

Abstract
Opioids are powerful pain relievers, but also potent inducers of dependence and tolerance. Chronic morphine administration (via subcutaneous pellet) induces morphine dependence in the nucleus accumbens, an important dependence region in the brain, yet the cellular mechanisms are mostly unknown. Toll-like receptor 2 (TLR2) plays an essential function in controlling innate and inflammatory responses. Using a knockout mouse lacking TLR2, we assessed the contribution of TLR2 to microglia activation and development of morphine dependence. We report here that mice deficient in TLR2 inhibit morphine-induced the levels of microglia activation and proinflammatory cytokines. Moreover, in TLR2 knockout mice the main symptoms of morphine withdrawal were significantly attenuated. Our data reveal that TLR2 plays a critical role in morphine-induced microglia activation and dependence.
Keywords: TLR2, morphine, microglia, cytokines, dependence
Opioids have been used as potent analgesics for centuries, but their abuse has deleterious physiological effects and can cause dependence. Opioid dependence, including morphine dependence, is a chronic, relapsing disease. Opioids such as morphine have different effects on the immune response based on the dosage and the number of exposures. We have reported that chronic morphine exposure, such as occurs in drug abuse, inhibits immune function and increases susceptibility to diseases [1–3]. Recent evidence indicates that the innate immune responses contribute to the development of opioid dependence and withdrawal [4–6]. Opioid dependence and withdrawal significantly increases the production of pro-inflammatory cytokines interleukin-1 (IL-1) and IL-6 [4;7]. These pro-inflammatory and immunoregulatory cytokines appear to be directly involved in the progression of opioid dependence and withdrawal [4;6;7].
Toll-like receptors (TLRs) are well known as recognition of pathogens in the innate immune system aimed as defending the survival of the host [8;9]. TLRs and their functions have been established in immune cells. Recent studies reveal that TLRs, including TLR2 and TLR4, are a key link between the innate immune system and the central nervous system (CNS) [10;11]. The functional role of TLRs in the CNS, however, remains to be elucidated. TLR2 is widely expressed in the CNS, particularly in microglial cells [11–14].
Microglial cells represent the resident immune host defense and are considered the major immune inflammatory cells of the CNS. TLR2 mediated signaling contributes to the impact on the CNS autoimmune diseases and inflammation [15]. However, roles of TLR mediated immune and inflammatory responses in glial activation during the development of opioid dependence remain to be elucidated. We investigated the role of TLR2 in microglia activation and morphine dependent. In present study we found that TLR2 is required for morphine-induced microglia activation and inflammatory responses. Furthermore, in TLR2 knockout mice the main symptoms of morphine withdrawal were significantly attenuated. Our data demonstrate that TLR2 plays a critical role in opioid dependence-mediated microglia activation.
Animals and reagents
TLR2 knockout mice on a C57BL/6 background and wild type C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). All mice were bred and housed pathogen-free in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU), a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). All aspects of the animal care and experimental protocols were approved by the ETSU Committee on Animal Care. Placebo pellets and morphine pellets were kindly provided by National Institutes of Drug Abuse (NIDA). The Quantative PCR kit was purchased from Invitrogen (Carlsbad, CA).
Drug administration
Morphine was administered chronically by subcutaneous pellet implantation. 2–3-month-old male wild type and TLR2 KO mice were anesthetized with inhaled isofluorane and implanted subcutaneously with a 25 mg morphine base pellet. Age and weight matched male mice with a placebo pellet implantation served as controls. Under isoflurane anesthesia, a small skin pocket was dissected in the animal’s back, where a single pellet was inserted and the skin was closed with surgical sutures [16–18]. At 1, 2, 3, and 4 days after pellet implantation, various regions of brains were harvested for molecular and cellular studies. In physical morphine dependence experiments, at 3 days after pellet implantation, morphine dependence was determined by assessing withdrawal responses after administration of naloxone (1.0 mg/kg body weight, intraperitoneally (i.p)) as described previously [16;17;19]. Briefly, 20 minutes before the naloxone treatment, mice will be placed in a transparent acrylic cylinder (20 cm diameter, 35 cm high) to habituate to their environment. The mice were weighed before naloxone injection. Immediately after the naloxone administration, the mouse was placed gently again in the cylinder and the frequency of naloxone-precipitated withdrawal signs (vertical jumping and wet-dog shakes) [16;17;19] were monitored for 30 min by trained observers who was blinded to all experimental treatments. The mice were then weighed again to express weight loss as a percentage of the pre-injection weight.
