

Abstract
A scalable method for screening and selection of peptide-specific monoclonal antibodies (mAbs) is described. To identify high affinity anti-peptide mAbs in hybridoma supernatants, antibodies were captured by magnetic affinity beads followed by binding of specific peptides from solution. After timed washing steps, the remaining bound peptides were eluted from the beads and detected by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS). This allowed measurement of monovalent interactions of peptides with single antigen binding sites on the antibodies, thus reflecting antibody affinity rather than avidity. Antibodies that were able to bind target peptides from solution phase and retain them during washing for a minimum of 10 minutes were identified by the strength of the appropriate m/z peptide MS signals obtained. This wash time reflects the minimum peptide dissociation time required for use of these antibodies in several current immuno-mass spectrometry assays. Kinetic analysis of antibody-peptide binding by surface plasmon resonance (SPR) showed that the selected antibodies were of high affinity and, most importantly, had low dissociation constants. This method, called MALDI immunoscreening (MiSCREEN), thus enables rapid screening and selection of high affinity anti-peptide antibodies that are useful for a variety of immunoproteomics applications. To demonstrate their functional utility in immuno-mass spectrometry assays, we used the selected, purified RabMAbs to enrich natural (tryptic) peptides from digested human plasma.
Keywords: MALDI-TOF, MiSCREEN, surface plasmon resonance, monoclonal antibodies, RabMAbs, anti-peptide, rabbit, high affinity
1. Introduction
There is currently a shortage of antibodies for use in a variety of proteomics applications. The lack of such reagents presents a serious bottleneck for measuring the large number of different proteins in biological samples including tissues and plasma, the major source of biomarkers used in clinical diagnostics. Several ambitious projects are underway in both Europe (http://www.hupo.org/research/hai/;www.proteomebinders.org) and the USA (http://antibodies.cancer.gov) to make and characterize antibodies for use in immunohistochemical assays and for immunoenrichment of proteins from complex mixtures. Such antibodies will be useful for expression analysis of proteins in tissues and cells and in “top-down” proteomics methods where intact protein targets are enriched before analysis by methods such as mass spectrometry, a method pioneered by Nelson and colleagues (1995).
Much less thought and effort has been focused so far on the development of anti-peptide antibodies suitable for quantitation of signature peptide surrogates of proteins from digests of complex biological materials. These kinds of antibody reagents are used in quantitative assays such as immuno-Matrix-Assisted Laser Desorption/Ionization (i-MALDI; Raska et al., 2003; Jiang et al., 2007) or Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA; Anderson et al., 2004). These applications use antibodies for specific enrichment of tryptic peptides from digests that are much more complex (in terms of number of different molecules present) than the protein mixtures from which they are derived. The unique requirements of such assays make it a challenge to derive and select antibodies with the desired characteristics. Essentially, such antibodies must be able to bind the target peptides from complex tryptic digests (typically from human plasma) and retain them through the washing steps prior to peptide elution and mass spectrometric analysis. Thus these antibodies must have high affinities, or more specifically, low off-rates for their peptide analytes. With current SISCAPA assays, the retention time required for effective peptide enrichment is a minimum of 10 minutes during which time unbound peptides are washed away. The MiSCREEN method described in this manuscript allows selection of such anti-peptide reagents.
A project to make quantitative assays for all human proteins, based on surrogate peptides, anti-peptide antibodies and mass spectrometry, has been proposed by Anderson et al. (2009). This human Proteome Detection and Quantitation Project (hPDQ) would function as an assay resource available worldwide. The project would require both MS-friendly proteotypic tryptic peptides for each target protein and renewable anti-peptide reagents specific for each. Currently, few such anti-peptide reagents are available and the capacity to produce them is limited. Although some commercial and academic research labs claim to be able to make antibodies (or other affinity reagents) with high throughput, this has not been widely demonstrated for anti-peptide reagents with the performance characteristics required for use in immuno-MS assays.
We previously developed a surface plasmon resonance (SPR) method that allows selection of monoclonal anti-peptide antibodies that are able to bind tryptic peptides in solution phase and that are suitable for immuno-MS assays (Pope et al., 2009). This method measures true antibody affinities (not avidities) and is useful for kinetic analysis of small numbers of mAbs. However, the assay is too slow, cumbersome and expensive for high throughput screening.
We therefore sought to develop a method that would allow selection of high affinity anti-peptide mAbs (more specifically, those with low off rates) in a more cost effective and high throughput fashion. To do this we have developed a method called MALDI-immunoscreening (MiSCREEN) for rapid screening of hybridoma supernatants. The method allows the identification of antibodies that are able to bind specific peptides in solution phase from complex mixtures and that have low dissociation constants (kd) suitable for ultimate use in immuno-MS assays.
2. Materials and Methods
2.1. Peptides
Synthetic tryptic peptides chosen as surrogates of protein biomarkers were used throughout. Peptides that occur in a single protein encoded within the human genome and that yield several, strong multiple reaction monitoring (MRM) transitions in a triple quadrupole mass spectrometer were selected (Anderson et al., 2004). Peptides were synthesized by solid-phase methods by either the Chinese Peptide Company (Hangzhou, China) or by the UVic-Genome BC Proteomics Centre (Victoria, BC) and were tested by the vendors for the correct masses by MALDI-TOF mass spectrometry and for purity by high performance liquid chromatography (HPLC). Stable-isotope-labeled versions of selected peptides were made by chemical synthesis at the UVic-Genome BC Proteomics Centre. A mass increment was added in each case through use of labeled C-term Arginine (6 or 10 amu) or Lysine (6 amu), providing mass shifts of m/z = 3 or 5 for typical doubly-charged peptide ions. All peptides were of greater than 80% purity, were quantified by amino acid analysis (Advanced Protein Technology Centre, The Hospital for Sick Children, Toronto, Ontario) and stored at −20 °C for short periods (2 weeks or less) at 4 °C in solution phase to prevent solubility problems that occur with some peptides after lyophilization. After thawing and/or just before use in MiSCREEN, all peptides were analysed by MALDI-TOF MS to determine their integrity and to assess the presence of altered forms.
Peptides were first synthesized with C-terminal cysteines to allow thiol coupling to keyhole limpet hemocyanin (KLH) carriers for immunization (Pierce Chemical Co., St Louis, MO). The same peptides synthesized without C-terminal cysteines were used in enzyme linked immunosorbent assays (ELISA; see peptide ELISA below) and in MiSCREEN and SPR assays for measuring antibody-peptide binding without interference from the linker cysteine. Although the peptides for this work were chosen as proteotypic surrogates of a variety of protein biomarkers, any peptide of interest that can be bound by an antibody and detected by MALDI-TOF mass spectrometry can be used. The peptides used in this work are described in Table 1.
Table 1.
Protein targets and their surrogate (proteotypic) tryptic peptides
| Protein Name | Surrogate Peptide ID | AA Sequence | Mass (Daltons) |
|---|---|---|---|
| Ferritin Light Chain | CPTAC-14b | KPAEDEWGK | 1059.14 |
| Ferritin Light Chain | CPTAC-14d | LGGPEAGLGEYLFER | 1607.78 |
| Alpha-fetoprotein precursor | CPTAC-23a | GYQELLEK | 979.10 |
| Alpha-fetoprotein precursor | CPTAC-23c | YIQESQALAK | 1150.30 |
| Receptor tyrosine-protein kinase | CPTAC-36c | NNQLALTLIDTNR | 1485.80 |
| Receptor tyrosine-protein kinase | CPTAC-36d | AVTSANIQEFAGC*K1 | 1495.72 |
| Mucin-16 (CA-125) | CPTAC-38b | ELGPYTLDR | 1063.54 |
| Mucin-16 (CA-125) | CPTAC-38c | VLQGLLGPIFK | 1184.74 |
| Thyroglobulin precursor | CPTAC-39c | FSPDDSAGASALLR | 1406.69 |
| Thyroglobulin precursor | CPTAC-39d | VIFDANAPVAVR | 1271.71 |
| WAP four-disulfide core domain protein | CPTAC-43c | C*C*SAGC*ATFC*SLPNDK1 | 1847.72 |
| LPS Binding Protein | PPI-1b | ITLPDFTGDLR | 1247.41 |
| LPS Binding Protein | PPI-1c | LAEGFPLPLLK | 1197.48 |
| Protein C Inhibitor (PCI; also known as PAI-3) | PPI-4d | EDQYHYLLDR | 1351.44 |
| Transferrin Receptor | PPI-6d | GFVEPDHYVVVGAQR | 1672.86 |
| Angiotensin-I2 | Ag-I | DRVYIHPFHL | 1296.49 |
The asterisk (*) denotes carbamidomethyl cysteine.
