and fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: Results of CALGB 89803. J Clin Oncol. 2007;25:3456–3461. doi: 10.1200/JCO.2007.11.2144. [DOI] [PubMed] [Google Scholar]
- 15.Piccart-Gebhart M, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 16.Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539–1544. doi: 10.1200/JCO.2006.09.6305. [DOI] [PubMed] [Google Scholar]
- 17.Miller KD, Chap LI, Holmes FA, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–799. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 18.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 19.Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21:2636–2644. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 20.Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 21.Cunningham D, Pyrhönen S, James RD, et al. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet. 1998;352:1413–1418. doi: 10.1016/S0140-6736(98)02309-5. [DOI] [PubMed] [Google Scholar]
- 22.Rougier P, Van Cutsem E, Bajetta E, et al. Randomized trial of irinotecan versus fluorouracil by continuous infusion after fluorouracil failure in patients with metastatic colorectal cancer. Lancet. 1998;352:1407–1412. doi: 10.1016/S0140-6736(98)03085-2. [DOI] [PubMed] [Google Scholar]
- 23.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 24.Cochran W. New York, NY: Wiley; 1977. Sampling Techniques (ed 3) [Google Scholar]
- 25.Gray R, Bhattacharya S, Bowden C, et al. Independent review of E2100: A phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol. 2009;27:4966–4972. doi: 10.1200/JCO.2008.21.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]