Real-time quantitative reverse transcription polymerase chain reaction (RT-PCR)
The real-time PCR detection technique was performed as described in our previous publication [3]. Briefly, first-strand cDNA was synthesized from 1μg of total RNA using a Reaction Ready first strand cDNA synthesis kit (Invitrogen, Carlsbad, CA). After incubation at 70 °C for 3 min and cooling down to 37 °C for 10 min, RT cocktail was added to the annealing mixture and further incubated at 37 °C for 60 min. 2μl of 1:2 diluted cDNA was subjected to real-time quantitative PCR using Bio-Rad iCycler iQ Multicolor Real-Time PCR Detection System (Bio-Rad). PCR was performed in a 25μl volume using SYBR GreenER qPCR Super Mix for iCycler (Invitrogen). All primers were purchased from SABiosciences (Frederick, MD). All PCR assays were performed in triplicate. The reaction conditions were: 50°C for 2 min, 95°C for 8 min 30s, followed by 40 cycles of 95°C for 15s, 60°C for 60s, and 72°C for 30s. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified from all samples on each plate as housekeeping genes. The reaction mixtures without template cDNA were used as negative controls.
All data were represented as means ± SEM. The data were analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni tests to determine where differences among groups existed. Differences were considered statistically significant for values of p < 0.05.
TLR2 is widely expressed in the CNS [11–13]. The cellular mechanisms that lead to opioid dependence are mostly unknown [16;20]. The nucleus accumbens (NAc) is a critical brain region related to the development of opioid dependence and withdrawal [16;20]. To investigate the mechanisms, we implanted wild type mice with a 25 mg sustained-release morphine pellet in the dorsal subcutaneous space, while control mice received a comparable placebo pellet for different time periods and determined the expression of TLR2 in the NAc by quantitative real time RT-PCR. TLR2 expression was significantly increased after 24 and 48 hours of morphine implantation (Fig. 1). However, morphine implantation could not alter the expression of TLR4 (data not shown). Our results demonstrated that chronic morphine administration significantly increases TLR2 expression.
Fig. 1.
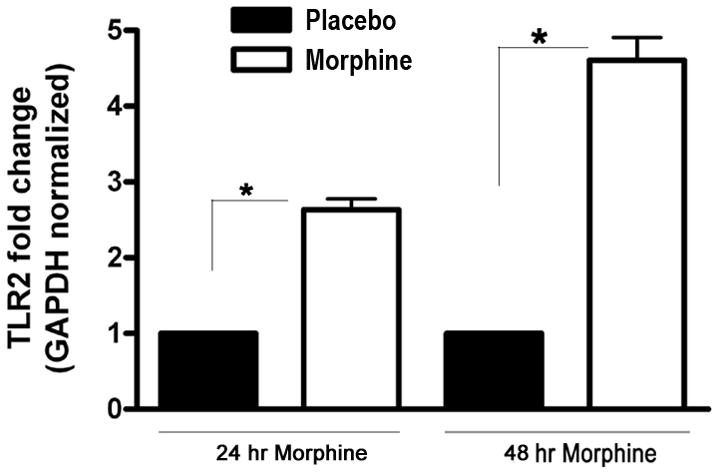
Chronic morphine administration induces the expression of TLR2. Wild type C57BL/6 male mice aged 9–10 week were implanted with a 25 mg sustained-release morphine pellet in the dorsal subcutaneous space, while control mice received a comparable placebo pellet as described under materials and methods. At 24 and 48 h after implantation, the mice were euthanized and the NAc was harvested and the expression of TLR2 was determined by real time quantitative RT-PCR. There were five mice per group. Means and SEs were calculated from 5 mice per group. * p < 0.01 compared with wild type placebo group.