Angiotensin-I was chosen as a positive control peptide since it ionizes extremely well and is often used as a standard in MALDI-TOF mass spectrometry.
2.2. Anti-peptide monoclonal antibodies
Rabbit monoclonal antibodies (RabMAbs) were produced by Epitomics Inc. (Burlingame, CA) using a proprietary, stabilized rabbit plasmacytoma cell line derived from the original parental myeloma 240-W (Spieker-Polet et al., 1995) as the fusion partner. To select hybridomas secreting anti-peptide antibodies, 4000 hybridoma supernatants from each fusion were tested by peptide ELISA (see below) using the immunizing peptides (without carrier or added C-terminal cysteine) dried onto ELISA plates. Positive rabbit hybridoma supernatants (usually obtained in small volumes of 400 μL after the initial peptide ELISA) were used for MiSCREEN and SPR assays. All hybridoma supernatants were stored at 4 °C before use to avoid freeze-thaw cycles.
Mouse monoclonal antibodies were also used in MiSCREEN assays. One of these, mAb 2A7 specific for peptide PPI-1b from LPS binding protein (see Table 1) was produced by Immunoprecise Antibodies Ltd (Victoria BC) using single-step selection and cloning with ClonaCell® (Cat No. 03800; StemCell Technologies Inc., Vancouver BC) and the other, mAb BGN/KA/4H, specific for angiotensin-I was purchased from Abcam Inc. (Cambridge, MA).
2.3. Peptide ELISA
In earlier work in our lab, many different RabMAbs and mouse mAbs raised against peptide-KLH conjugates showed reactivity in ELISA with unrelated peptide-carrier conjugates, even when these peptides were coupled to a carrier protein different from the one used in immunization (e.g., bovine serum albumin instead of KLH; unpublished observations). Such antibodies appeared to bind to linker structures that comprise at least part of the recognized epitopes. For this reason, we modified a standard indirect ELISA method (Tolson et al, 1989) to use unconjugated peptide antigens (i.e. not coupled to protein carriers) to coat polystyrene microtitre ELISA plates (Griener Bio-One Microlon™ 600, high binding, flat well, Cat. No. 655081; VWR, Mississauga ON). In this specialized peptide ELISA, peptides were dissolved in distilled water to a final concentration of 0.1 to 5.0 μg/mL (each peptide was first titrated to select the optimum concentration to give good signal to noise ratios) and 100 μL of this solution were dried onto each well by overnight incubation at 37 °C in a dry incubator. This use of free peptides as antigen in ELISA is important since the cysteine-coupled peptides used for immunization often induce antibodies that recognize cysteine as part of the epitopes (unpublished observations). We were interested only in selecting antibodies that bind strongly to free, natural peptides that are released by trypsin digestion of human plasma. In addition, the simplicity of peptide ELISA allows us to screen all 4000 hybridoma supernatants (obtained in a single fusion) for RabMAbs that bind the peptide of interest, prior to further selection for high affinity (low off-rates). This initial selection ensures that only a small number of positive mAbs must undergo more complex testing, for example kinetics analysis by surface plasmon resonance. Nevertheless it is important to note that the mAbs selected by peptide ELISA screening may not be suitable for binding of peptides from solution phase since antibody avidity (i.e., two site binding for IgG) may lead to positive ELISA signals for mAbs with low individual site affinities (single site interactions). For this reason, peptide ELISA was used only to select peptide-specific RabMAbs before they underwent further selection based on dissociation rate.
2.4. Measurement of solution-phase peptide binding by MiSCREEN assay
To identify high affinity, anti-peptide mAbs from among the candidates selected by peptide ELISA, mAbs in hybridoma supernatants were first captured by magnetic affinity beads (see below) followed by binding of specific peptides from solution. After a series of washing steps, remaining bound peptides were eluted from the beads for analysis by MALDI-TOF MS. Our hypothesis was that if a given monoclonal antibody were able to bind peptide from solution phase and retain it through the timed washing procedures (i.e. if it had a low dissociation rate, kd), then the eluted peptide should be detectable by MALDI-TOF MS in the bead eluate. Antibodies with slower off-rates should yield more peptide than antibodies with faster off-rates and this would be reflected in the MS signal strengths obtained.
A variety of parameters (incubation times, washing steps, elution conditions, etc.) were tested to develop the optimized MiSCREEN assay presented here. All washing and elution steps were performed using a magnetic particle processor (KingFisher 96, Thermo Fisher Scientific, MA, USA). Briefly, six 96-well polypropylene microplates (Cat. No. CA83007-596, Thermo KingFisher 96 KF plate, 200 μl; Thermo Scientific) were first loaded into the Kingfisher particle processor. Wells of the first plate contained 10 μL of sheep anti-rabbit IgG Dynabeads (Cat. No. 112.03D, Invitrogen, Oslo, Norway; the volume corresponding to the original concentration in the bottle) suspended in 200 μL of phosphate buffered saline (PBS)/0.03% (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate) (CHAPS) detergent. In MiSCREEN assays using mouse mAbs, Dynabeads protein G (Cat No. 100.04D) or goat anti-mouse IgG magnetic beads (Cat No. 110.33) were used. After washing by agitation for 5 minutes, the beads were transferred by the Kingfisher magnet array into wells of the second plate, each containing 50 μL of hybridoma supernatant. After 2 hours incubation at room temperature the magnetic beads and captured antibodies were transferred to the third plate, each well containing 1 picomole of target peptide in 100 μL of PBS/0.03% CHAPS. After another 2-hour incubation, the magnetic bead-antibody-peptide complexes were transferred sequentially to three microplates for washing (3 times for 1-minute in 200 μL of PBS/0.03% CHAPS). The washed beads were finally transferred to an elution plate (96-well Hard-Shell® PCR plate, Bio-Rad, Ca, USA) each well containing 25 μL of 5% acetic acid, and incubated for 5 minutes to release any bound target peptide and possibly some bound antibody. During each of these incubation steps, bead suspensions were agitated by a reciprocal motion of the Kingfisher magnet head. The eluted peptides from all 25 μL were desalted and concentrated using ZipTip C18 tips (Cat. No. ZTC18S960, Millipore, MA, USA) before spotting the entire sample in a 2 μL volume onto a MALDI plate. In some experiments, to test whether or not Zip-Tip C18 cleanup was necessary for achieving sensitivity, the peptides were eluted in 12 μL of 5% acetic acid and 2 μL of this were directly spotted onto MALDI plates for mass spectrometric analysis. After drying, 1 μL of the matrix alpha-cyano-4 –hydroxycinnamic acid (CHCA) was added to each spot. A Voyager DE™ STR (Applied Biosystems, Foster City, CA) was used to analyze the eluted samples from MiSCREEN experiments. The instrument was set to reflectron mode with laser intensity of 2800, accelerating voltage of 20 kV, delay time of 220 nsec and the mass range was set to 800–3000 Daltons. One hundred laser shots were accumulated per spectrum and 5–10 spectra were accumulated for each sample spot. All experiments were performed in duplicate.
In the MiSCREEN assay, the total elapsed time between the end of peptide binding and peptide elution (including the bead wash and transfer steps) was approximately 10 min, the minimum peptide dissociation time useful for SISCAPA assays. Prior to MiSCREEN analysis, a dilution series of each peptide was made and known amounts were spotted onto MALDI plates to establish a standard curve. In this way, the signal intensity for each peptide could be gauged as a measure of peptide performance.
Multiplexing of the MiSCREEN assay was tested by mixing supernatants from a number of different hybridomas (each secreting different peptide-specific RabMAbs) and measuring the enrichment of the relevant peptides from mixtures that also contained irrelevant tryptic peptides as specificity controls. In addition, multiplexing was tested using a different set of five peptides, two of which were specificity controls. Multiplexing experiments were performed with three replicates per sample.
In MiSCREEN assays (and SISCAPA assays) the natural tendency of the beads to adhere to plastic and other surfaces was overcome by addition of low concentrations (0.03%) of CHAPS detergent to most solutions. In MALDI-TOF MS, CHAPS appears as a single peak at 1229.7 Daltons. This detergent was also chosen for SISCAPA assays employing triple quadrupole mass spectrometers since CHAPS elutes as a single major hydrophobic peak late in the reverse phase chromatographic separation, after most peptides. It is thus more practical than other detergents, most of which are polymeric, yielding many peaks revealed by MS, some of which may interfere with peptide ionization and MS spectra analysis.
2.5. Measurement of solution-phase peptide binding by SPR
Kinetic screening of anti-peptide RabMAbs was performed by SPR using a Biacore 3000 optical biosensor (Biacore, Uppsala, Sweden) according to a previously published method designed specifically for measuring peptide binding affinities of monoclonal antibodies (Pope et al., 2009). Research-grade CM5 chips (Order Code BR-1003-99) were used for all experiments and were obtained from Biacore Life Sciences (Piscataway, NJ). Preselected, affinity-purified goat anti-rabbit IgG (Fc fragment specific; Jackson ImmunoResearch Laboratories, West Grove PA; Cat No. 111-005-008) was covalently bound to the CM-5 chips using an amine coupling method with 100 nM N-hydroxysuccinimide/390 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride as crosslinker. The affinity chip was then used for capture of RabMAbs. The method has also been adapted for screening of murine mAbs by using affinity purified goat anti-mouse IgG (Fc specific) capture antibodies. This general method allows determination of antibody affinity (equilibrium dissociation constant, KD) by measuring binding and release of peptide in solution, without avidity considerations. Most importantly, since low peptide off-rates are critical for anti-peptide antibodies used in current immuno-mass spectrometry methods, the dissociation rates (kd) for each antibody were calculated. This allowed us to determine the off-rates for all mAbs selected by the MiSCREEN assay.
2.6. Enrichment of peptides from human plasma digests
RabMAbs selected by MiSCREEN and confirmed by SPR analysis were tested for their ability to enrich specific “natural” surrogate peptides from trypsin-digested human plasma.
a) Trypsin digestion of human plasma
Pooled human potassium-EDTA treated plasma was purchased from Bioreclamation Inc., Westbury NY (Cat No. HMPLEDTA). The final protein concentration of the pooled plasma was 73.13 mg/mL as determined using a bicinchoninic acid colorimetric assay kit (Sigma-Aldrich Ltd., St. Louis, MO) and using a 1% solution of BSA as a standard. Plasma was added to a lyophilized mixture of urea (Ultra Urea, Sigma-Aldrich Ltd., St. Louis, MO), tris (2-carboxyethyl) phosphine) (TCEP) (Bondbreaker neutral TCEP solution, Thermo Scientific, Rockford, IL) and Tris (Trizma preset crystals pH 8.1, Sigma-Aldrich) to yield a final concentration of 9 M urea, a 2 fold molar excess of TCEP over plasma cysteines (based on a cysteine concentration in plasma of 2.68E-2 M) and 0.2 M Tris pH 8.1. The plasma proteins were denatured and reduced at room temperature for 30 min. Iodoacetamide was added at a 3 fold molar excess over plasma cysteines and allowed to alkylate the sample for 30 min at room temperature in the dark. Prior to trypsin addition, the urea concentration was reduced to 1 M by dilution with 0.2 M Tris pH 8.1 (Trizma preset crystals pH 8.1). Tosyl phenylalanyl chloromethyl ketone (TPCK; an inhibitor of chymotrypsin) - treated trypsin (Worthington Biochemical Corp., Lakewood, NJ) was prepared in 10 mM HCl and added at a 20:1 protein to enzyme ratio and allowed to digest overnight in a 37 °C dry incubator. The sample was cooled to room temperature and tosyl lysyl chloromethyl ketone (TLCK; an irreversible inhibitor of trypsin; Fluka Biochemica, Buchs, Switzerland) was added at 2-fold excess over trypsin and incubated for 30 min at room temperature to arrest digestion. The digested sample was concentrated and desalted using 150 mg solid-phase extraction cartridges (Waters Oasis HLB, Milford, MA). The digest was acidified using 0.2 M formic acid, gravity loaded onto a pre-equilibrated cartridge, desalted with 5 mL 0.1% formic acid, eluted in 3 mL 50% ACN, 0.1% FA and the eluate was lyophilized. Finally, the mixture of lyophilized peptides was reconstituted to the original plasma volume with PBS and brought to pH 7.0 using 6 M NaOH. Aliquots of the reconstituted plasma digest were stored at −80° C and thawed immediately prior to use.
b) Peptide enrichment and analysis
We used the SISCAPA method (Anderson et al., 2004) for enrichment and detection of peptides from trypsin digested human plasma. As described above for MiSCREEN, all washing and elution steps involved in this procedure were performed using a Kingfisher magnetic particle processor. However, special custom produced, 1 micron diameter, protein G Dynabeads specifically designed for SISCAPA were used (MyOne OK432 Protein G Dynabeads, Invitrogen). These beads offer a greater surface area to allow increased binding capacity and have special surface chemistry to reduce background peptide binding. To capture 1.0 μg of RabMAb, 1.43 μL of beads are required. Therefore the wells of the first microplate contained this amount of beads in 200 μL PBS/0.03% CHAPS detergent. After washing the beads, they were transferred to a second microplate containing 1 μg of purified RabMAb in each well. After 2 hours incubation at room temperature, the magnetic beads and captured antibodies were transferred to a third plate, with each well containing 100 μL of a 1 in 10 plasma digest (i.e. 10 μL of digest diluted in 90 μL of PBS/0.03% CHAPS). After 2 hours incubation, the magnetic bead-antibody-peptide complexes were transferred sequentially to three microplates for washing (3 × 1-minute in 200 μL of PBS/0.03% CHAPS). Dynabeads were also incubated with a 1 in 10 dilution of plasma digest (without added antibodies) to control for any possible non-specific background binding of irrelevant peptides. The bead-antibody-peptide complexes were incubated in 25 μL of 5% acetic acid for 5 minutes to release any bound target peptides. Finally, the eluted samples were concentrated using ZipTips as described for MiSCREEN above and manually spotted onto a 384 well OptiTOF MALDI target plate (Applied Biosystems, Foster, CA.) After the spots had dried, 1 μL of CHCA matrix was spotted onto each sample. MALDI-MS data were automatically acquired over a mass range of 800–4000 Da in the positive-ion reflector mode on a 4800 MALDI-TOF/TOF Analyzer with 4000 Series Explorer v3.5 installed (Applied Biosystems/MDS Sciex, Framingham, MA), using a fixed laser intensity of 3400 at 500 shots/spectrum and using a random spot search pattern. Both MALDI-MS and MS/MS analyses were performed on the samples. Selected precursor ions were fragmented using an MS/MS acquisition method set to 2 kV collision energy. The relative precursor mass window was set at 300 (FWHM) and MS/MS was acquired with CID turned on and with metastable suppression enabled. MS/MS acquisition of selected precursors was set to 1,250 shots per spectrum. The sequence of related peptides that generated acceptable MS/MS spectra was manually verified using MS-Product tool from the ProteinProspector freeware v5.5.0 (University of California, San Francisco, USA).