It has been reported that microglia have a higher expression level of TLR2 [11–13]. We next investigated the role of TLR2 in the activation of microglia following chronic morphine administration. At 2 and 4 days after implantation of morphine pellets or placebo pellets in wild type mice and TLR2 knockout (TLR2 KO) mice, the NAc was harvested and total RNA was isolated. The activation level of microglia was quantified by measuring the transcript levels of CD11b. Morphine implantation after 2 days could not alter the level of CD11b in wild type and TLR2 KO mice (data not shown). At 4 days after morphine implantation, the CD11b transcript level was significantly increased > 9-fold in the NAc of wild type mice. Notably, the transcript level of CD11b in TLR2 KO mice was significantly decreased compared with wild type mice (p < 0.01) (Fig. 2). Thus, our data indicates that TLR2 is required for the activation of microglia following chronic morphine administration.
Fig. 2.
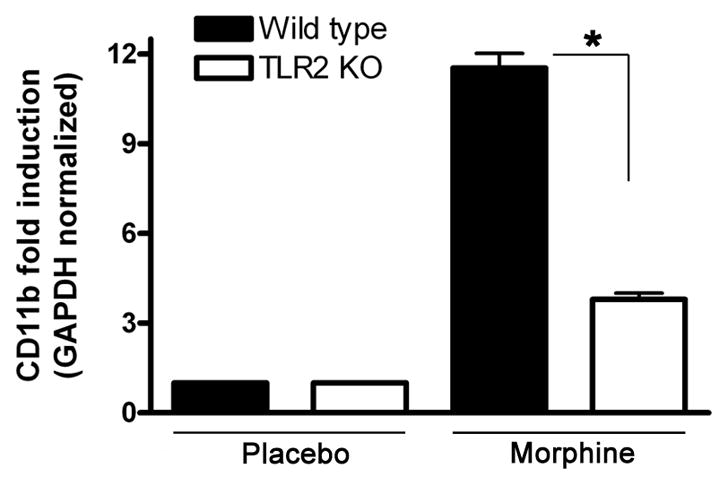
TLR2 is required for the activation of microglia following chronic morphine administration. Wild type (WT) and TLR2 KO male mice aged 9–10 week were implanted with a 25 mg morphine pellet or a placebo pellet subcutaneously under isoflurane anesthesia as described under materials and methods. Four days after pellet implantation, the NAcs were harvested and total RNA was isolated. The activation level of microglia was quantified by measuring the transcript levels of CD11b by real time RT-PCR. Means and SEs were calculated from 5 to 7 mice per group. * p < 0.01 compared with indicated groups.
Activation of TLR2 stimulates the production of inflammatory cytokines [21]. We examined the expression levels of proinflammatory cytokines in TLR2 KO and wild type mice following chronic morphine administration. As shown in Fig. 3, chronic administration of morphine significantly enhanced the expression of TNF-α and IL-6 in the wild type mice. Interestingly, significantly (3- to 5-fold) lower expression of TNF-α and IL-6 was observed in the TLR2 KO mice as compared with the wild type control group (Fig. 3). These data suggest that a pro-inflammatory role for TLR2 in opioid-mediated immune responses.
Fig. 3.