3. Results and Discussion
3.1. Synthetic peptides
It was important that all peptides synthesized for this work be of high purity and that they be handled in a way that would preserve their integrity and prevent post synthesis modifications. Initially, peptide vendors were instructed that 80% purity was required and that both MALDI-TOF traces and HPLC traces be supplied. For almost all peptides, these criteria were met. However, further quality control was undertaken whereby after thawing and just before use of the peptides, they were examined by MALDI-TOF MS to ensure that the appropriate peptide mass was the predominant species. Examples of two peptides, one highly pure the other not, are shown in Figure 1. A typical high-purity peptide, CPTAC-39d, is shown in Figure 1A. One peptide, CPTAC-43c (Figure 1B) did show an unacceptable, alternative form of the peptide, indicating a sub-optimal synthesis, although this was an unusual peptide since it contained 4 cysteines that required special blocking chemistry for synthesis. Despite the presence of impurities, this peptide was used in MiSCREEN analysis and revealed an unanticipated benefit of the method (see below). The parent proteins and sequences and masses of the peptides used in the work reported here are shown in Table 1.
Figure 1.

Examples of MALDI spectra of purified peptides. Panel A: 1 picomole of peptide CPTAC-39d; Panel B: 1 picomole of peptide CPTAC-43c. The peptides were spotted directly onto a MALDI plate. The peptides were chosen as examples of both high and low purity.
3.2. Peptide titration curves
The peptides used in this work were originally chosen on the basis of their performance in electrospray ionization - triple quadrupole mass spectrometry. Therefore, it was important to test each peptide for its ability to ionize effectively by MALDI since not all peptides ionize equally well under different ionization conditions. For each MiSCREEN analysis we performed a standard curve by directly spotting varying amounts of the appropriate peptide onto MALDI plates. To illustrate this, the results obtained from three selected peptides are shown in Figure 2. Panel A shows the strong MS signals obtained with varying amounts of angiotensin-I, a peptide that is often used as a standard in MALDI-TOF MS. Even low femtomole amounts of this model peptide yielded measurable MS signals and the linearity of the response was excellent. Panel B shows the more typical MS signals obtained with tryptic peptides that we use in our immuno-MS assays. One peptide, (CPTAC-38b) ionized well whereas the other (CPTAC-23c) showed significantly lower signal. Because of these differences among peptides, the ability of the MiSCREEN assay to distinguish the best mAbs is somewhat better for peptides with high MALDI response. The fact that some peptides do not ionize well by MALDI must be taken into consideration in designing the peptides for MiSCREEN or SISCAPA analyses using MALDI-TOF. By testing a variety of synthetic tryptic peptide surrogates, those that ionized well by MALDI could be selected. The same consideration holds for electrospray ionization and MS analysis using triple quadrupole mass spectrometers.
Figure 2.

Peptide titration by MALDI-TOF MS. Panel A: Angiotensin-I (positive control) Panel B: CPTAC-38b and CPTAC-23c. Angiotensin-I was chosen as a model peptide since it ionizes extremely well by MALDI whereas the other peptides were more typical of tryptic peptides chosen as surrogates for protein targets and ionize moderately well (CPTAC-38b) or poorly (CPTAC-23c).
3.3. Initial screening of hybridoma supernatants by peptide ELISA
Since 4000 hybridoma supernatants were obtained from each fusion, these were first screened for the presence of anti-peptide antibodies using the novel peptide ELISA. This allowed us to identify supernatants containing specific anti-peptide mAbs that were then subjected to more definitive kinetic analysis by MiSCREEN and SPR for ultimate selection of RabMAbs with low peptide off-rates. Using peptide ELISA, in each fusion we identified between 40 and 150 individual hybridomas that secreted anti-peptide antibodies (data not shown). These subsets were tested by MiSCREEN and SPR analysis in order to identify mAbs that were able to bind peptides from solution and that had low peptide off-rates.
3.4. Screening of hybridoma supernatants by MiSCREEN
Representative examples of MALDI-TOF MS spectra obtained in a typical MiSCREEN experiment are shown in Figure 3. In such experiments, a single peptide is used and different hybridoma supernatants are tested for peptide binding. For illustrative purposes, mAbs with poor (panel A), medium (panel B) and good (panel C) binding activity against a proteotypic peptide CPTAC-39d from human thyroglobulin, are shown with peak intensities of 113, 1275 and 2623 respectively (shown on the vertical axis at the top right of each spectrum).
Figure 3.

Peptide enrichment by MiSCREEN and surface plasmon resonance plots of low, medium and high affinity RabMAbs. Selected RabMAbs specific for the same peptide and having low (Panel A) medium (Panel B) and high (Panel C) affinities were compared using MiSCREEN assays for their ability to bind peptides from solution. On the left hand side of the figure are shown single replicate spectra representing binding of peptide CPTAC-39d (1271.71 Daltons; a proteotypic surrogate of thyroglobulin) by RabMAbs in different hybridoma supernatants as tested by MiSCREEN. Panel A: RabMAb 35-25; Panel B: RabMAb 35–36; Panel C: RabMAb 35–41. Note the differences in peak intensities shown on the right hand vertical axes. On the right hand side of the figure are shown the corresponding SPR peptide binding and dissociation curves for the same antibodies. The half off-times of these antibodies were: Panel D: 8 minutes, Panel E: 22 min and Panel F: 174 min, respectively.
More than 500 hybridoma supernatants producing RabMAbs against fifteen different tryptic peptides were screened for peptide binding in the MiSCREEN assay. A summary of the results obtained is shown in Table 2. Although all of the supernatants tested contained anti-peptide mAbs detected by peptide ELISA, only a fraction of them were positive by MiSCREEN suggesting that the latter assay detected only high affinity antibodies and that with ELISA there are complicating avidity considerations. However, if some supernatants contained very low amounts of antibodies, then the small amounts of captured antibodies would not bind enough peptide for measurement and would be considered negative, even if the antibodies were of high affinity. This is one failing of the MiSCREEN method (and perhaps some other screening methods such as ELISA) since such hybridomas would be considered negative and thrown away. However, we decided that such low-secreting hybridomas are problematic so we were not interested in these and only selected those that are good secretors and of course that had low off-rates. For a single peptide target measured with multiple hybridoma supernatants, comparison of the accumulated MALDI-TOF peak intensities for peptides bound by each mAb allowed selection of those that were able to bind and retain peptides with a higher affinity. Such mAbs have low dissociation rate constants (kd) and half off-times as determined by kinetic analysis using SPR.
Table 2.
Summary of MiSCREEN vs. SPR for a variety of anti-peptide mAbs
| Surrogate Peptide ID | Selected Hybridoma Sup ID | Accumul ated MALDI Peak Intensity | SPR KD (μM) | SPR ka (1/Ms) | SPR kd (1/s) | SPR Half Off-time (min) | mAb Selected (Yes/No)1 |
|---|---|---|---|---|---|---|---|
| CPTAC-14b | 10–10 | 465 | 172.4 | 4.75E5 | 8.19E-2 | 0.14 | No |
| CPTAC-14d | 10–58 | 8014 | 0.18 | 1.27E6 | 2.25E-4 | 51.3 | Yes |
| CPTAC-23a | 19–40 | 2012 | 9.60 | 9.01E4 | 8.65E-4 | 13.3 | Yes |
| CPTAC-23c | 19–44 | Peptide ionizes poorly | 3.99 | 2.71E5 | 1.08E-3 | 10.7 | Yes |
| CPTAC-36c | None2 | None | No binding of peptide detected by SPR | No | |||
| CPTAC-36d | 32–27 | 3802 | 1.16 | 1.36E5 | 1.58E-4 | 73.1 | Yes |
| CPTAC-38b | 34–102 | 33774 | 0.22 | 1.23E5 | 2.71E-5 | 426.3 | Yes |
| CPTAC-38c | 34–15 | 6174 | 1.72 | 4.32E5 | 7.41E-4 | 15.6 | Yes |
| CPTAC-39c | 35–31 | 2966 | 0.04 | 6.16E5 | 2.44E-5 | 473.5 | Yes |
| CPTAC-39d | 35–41 | 2842 | 1.16 | 7.71E4 | 6.64E-5 | 174.0 | Yes |
| CPTAC-43c3 | 38–12 | 2108 | 0.82 | 7.26E5 | 5.93E-5 | 194.8 | Yes |
| PPI-1b | 2A74 | 12300 | ND7 | ND | ND | ND | Yes |
| PPI-1c | 109–4 | 1000 | ND | ND | ND | ND | Yes |
| PPI-4d | 58–45 | 13000 | ND | ND | ND | ND | Yes |
| PPI-6d | 79–9 | 6739 | ND | ND | ND | ND | Yes |
| Angiotensin-I | BGN/KA/4H6 | 3464 | ND | ND | ND | ND | Yes |
Only antibodies that demonstrated a half off-time of greater than 10 minutes were selected
None of 12 hybridoma sups selected by ELISA showed any peaks detected by MiSCREEN.