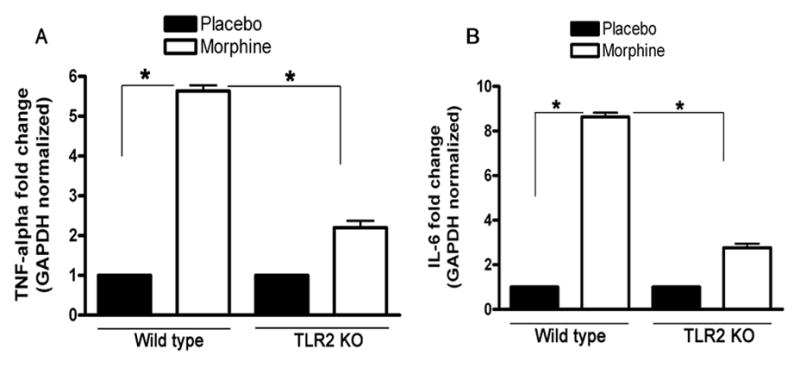
Effect of chronic administration of morphine on the expression of TNF-α, IFN-γ and IL-6 in wild type mice and TLR2 KO mice. 9–10-week-old TLR2 KO male mice (n = 5) and age-matched wild type male mice (n = 6) were implanted with a 25 mg morphine pellet or a placebo pellet subcutaneously under isoflurane anesthesia as described under materials and methods. At 72 h after pellet implantation, the NAcs were harvested and total RNA was isolated. The expression of TNF-α (A) and IL-6 (B) was determined by real time quantitative RT-PCR. The level of gene expression is presented as relative mRNA expression units. Means and SEs were calculated from 5 mice per group. ** p < 0.01 compared with indicated groups.
Prominent consequence of chronic opioid exposure is the development of physical dependence, which is manifested by a characteristic withdrawal syndrome upon the administration of an opioid receptor antagonist. To determine whether TLR2 blockade or gene deletion would reduce or eliminate physical dependence on morphine, TLR2 KO and the wild type mice were implanted with subcutaneous morphine pellets. After 72 h of morphine pellet implantation, mice were injected with the opioid receptor antagonist naloxone, and opioid withdrawal signs were monitored. Behavioral signs of morphine withdrawal (wet-dog shakes, vertical jumps, and weight loss) were monitored for 30 minutes [16;17;19]. Naloxone-precipitated withdrawal in wild type mice induced classic behavioral signs of withdrawal. Notably, in TLR2 KO mice the main behavioral signs of morphine withdrawal were markedly attenuated, which indicates that these mice do not become dependence on morphine during the 72-h treatment (Fig. 4). The TLR2 KO mice implanted with a placebo pellet appeared healthy and their spontaneous behavior was normal compared with control wild type mice. These results suggest that TLR2 plays an important role in response to morphine dependence.
Fig. 4.
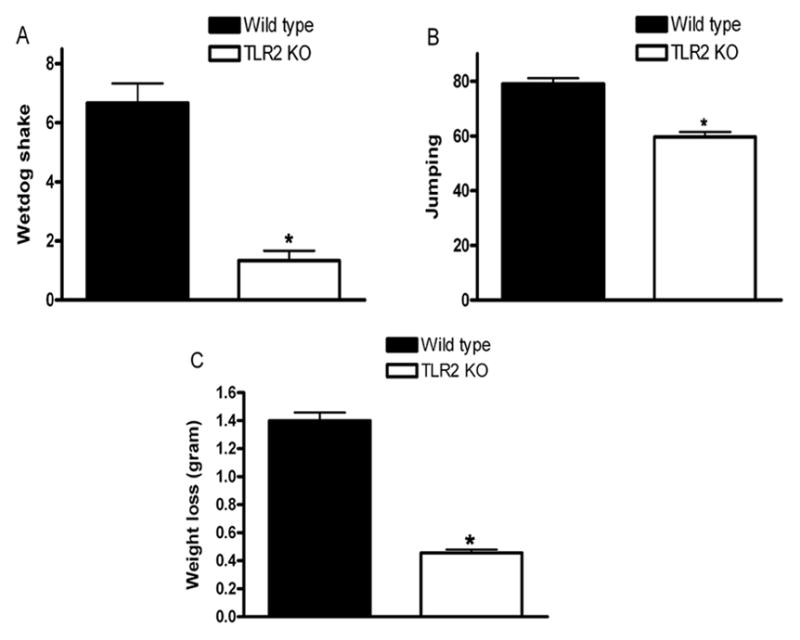
Naloxone-precipitated withdrawal after 72 h of morphine pellet implantation. 9–10-week-old TLR2 KO male mice and age-matched wild type male mice were implanted with a 25 mg morphine pellet subcutaneously or received a placebo pellet under isoflurane anesthesia as described under materials and methods. At 72 h after pellet implantation, mice received naloxone (1.0 mg/kg, i.p.) and morphine withdrawal symptoms for wet dog shakes (A), jumps (B), and weight loss (C) were monitored for 30 min. The weight of each mouse was determined before and 30 min after the naloxone injection. Means and SEs were calculated from 6 to 8 mice per group. * p < 0.01 compared with WT mice.