Peptide impurities were present.
Mouse mAb (Immunoprecise Antibodies Ltd)
Recombinant rabbit mAb
Mouse mAb (AbCAM)
ND=Not Done
It is important that the ligand chosen for capturing monoclonal antibodies out of solution is itself a strong binder so that the dissociation of the captured antibody during subsequent peptide binding and washing steps is minimized. Therefore, selection of high quality, affinity purified capture antibodies is crucial. In previous work (Pope et al, 2009) we measured the binding kinetics of a selected affinity-purified anti-rabbit IgG capture antibody with a variety of RabMAbs and compared off-rates with RabMAb-peptide binding. The off-rates observed for RabMAb-peptide binding were always much faster than the slow off-rates observed with the captured RabMAbs themselves. This can perhaps be explained by a combination of high affinity, and especially avidity, of the capture antibody since multiple epitopes on the Fc region of the RabMAbs are available for binding. We used the same capture antibodies in the current work with the MiSCREEN assay and did not detect any release of the captured monoclonal antibodies.
Although the CHCA matrix has been shown to form homogenous crystals, formation of occasional `hot spots' (i.e. variations in crystallization) is inevitable (Tholey et al., 2006). Therefore, comparing samples based on accumulated MALDI peak intensity can have coefficient of variations (CV) as high as 30% depending on the number of spectra accumulated. To overcome the latter limitation, known amounts of stable isotope-labeled peptides (heavy peptides) are `spiked' into the reaction mixture to control for variations in matrix formation (Szájli et al., 2008; Zabet-Moghaddam et al., 2004). In MiSCREEN assays, we accumulated multiple spectra for each RabMAb over two replicates and thus inconsistencies due to variations in crystallization are deemed to be minimal. However, to test whether `hot spots' significantly affect the selection of RabMAbs by MiSCREEN, we repeated the screening for one of the surrogate peptides (CPTAC-39d) with and without spiked internal standards (Figure 4). The MALDI peak intensities (without heavy spike) and the light:heavy peak ratios were remarkably similar for each RabMAb, demonstrating that the peptide peak intensities in MiSCREEN are representative of the amount of peptide bound by the individual antibodies and that RabMAb selection based on accumulated MALDI peak intensity has not been significantly affected by `hot spot' variations.
Figure 4.
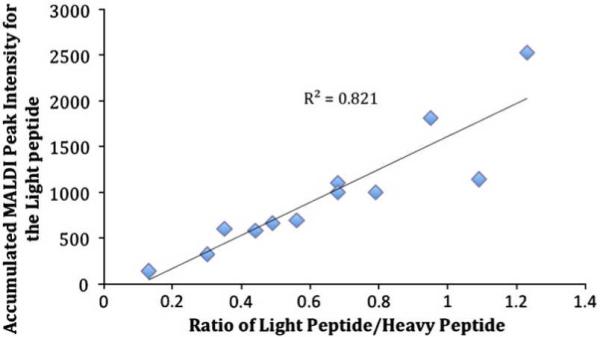
MALDI-TOF analysis of eluted peptides using internal stable isotope labeled standards to control for variations in matrix crystallization. After capture of peptide from solution (PBS) by each RabMAb, bound peptides were eluted in 25 μL of 5% acetic acid. Stable isotope labeled peptide (500 femtomoles) was added to each eluate and after Zip-Tipping, the light and heavy peptides were spotted onto MALDI plates. All samples were ionized using a fixed laser intensity of 3400 at 500 shots/spectrum.
One notable weakness of the MiSCREEN method is illustrated by some of the results shown in Table 2. SPR analysis of mAb 19-44, specific for peptide CPTAC-23c, showed that this mAb had a half off-time of 10.7 minutes and is probably useful for immuno-MS assays. However, this peptide was not detected by the MiSCREEN assay since the peptide does not ionize well by MALDI-TOF MS (shown in Figure 2). On the other hand, a useful and positive attribute of the MiSCREEN method was demonstrated with the impure peptide CPTAC-43c. This peptide has four cysteines and after synthesis contained a contaminating form as shown in Figure 1B. By SPR, one could not differentiate between antibodies that capture the `wrong' peptide and the ones that bind to the `correct' form of the peptide, whereas MiSCREEN analysis revealed that mAb 38-12 recognized the appropriate peptide (containing four S-carbamidomethyl cysteines) more readily than the `wrong' form of the peptide and is therefore likely a useful reagent for peptide enrichment.
Not surprisingly, we were also able to select high affinity recombinant RabMAbs (eg mAb 58-4 in Table 2) using the rabbit antibody capture system. This is now our preferred method for screening recombinant clones. In addition, high affinity murine mAbs (eg. mAbs 2A7 and BGN/KA/4H, see Table 2) were detected by substituting either protein G Dynabeads or anti-mouse IgG Dynabeads as capture agents, illustrating the general utility of the MiSCREEN method.
To further illustrate selection of anti-peptide mAbs by MiSCREEN, the results of screening assays for selection of hybridoma supernatants are shown in Figure 5. In one experiment, MiSCREEN was performed using a 10 minute duration of wash steps after peptide binding. The same supernatants were then tested by SPR for peptide binding. The results are shown in Figure 5A. Antibodies that had dissociation constants (kd) of less than 1.0E-03 s−1 (i.e. half off-time of 10 minutes or more) as determined by SPR retained more of the target peptide during the 10 minute MiSCREEN wash steps and thus yielded stronger MALDI signals. The 10 minute threshold we used was arbitrary and was chosen for this MiSCREEN assay to resemble the wash steps in SISCAPA assays (8–12 minutes). In another experiment (Figure 5B), the RabMAbs were washed for approximately 4 minutes after peptide binding. Antibodies that performed well in MiSCREEN had a kd of approximately 2–3E-03 s−1 (i.e. half off-time of approximately 4 minutes). These results demonstrate that for each target peptide, MiSCREEN selects anti-peptide antibodies with slow off-rates. It is important to note that there is not a perfect linear relationship between SPR and MiSCREEN - some antibodies have very similar off-rates (as determined by SPR) yet their performance in the MiSCREEN assay is different from one another, and vice versa. These differences may be attributed to the inherent differences in biophysical aspects of peptide capture and washing between these two assays, and highlights the importance of screening of mAbs in assays that resemble the assay of ultimate use as closely as possible, for example MiSCREEN and SISCAPA. Nevertheless, the strong overall trend towards increasing SPR off-rate with increasing MiSCREEN signal, with strongest MiSCREEN response observed above the SPR off-rate equivalent of the MiSCREEN wash time, supports a model in which antibody off-rate is the primary determinant of success in MiSCREEN and SISCAPA assays.
Figure 5.
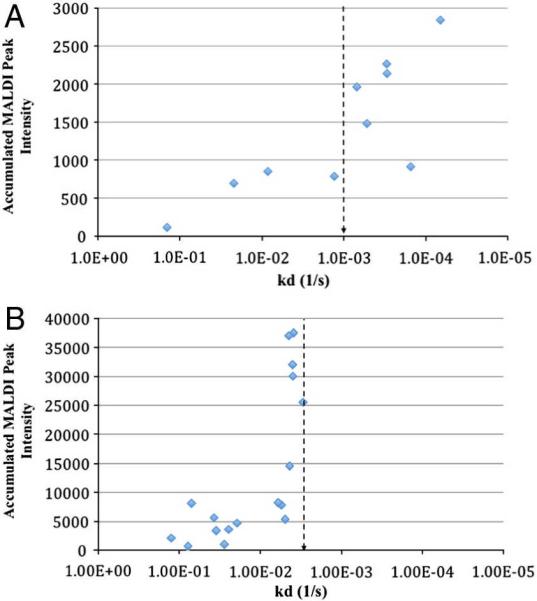
RabMAb selection by MiSCREEN and comparison to SPR. Panel A: Hybridoma supernatants containing RabMAbs specific for thyroglobulin peptide CPTAC-39d were screened by MiSCREEN using a wash time of 10 minutes. The same supernatants were also tested for peptide binding kinetics by SPR. The vertical axis represents the accumulated MALDI peak intensity for each RabMAb while the horizontal axis represents the dissociation constant (kd) for the same antibody. Antibodies that had a high enough affinity to retain the target peptide during the wash steps (approximately 10 mins; kd of ~1.0E-03) showed a high MALDI signal by MiSCREEN. Panel B: Hybridoma supernatants containing RabMAbs specific for thryoglobulin peptide CPTAC-39c were screened by MiSCREEN using a wash time of approximately 4 minutes (kd of ~3.5E-03). Antibodies with off-rates below this threshold performed weakly in the MiSCREEN assay while high affinity RabMAbs capable of retaining peptides beyond the wash period yielded a strong MALDI signal. The vertical lines in each panel represent the half off-times (10 minutes in Panel A and 4 minutes in Panel B).
It is also worth noting that by direct spotting of peptide eluates onto MALDI plates, we could avoid Zip-Tipping of samples. Although the peak intensities were reduced when only 2 μL of the 12 μL sample were used, signals were still easily detected for all peptides tested (data not shown). This is an important point since by avoiding sample concentration using C18 ZipTips, spotting of MALDI plates is more easily amenable to automation and the resultant increase in throughput.
3.5. Multiplexed binding of peptides by MiSCREEN
Two sets of multiplexing experiments were performed, each using different combinations of RabMAbs and peptides. Representative results from one of these experiments are shown in Figure 6. In this case, MALDI-TOF analysis showed that all five peptides were resolved after directly spotting the mixture onto a MALDI plate (Panel A). A mixture of four hybridoma supernatants containing RabMAbs specific for four of the peptides captured them, whereas the control peptide (for which no specific antibody was included) was not captured (Panel B). Indeed, the control peptide CPTAC-28d that ionizes well, was not detected at all (panel C) showing that specificity was retained by the multiplexed antibodies. Similar results were seen with all three replicates in this experiment. In addition, in a second set of multiplexing experiments also performed in triplicate, specific RabMAbs selectively enriched 3 (of 3) of their cognate peptides whereas the two control peptides were not captured. In this latter experiment we also used two control RabMAbs (of “wrong” specificity) and none of the 5 peptides were bound (data not shown). These experiments demonstrated the ability of the selected RabMAbs to pull peptide analytes out of solution (in this case, PBS) and show the specificity of the capture reagents.
Figure 6.

MiSCREEN Multiplexing. Panel A: The MALDI-TOF MS spectrum of a mixture of 5 peptides (1picomole each) that was directly spotted onto a MALDI plate. Peptide CPTAC-28d was added as a specificity control. Panel B: Forty microliters of each of four hybridoma supernatants containing RabMAbs specific for peptides CPTAC-14d, 36d, 38b and 39d were pooled and MiSCREEN was performed using this mixture. The spectrum shows that all four specific peptides were captured. Notably, the negative control peptide CPTAC-28d was not bound. Panel C. An expanded view of the spectrum shown in Panel B shows that that the control peptide was not present, even at background levels. This spectrum is representative of three independent experiments each performed with three technical replicates. Similar results were obtained in three independent experiments performed using a different peptide mixture as described in Materials and Methods (data not shown).
3.6. Detection of surrogate peptides in human plasma
All of the 8 RabMAbs that we tested were able to bind and enrich low femtomole levels of spiked heavy-labeled peptides out of trypsin digested human plasma. Three of the eight selected recombinant RabMAbs were also able to enrich “natural” (not spiked) cognate peptides out of 10 μl of trypsin digested human plasma to levels that were detectable by MALDI-TOF MS (results summarized in Table 3). Examples of the MALDI spectra showing specific peptide enrichment are shown in Figure 7. All surrogate peptides from higher abundance proteins were detected except one, CPTAC-14d, corresponding to ferritin light chain. In this case the surrogate peptide does not ionize well by MALDI, perhaps explaining why this peptide was not easily detected. This observation again illustrates that MiSCREEN may not be useful for all peptides, as occurs with electrospray methods for peptide detection. The SISCAPA peptide enrichment experiments were performed on three separate occasions. In each case we detected the anticipated masses for those surrogate peptides that we were able to enrich. Since peptide masses do not unequivocally identify the peptide, for the three most abundant protein targets, protein C inhibitor (PCI; also known as PAI-3), LPS binding protein and transferrin receptor, the identity of peptides were also verified by manual analysis of MS/MS spectra (data not shown). Although we detected correct m/z signals from three low abundance plasma proteins (CA-125, receptor tyrosine-protein kinase and thyroglobulin), we could not obtain accurate MS/MS analysis for these targets due to their weak MALDI signal (Figure 7 panels C and D). Therefore, we are hesitant to report unequivocal enrichment of these targets.
Table 3.
Detection of peptides in plasma digest1
| Protein Target | Peptide ID | Plasma concentratio n according to literature (ng/mL)2 | RabMAb ID | RabMAb half- off time (min)3 | Peptide detection limit (fmol)4 | Peptide enriched from plasma5 |
|---|---|---|---|---|---|---|
| Protein C Inhibitor (PCI) | PPI-4d | 5300 | 58–4 | ND | <10 | Yes |
| LPS Binding Protein | PPI-lc | 5000 | 109–4 | ND | <10 | Yes |
| Transferrin Receptor | PPI-6d | 3200 | 79–9 | ND | <10 | Yes |
| Ferritin light chain | CPTAC-14d | 50 | 10–58 | 51.3 | 10–50 | No |
| Receptor tyrosine-protein kinase | CPTAC-36d | 11.2 | 32–7 | 73.1 | <10 | Yes* |
| Mucin-16 (CA-125) | CPTAC-38b | 7.5 | 34–102 | 426.3 | <10 | Yes* |
| Thyroglobulin precursor | CPTAC-39c | 1 | 35–31 | 473.5 | <10 | Yes* |
| WAP four-disulfide core domain protein | CPTAC-43c | 1 | 38–12 | 194.8 | <10 | No |
No accurate MS/MS analysis due to low intensity signals on MALDI-TOF MS.
Trypsin digested human plasma
Values taken from published literature
Determined by SPR (See also Table 2)
Varying levels of heavy (Stable Isotope Standard) peptide were spotted directly onto the MALDI plate.
Enrichment of “natural” tryptic peptide from 10 microliters of plasma digest. One microgram of the relevant RabMAb was used to enrich the peptides.
Figure 7.

MALDI-TOF spectra of peptides enriched from human plasma digest using RabMAbs selected by MiSCREEN. For each protein target tested, one microgram of specific anti-peptide RabMAb was used to enrich the surrogate peptide of interest from 10 μL of trypsin-digested human plasma. The spectra on the left side of the figure show the presence of enriched peptides. Panel A: Surrogate peptide EDQYHYLLDR (1351.44 Daltons) from the parent protein PCI (also known as PAI-3). Panel B: Surrogate peptide GFVEPDHYVVVGAQR (1672.86 Daltons) from the parent protein transferrin receptor. Panel C: Surrogate peptide ELGPYTLDR (1063.54 Daltons) from the parent protein mucin-16 (CA-125). Panel D: Surrogate peptide FSPDDSAGASALLR (1406.69 Daltons) from the parent protein thyroglobulin. As shown on the right side of the figure, no peaks corresponding to the surrogate peptides were observed in the negative controls, which lack the specific RabMAb (i.e. Dynabeads incubated with 1/10 dilution of plasma digest to test for non-specific background binding of peptides).