Our results shed light on the relationship between TLR-mediated microglia activation in response to an agonist such as morphine and the development of physical dependence on the drug. The data presented herein demonstrated for the first time, to our knowledge, a key role for TLR2 in the induction of morphine-mediated immune responses and involvement of opioid dependence. Our studies show that chronic morphine treatment regulates TLR2-mediated immunity and results in microglia activation. It is widely accepted that TLR2 signaling mainly activates inflammatory responses, including proinflammatory cytokines [22]. Most studies evaluated the effects of TLR2 on the CNS functions through immune cell mediated mechanisms [13;23]. We found in this study that deficiency of TLR2 activity inhibited morphine-induced microglia activation.
Recently, studies have revealed that TLRs signaling was involved in morphine-induced detrimental and beneficial effects, however, most studies evaluated morphine-induced effects only in TLR4-mediated pathways [24]. In terms of TLRs signaling, TLR2-mediated immune responses play a critical role in clinical consequences. Inhibition of TLR2 in vivo showed dramatically decreased the percentage of apoptosis in microglia cells induced by virus infection [25]. In this study, deficiency of TLR2 in mice cause a significantly decrease in the activation of microglia after chronic morphine treatment. Thus, our results suggest that opioids trigger microglia activation through TLR2.
We have recently reported that chronic stress-induced immune suppression is mediated through TLRs-dependent pathway [26]. In the present study, we extended these observations in opioid-mediated inflammatory responses to demonstrate that morphine dramatically induces inflammatory cytokine levels in the CNS. Of great significance, deficiency of TLR2 in mice significantly attenuated morphine-induced the levels of inflammatory cytokines compared with the wild type mice. These data indicate that TLR2 is required for morphine-induced inflammatory responses in the CNS.
TLR2 plays a critical role in neuropathic pain [11;27]. These findings strengthen the case for the sharing of signals between the immune and nervous system [15;28;29]. Recently Hutchinson and Watkins et al. have reviewed the role of glial activation in TLRs-mediated neuropathic pain and opioid analgesia [6]. They have speculated that opioids such as morphine “may be as-yet-unrecognized agonists of TLRs” [6]. Our studies found that morphine-increased the expression of TLR2 is mediated through the μ-opioid receptor (MOR) (our unpublished data). In the morphine dependence behaviors in rodents, wet-dog shaking and vertical jumping are widely considered the most sensitive and reliable index of withdrawal intensity and is the most commonly used [17;19;30]. The use of TLR2 knockout mouse, we found that a deficiency of TLR2 strongly reduced the behavior characteristic of opioid physical dependence than their wild type littermates, indicating TLR2 plays a critical role in the development of opioid dependence. In summary, our studies reveal that TLR2 plays a critical role in opioid-induced microglia activation and dependence. Furthermore, our data will enhance the understanding of TLRs in the nervous and immune systems. Inhibition of TLR2 may find application in the development of novel therapeutics to treat opioid dependence and addiction.
Research highlights.
Chronic morphine treatment promotes morphine dependence in the nucleus accumbens.
TLR2 deficiency inhibits morphine-induced microglia activation.
TLR2 deficiency displays attenuated behavioral signs of morphine withdrawal.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grant DA020120-03A1and ETSU RDC grant 82061 to D. Yin. We thank NIDA for providing us with both placebo and morphine pellets.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yin D, Mufson RA, Wang R, Shi Y. Fas-mediated cell death promoted by opioids. Nature. 1999;397:218. doi: 10.1038/16612. [DOI] [PubMed] [Google Scholar]
- 2.Yin D, Woodruff M, Zhang Y, Whaley S, Miao J, Ferslew K, Zhao J, Stuart C. Morphine promotes Jurkat cell apoptosis through pro-apoptotic FADD/P53 and anti-apoptotic PI3K/Akt/NF-kappaB pathways. J Neuroimmunol. 2006;174:101–107. doi: 10.1016/j.jneuroim.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Moorman J, Zhang Y, Liu B, LeSage G, Chen Y, Stuart C, Prayther D, Yin D. HIV-1 gp120 primes lymphocytes for opioid-induced, beta-arrestin 2-dependent apoptosis. Biochim Biophys Acta. 2009;1793:1366–1371. doi: 10.1016/j.bbamcr.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holan V, Zajicova A, Krulova M, Blahoutova V, Wilczek H. Augmented production of proinflammatory cytokines and accelerated allotransplantation reactions in heroin-treated mice. Clin Exp Immunol. 2003;132:40–45. doi: 10.1046/j.1365-2249.2003.02103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avila AH, Alonzo NC, Bayer BM. Immune cell activity during the initial stages of withdrawal from chronic exposure to cocaine or morphine. J Neuroimmunol. 2004;147:109–113. doi: 10.1016/j.jneuroim.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 6.Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. Scientific World Journal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson PK, Molitor TW, Chao CC. The opioid-cytokine connection. J Neuroimmunol. 1998;83:63–69. doi: 10.1016/s0165-5728(97)00222-1. [DOI] [PubMed] [Google Scholar]
- 8.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 10.Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, Choi SY, Park K, Kim JS, Akira S, Na HS, Oh SB, Lee SJ. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282:14975–14983. doi: 10.1074/jbc.M607277200. [DOI] [PubMed] [Google Scholar]
- 12.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 14.Zhou S, Halle A, Kurt-Jones EA, Cerny AM, Porpiglia E, Rogers M, Golenbock DT, Finberg RW. Lymphocytic Choriomeningitis Virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J Neuroimmunol. 2008;194:70–82. doi: 10.1016/j.jneuroim.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owens T, Babcock AA, Millward JM, Toft-Hansen H. Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Brain Res Rev. 2005;48:178–184. doi: 10.1016/j.brainresrev.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 17.Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D, Nestler EJ. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci. 2002;22:3663–3672. doi: 10.1523/JNEUROSCI.22-09-03663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 19.Raehal KM, Lowery JJ, Bhamidipati CM, Paolino RM, Blair JR, Wang D, Sadee W, Bilsky EJ. In vivo characterization of 6beta-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. J Pharmacol Exp Ther. 2005;313:1150–1162. doi: 10.1124/jpet.104.082966. [DOI] [PubMed] [Google Scholar]
- 20.Russo SJ, Bolanos CA, Theobald DE, DeCarolis NA, Renthal W, Kumar A, Winstanley CA, Renthal NE, Wiley MD, Self DW, Russell DS, Neve RL, Eisch AJ, Nestler EJ. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci. 2007;10:93–99. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- 21.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 22.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 23.Carpentier PA, Duncan DS, Miller SD. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun. 2008;22:140–147. doi: 10.1016/j.bbi.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008 doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aravalli RN, Hu S, Lokensgard JR. Toll-like receptor 2 signaling is a mediator of apoptosis in herpes simplex virus-infected microglia. J Neuroinflammation. 2007;4:11. doi: 10.1186/1742-2094-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Zhang Y, Miao J, Hanley G, Stuart C, Sun X, Chen T, Yin D. Chronic restraint stress promotes immune suppression through toll-like receptor 4-mediated phosphoinositide 3-kinase signaling. J Neuroimmunol. 2008;204:13–19. doi: 10.1016/j.jneuroim.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Sun X, LeSage G, Zhang Y, Liang Z, Chen J, Hanley G, He L, Sun S, Yin D. Beta-Arrestin 2 regulates toll-like receptor 4-mediated apoptotic signalling through glycogen synthase kinase-3beta. Immunology. 2010;130:556–563. doi: 10.1111/j.1365-2567.2010.03256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang DY, Shi X, Li X, Li J, Clark JD. The beta2 adrenergic receptor regulates morphine tolerance and physical dependence. Behav Brain Res. 2007;181:118–126. doi: 10.1016/j.bbr.2007.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]