To demonstrate that antibodies selected by MiSCREEN can be used for peptide (thus protein target) quantitation in human plasma digests, RabMAb 58-4, specific for peptide PPI-4d, a surrogate for protein C inhibitor (PCI) was used for peptide enrichment in experiments using stable isotope labeled peptide as internal standard. The results of a titration of labeled peptide in comparison with the constant amount of endogenous PPI-4d peptide are shown in Figure 8. The average amount of this peptide (calculated from the standard curve determined in duplicate at 8 different dilutions) was determined from this titration to be 225 fmol (1 μg/mL) in 10 μL of plasma. Given that the PPI-4d peptide is unique to protein C inhibitor, there should be a one-to-one ratio between peptide levels and protein levels in plasma if the tryptic digestion is complete and there is no degradation of the peptide prior to capture (the basic requirements of peptide-based MRM assays). If these conditions are met, the concentration of this protein in the test plasma sample is computed to be 1 mg/mL, in very close agreement with 0.83 μg/ml measured in the equivalent SISCAPA assay using the same antibody and sample but with triple-quadrupole MS quantitation (data not shown) and reasonably good agreement with the literature value from immunoassay of 5 μg/ml (Hortin et al., 2008). For absolute quantitation of target proteins using peptide surrogates in plasma, it is necessary that complete digestion of the analyte be achieved. Studies are underway in many laboratories to address this issue.
Figure 8.
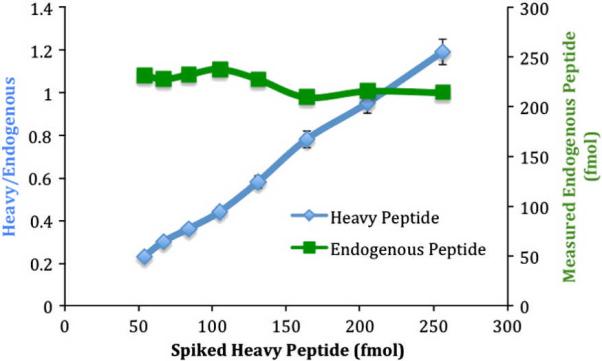
Quantitation of a protein C inhibitor (PCI) specific peptide in trypsin-digested human plasma. Stable isotope labeled (heavy) peptide with the same sequence as the endogenous PCI peptide was spiked into the reaction mixture at different levels. One μg of purified RabMAb 58-4 was used to capture both heavy and endogenous peptides from 10 mL of digested plasma. The captured peptides were eluted using 25 μL of 5% acetic acid and were analyzed by MALDI-TOF MS. The ratio of accumulated peak intensity of heavy:endogenous peptide was used to determine the amount of endogenous peptide. The coefficient of variation from 16 samples was determined to be 5.3%.
Conclusions
Although anti-peptide mAbs are made worldwide, very few laboratories or companies have established methods to select those that bind short (e.g., tryptic) peptides with high affinity. Such antibodies are required for several different immuno-MS assays used for measurement of protein biomarkers in complex peptide mixtures. Often ELISA assays using peptides coupled to carrier proteins are used for anti-peptide antibody screening, although such assays measure avidity rather than true affinity (binding of a single peptide epitope to a single antigen binding site of an antibody). In addition, we have often observed mAbs that bind, at least in part, to linker moieties used to couple peptides to carrier proteins used as antigens in ELISA. We were able to avoid selecting such antibodies in our study by developing a “peptide ELISA” in which free peptide antigens are dried onto the ELISA wells. These ELISAs, although not useful for measuring antibody affinity, were useful for initial selection of peptide-binding mAbs prior to further selection by MiSCREEN (this manuscript) designed to identify mAbs with high affinity, or more specifically, low off-rates.
Screening of anti-peptide mAbs using SPR has proven to be a useful technique for identification of high affinity anti-peptide antibodies (Pope et al., 2009) and yields a quantitative result capable of ranking antibody clones. However, this SPR protocol, with a cycle time of approximately 30–45 minutes, is too slow for processing large numbers of hybridoma supernatants. For this reason, we developed the MiSCREEN method that combines the same peptide enrichment procedures used in SISCAPA assays with robust MALDI-TOF MS for analysis of bound and eluted peptides to produce a semi-quantitative test of antibody off-rate. In MiSCREEN, both peptide enrichment and MALDI-TOF MS analysis of the bound-eluted peptides can be automated so that high throughput is possible. First, peptide capture is performed using a Kingfisher magnetic particle handling robot and second, since we have demonstrated that peptide eluates do not need to be desalted or concentrated (data not shown), MALDI plates can potentially be spotted with a liquid handling robot. We can now routinely screen 100 hybridoma supernatants in 4 hours. In addition, we have demonstrated that multiplexing of the MiSCREEN assay using a mixture of different peptides is possible. This facilitates the screening of large numbers of hybridomas for the presence of a variety of high affinity-peptide specific antibodies. Hybridoma supernatants can thus be screened for the presence of a different specific anti-peptide antibody using a single MiSCREEN peptide mixture. Alternatively, screening of antibodies from clones selected from recombinant libraries may be performed using multiple peptides in MiSCREEN assays.
We have shown that rabbit mAbs (including recombinant antibodies) and importantly, mouse mAbs, of sufficiently high affinity for use in peptide enrichment can be selected by MiSCREEN. High affinity murine mAbs (produced against four of the same peptides as RabMAbs) could also be selected, although these were more infrequently found (unpublished observations). Regardless of the species of origin, mAbs that bound peptides yielding high MALDI-TOF peak intensities (and low off-rates as determined by SPR) are clearly able to retain peptides long enough for use in immuno-MS methods such as SISCAPA under current configurations. Whether or not an absolute affinity ranking of different antibodies specific for a given peptide is possible remains to be determined. However, it is clear that MiSCREEN can discriminate qualitatively between those antibodies that will function in current immuno-MS assays and those that will not. Progress towards development of methods for quantitative prediction of peak intensities in MALDI-TOF MS (Timm et al., 2008), coupled with the use of stable isotope labeled internal standards should allow more quantitative multiplexed MiSCREEN applications as has been achieved with SISCAPA (Anderson et al., 2004; Whiteaker et al., 2009). This should allow accurate affinity ranking of antibodies by MiSCREEN thus facilitating selection of antibodies with desired off-rates for each specific application.
Since the association rates (ka; on-rate) that we observed for a given tryptic peptide binding to antibodies were roughly similar (generally within a 10-fold or smaller range) the dissociation rates (kd; off-rate) are the major determinants of overall antibody affinity (KD). For a chemically homogeneous mAb reagent, half the peptide dissociates in ln2/kd. To retain 75% or more of the captured peptide, and avoid losing assay sensitivity, a half off-time greater than twice the washing period is desirable. Current quantitative SISCAPA protocols involve serial washes over a total elapsed time of 8–12 minutes after removal of antibody-coated magnetic beads from the sample digest, suggesting that antibodies with half off-times of at least 10 minutes and ideally 20–30 minutes are desirable. Antibodies of even higher affinity (longer half off-times) allow extended washing and removal of non-specifically bound peptides. We have frequently selected antibodies with half off-times of several hours, yet despite such high affinities, we have so far not observed instances in which 5% acetic acid eluent fails to elute the bound peptides.
We believe that a method similar to the multiplexed MiSCREEN technique presented here can be developed as a quantitative assay for measuring several biomarkers simultaneously. Such assays, and the previously described SISCAPA assays depend on antibody quality. In the recent literature, there has been some skepticism about the prospect of using antibodies for proteomics from a “high content” approach to biomarker discovery and use (Zichi et al., 2008). The argument has been made that antibodies do not have the requisite specificity, that off-target proteins would interfere with detection and with multiplexing and that autoantibodies present in many people confound the problem. Many of these criticisms will hold true for top down proteomics approaches to measurement of intact proteins, as has been observed for standard sandwich immunoassays. Such criticisms do not apply to anti-peptide antibodies, however, since protein-protein interactions are destroyed during sample digestion to peptides, and we believe they should receive much more attention. It is important to emphasize that anti-peptide antibodies of high specificity and with low off-rates can be selected by SPR (Pope et al, 2009) or by MiSCREEN (this manuscript). We have shown that such antibodies are functional in SISCAPA assays, i.e. they are able to enrich natural (non-spiked) surrogate peptides from complex tryptic digests of human plasma. Three of eight RabMAbs that we tested were able to enrich their cognate peptides to detectable levels from the digest of 10 μl of plasma (Table 3). We also observed correct m/z signals for three low abundance biomarkers (CA-125, receptor tyrosine-protein kinase and thyroglobulin) although we were unable to verify their identity by MS/MS analysis due to weak MALDI signals. However, the utility of the selected antibodies for these targets in digested plasma was confirmed using spiked heavy labeled peptides and identification by triple quadrupole MS in which case low femtomole levels of the spiked peptides in tryptic plasma digest were detected (data not reported here). Enrichment of such low abundance endogenous peptides will possibly be achieved by increasing the amount of plasma used for immunocaptures.
The two targets that we could not detect were ferritin light chain and WAP four-disulfide core domain protein (HE-4). Although ferritin light chain is relatively abundant in human plasma (50 ng/mL), the surrogate peptide from this target did not ionize well by MALDI. Performing SISCAPA by measuring MRM transitions after electrospray ionization of captured peptides may solve this issue for many surrogate peptides that do not ionize well by MALDI (Whiteaker et al., 2009), although this is not an approach that is amenable to high throughput (the strength of MiSCREEN). The other peptide that we failed to sufficiently enrich to detectable levels, was a specific surrogate for WAP four-disulfide core domain protein (HE-4), a very low abundance molecule in human plasma. Perhaps this protein is not digested well with trypsin or the parent protein is at a level too low for detection. By performing capture from larger amounts of plasma digest this problem may be solved.
To show that antibodies selected by MiSCREEN are able to perform in assays for measurement of potential biomarker targets, a quantitiative MALDI-TOF assay based on stable isotope standards was demonstrated using protein C inhibitor as a model target. In addition, several of the selected RabMAbs reported in this study have also been used in SISCAPA assays using triple quadrupole MS for peptide identification and quantitation by monitoring MRM transitions (data not reported here). In these latter assays, unlike those employing top-down immuno-proteomics methods (coupled with MALDI-TOF MS analysis) or standard immunoassays, the analytes are absolutely identified by analysis of MRM transitions. Thus, even if off target binding occurs, this will not seriously interfere with measurement of the target peptide.
Derivation and affinity characterization of a large number of selected high affinity anti-peptide antibodies will be necessary for more ambitious approaches to studying the human proteome. One such strategy is to develop tools for measuring all human proteins by utilizing proteotypic peptides and anti-peptide antibodies specific for each of them. This has been described as the human proteome detection and quantitation (hPDQ) project (Anderson et al., 2009). The MiSCREEN method described here should aid in this endeavor, particularly since the method is amenable to automation, thus capable of high throughput.
Acknowledgements
We thank Immunoprecise Antibodies Ltd and Jennifer Reid for the gifts of anti-peptide mouse mAbs and Leanne Ohlund for helping us with the Applied Biosystem-MDS SCIEX 4800 MALDI-TOF/TOF instrument. The authors acknowledge support from the US National Cancer Institute's Clinical Proteomic Technology Assessment for Cancer program (grant U24-CA126476-01) and from the Canadian Institutes for Health Research (grant MOP 81267 to TWP). The Kingfisher magnetic particle processor was supplied by Anderson Forschung Group LLC (Washington DC).
Abbreviations
- MiSCREEN
MALDI immunoscreening
- mAbs
monoclonal antibodies
- MALDI-TOF
matrix-assisted laser desorption/ionization time-of-flight
- MS
mass spectrometry
- SPR
surface plasmon resonance
- SISCAPA
stable isotope standards and capture by anti-peptide antibodies
- hPDQ
human proteome detection and quantitation project
- MRM
multiple reaction monitoring
- HPLC
high performance liquid chromatography
- KLH
keyhole limpet hemocyanin
- ELISA
enzyme linked immunosorbent assay
- RabMAbs
rabbit monoclonal antibodies
- PBS
phosphate buffered saline
- CHAPS
3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate
- KD
equilibrium dissociation constant
- ka
association rate constant
- kd
dissociation rate constant
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA) J. Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- Anderson NL, Anderson NG, Pearson TW, Borchers CH, Paulovich AG, Patterson SD, Gillette M, Aebersold R, Carr SA. A human proteome detection and quantitation project. Mol Cell Proteomics. 2009;8:883–886. doi: 10.1074/mcp.R800015-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortin GL, Sviridov D, Anderson NL. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clinical Chemistry. 2008;54:1608–1616. doi: 10.1373/clinchem.2008.108175. [DOI] [PubMed] [Google Scholar]
- Jiang J, Parker CE, Fuller JR, Kawula TH, Borchers CH. An immunoaffinity tandem mass spectrometry (iMALDI) assay for detection of Francisella tularensis. Analytica Chimica Acta. 2007;605:70–79. doi: 10.1016/j.aca.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RW, Krone JR, Bieber AL, Williams P. Mass spectrometric immunoassay. Anal. Chem. 1995;67:1153–1158. doi: 10.1021/ac00103a003. [DOI] [PubMed] [Google Scholar]
- Pope ME, Soste MV, Eyford BA, Anderson NL, Pearson TW. Anti-peptide antibody screening: Selection of high affinity monoclonal reagents by a refined surface plasmon resonance technique. J. Immunol. Methods. 2009;341:86–96. doi: 10.1016/j.jim.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Raska CS, Parker CE, Sunnarborg SW, Pope RM, Lee DC, Glish GL, Borchers CH. Rapid and sensitive identification of epitope-containing peptides by direct matrix-assisted laser desorption/ionization tandem mass spectrometry of peptides affinity-bound to antibody beads. J. Am. Soc. Mass Spectrom. 2003;14:1076–1085. doi: 10.1016/S1044-0305(03)00405-7. [DOI] [PubMed] [Google Scholar]
- Sza'jli E, Fehe'r T, Medzihradszky KF. Investigating the quantitative nature of MALDI-TOF MS. Mol Cell Proteomics. 2008;7:2410–2418. doi: 10.1074/mcp.M800108-MCP200. [DOI] [PubMed] [Google Scholar]
- Spieker-Polet H, Sethupathi P, Yam PC, Knight KL. Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit-rabbit hybridomas. Proc. Natl. Acad. Sci. (USA) 1995;92:9348–9352. doi: 10.1073/pnas.92.20.9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolson DL, Turco SJ, Beecroft RP, Pearson TW. The immunochemical structure and surface arrangement of Leishmania donovani lipophosphoglycan determined using monoclonal antibodies. Mol. Biochem. Parasitol. 1989;35:109–118. doi: 10.1016/0166-6851(89)90113-8. [DOI] [PubMed] [Google Scholar]
- Tholey A, Heinzle E. Ionic (liquid) matrices for matrix-assisted laser desorption/ionization mass spectrometry-applications and perspectives. Anal. Bioanal. Chem. 2006;386:24–37. doi: 10.1007/s00216-006-0600-5. [DOI] [PubMed] [Google Scholar]
- Timm W, Scherbart A, Böcker S, Kohlbacher O, Nattkemper TW. Peak intensity prediction in MALDI-TOF mass spectrometry: a machine learning study to support quantitative proteomics. BMC Bioinformatics. 2008;35:443–461. doi: 10.1186/1471-2105-9-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker JR, Zhao L, Anderson NL, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics. 2009 Oct 20; doi: 10.1074/mcp.M900254-MCP200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabet-Moghaddam M, Heinzle E, Tholey A. Qualitative and quantitative analysis of low molecular weight compounds by ultraviolet matrix-assisted laser desorption/ionization mass spectrometry using ionic liquid matrices. Rapid Commun. Mass Spectrom. 2004;18:141–148. doi: 10.1002/rcm.1293. [DOI] [PubMed] [Google Scholar]
- Zichi D, Eaton B, Singer B, Gold L. Proteomics and diagnostics: Let's Get Specific, again. Curr. Opin. Chem. Biol. 2008;12:78–85. doi: 10.1016/j.cbpa.2008.01.016. [DOI] [PubMed] [Google Scholar]